Introduction
Breast cancer is a leading cause of death in women
worldwide (1). Tamoxifen, a
selective estrogen receptor modulator (SERM), is one of the most
effective endocrine therapy for estrogen receptor (ER)-positive
breast cancer (2). However,
approximately 30–40% of ER-positive breast cancer patients do not
respond to tamoxifen endocrine therapy, and moreover, tumors that
initially respond to tamoxifen treatment develop resistance to this
drug over time (3,4). The mechanisms underlying tamoxifen
resistance are complex and remain unclear, although loss of ER
expression or dysregulation of ER co-regulators, and activation of
many kinases such as the receptor tyrosine kinase have been found
to contribute to tamoxifen resistance (5).
Enhancer of zeste homologue 2 (EZH2), as a histone
methyltransferase, is a catalytic subunit of polycomb repressive
complex 2 (PRC2) which induces transcriptional inhibition through
the tri-methylation of lysine residue 27 on histone H3 (H3K27m3)
(6). Abnormal activities of DNA
methyltransferases lead to epigenetic changes of many genes that
contribute to carcinogenesis (7).
EZH2 plays an important role in many cellular processes such as
cell cycle regulation, proliferation, apoptosis, tumorigenesis, and
drug resistance (8–10). EZH2 has been found to be
overexpressed in a wide range of tumors such as osteosarcoma
(11), breast cancer (12), and prostate cancer (13). EZH2 overexpression is associated
with poor clinical outcomes (14,15).
Inhibition of EZH2 may represent a promising therapeutic strategy
for anticancer treatment (16).
Breast cancer is a heterogeneous disease that
includes different subtypes defined by ER, progesterone receptor
(PR), and human epidermal growth factor receptor 2 (HER2). In
addition, Jang et al (17)
reported that high EZH2 protein expression was associated with poor
survival in patients with Luminal A breast cancer. Moreover, high
EZH2 expression has been reported to be associated with unfavorable
outcome in the ER-positive breast cancer patients following
tamoxifen treatment (18).
Therefore, it appears that EZH2 overexpression may contribute to
tamoxifen resistance in ER-positive breast cancer. However, the
role of EZH2 in tamoxifen resistance has not been investigated
yet.
In the present study, we established a tamoxifen
resistant MCF-7 breast cancer cell line, and investigated the role
of EZH2 in tamoxifen resistance. The purpose of this study was to
explore the mechanisms by which EZH2 mediated tamoxifen resistance
in breast cancer cells.
Materials and methods
Cell culture
The human breast cancer cell line MCF-7 was obtained
from the American Type Culture Collection (ATCC, Rockville, MD,
USA). MCF-7 cells were cultured in Dulbecco's Modified Eagle Medium
(DMEM; Biological Industries, Beit-Haemek, Israel) supplemented
with 10% fetal bovine serum (FBS; Biological Industries), 100 IU/ml
penicillin, and 100 IU/ml streptomycin (Beyotime, Shanghai,
China).
MCF-7 tamoxifen resistant (MCF-7 TamR) cells were
selected from MCF-7 parental cells after treatment with
4-hydroxytamoxifen (4-OH TAM; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) for 10 months as previously reported (19) with some modifications. Briefly,
MCF-7 cells were cultured in the phenol red-free RPMI 1640 medium
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA). Cells
were treated with 1 µM 4-OH TAM for 21 days, followed by incubation
with TAM-free medium for 7 days. Survived cells were diluted to
obtain the monoclonal cells, which were further cultured in 1 µM
4-OH TAM for 10 months. MCF-7 parental cells grown in the RPMI 1640
medium with 0.1% ethanol (vehicle) for 10 months were used as
control cells. All cultures were maintained in a 5% CO2,
37°C, and 95% humidity cell culture incubator.
Cell viability assays
Cell viability was assessed using the 3-(4,
5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT)
assay. Briefly, cells were plated into 96-well plates at a density
of 500 cells/well. Cells were cultured for 24 h, and then were
treated with different concentrations of 4-OH TAM (0–18 µM) for 2
days. MTT solution (20 µl; Solarbio, Beijing, China) was added to
each well at a final concentration of 0.05 mg/l and incubated for 4
h. After MTT solution was removed, 150 µl DMSO was added to each
well, and mixed carefully. The plate was read at a wavelength of
570 nm in a microplate reader (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). All experiments were performed in triplicated
and repeated at least three times.
RNA isolation and RT-qPCR assays
Total RNA was isolated from cells using Trizol
reagent (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA,
USA). Complementary DNA was synthesized with 1 µg of total RNA in a
10 µl of a reaction mixture (Promega Corporation, Madison, WI, USA)
according to the manufacturer's instruction. Quantitative RT-PCR
was performed using SYBR Green real-time qPCR kit (Toyobo Life
Science, Osaka, Japan) in the Agilent Technologies Stratagene
Mx3000P (Agilent Technologies, Inc., Santa Clara, CA, USA). The
primer sequences were as follows: 5′-AGGACGGCTCCTCTAACCAT-3′
(sense) and 5′-CTTGGTGTTGCACTGTGCTT-3′ (antisense) for EZH2; and
5′-TGACGTGGACATCCGCAAAG-3′ (sense) and 5′-CTGGAAGGTGGACAGCGAGG-3′
(antisense) for β-actin. The PCR amplification conditions were 10
min at 95°C, followed by 40 cycles at 94°C for 15 sec, 60°C for 45
sec and 72°C for 20 sec. Quantitative RT-PCR assays were conducted
on a MxPro-Mx3000P (Standalone) Comparative Quantitation (Agilent
Technologies, Inc.). All quantitative RT-PCRs were performed in
triplicate.
Plasmid construction and
transfection
The human genomic cDNA of the EZH2 (NM_004456.4) was
amplified by PCR and was subcloned into the XhoI/KpnI site of
pcDNA3.1 (+) vector (cat. no. V79020; Invitrogen; Thermo Fisher
Scientific, Inc.). The primer sequences were shown in Table I. The construct of the EZH2
expression vector was confirmed by DNA sequencing. MCF-7 cells were
transfected with the EZH2 expression vector or control vector using
Lipofectamine 2000 Transfection Reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions. The
transfection efficiency was confirmed by western blot analysis.
After transfection for 72 h, cells were used for MTT assay, western
blot analysis, and flow cytometry.
 | Table I.Primer sequences used in the present
study. |
Table I.
Primer sequences used in the present
study.
| Oligo name | Primer
sequence | Product size
(bp) |
|---|
| EZH2-F |
5′-TTTAAACTTAAGCTTGGTACCATGGGCCAGACTGGGAAG-3′ | 2,256 |
| EZH2-R |
5′-AACGGGCCCTCTAGACTCGAGTCAAGGGATTTCCATTTCTC-3′ |
|
| p16 MF |
5′-TTATTAGAGGGTGGGGCGGATCGC-3′ | 150 |
| p16 MR |
5′-GACCCCGAACCGCGACCGTAA-3′ |
|
| P16 UF |
5′-TTATTAGAGGGTGGGGTGGATTGT-3′ | 151 |
| P16 UR |
5′-CAACCCCAAACCACAACCATAA-3′ |
|
siRNA transfection
Cells were seeded into six-well plates at a density
of 1.5×105 cells/well. Then, siRNAs against EZH2 (target
sequence: 5′-CAGACGAGCTGATGAAGTAAA-3′; cat. no. SI00063966; Qiagen
GmbH, Hilden, Germany) or negative control siRNAs (target sequence:
5′-AATTCTCCGAACGTGTCACGT-3′; cat. no. 1022076; Qiagen, Hilden,
Germany) (20) were transfected
into cells using Lipofectamine 2000 Transfection Reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's recommendations. The transfection efficiency was
confirmed by western blot analysis. After transfection, cells were
used for MTT assay, western blot analysis, and flow cytometry.
Western blot analysis
Cells were homogenized in ice-cold RIPA lysis buffer
(Beyotime, Shanghai, China) supplemented with 1 mM
phenylmethylsulfonyl fluoride (PMSF) (Solarbio) as previously
described (21). Protein
concentrations were determined using Bicinchoninic Acid Kit
(Beyotime). Then, equal amounts of protein (10–20 µg) were
separated by 12% SDS-polyacrylamide gel electrophoresis (SDS-PAGE)
and electrically transferred to PVDF membranes (GE Healthcare,
Chicago, IL, USA). The membranes were blocked with 5% fat-free milk
followed by incubation with primary antibodies against EZH2 (mouse
anti-human polyclonal antibody, dilution 1:2,500; cat. no.
ab168764; Abcam, Cambridge, MA, USA), cyclin D1 (rabbit anti-human
polyclonal antibody, dilution 1:1,000; cat. no. AB32262; Absci,
Vancouver, WA, USA), p16ink4a (rabbit anti-human
polyclonal antibody, dilution 1:1,000; cat. no. 10883-1-AP; Wuhan
Sanying Biotechnology, Wuhan, China), ER (rabbit anti-human
polyclonal antibody, dilution 1:500; cat. no. 21244-1-AP; Wuhan
Sanying Biotechnology), phosphorylated AKT (rabbit anti-human
monoclonal antibody, dilution 1:2,000; cat. no. 4060; Cell
Signaling Technology, Inc., Danvers, MA, USA), AKT (mouse
anti-human monoclonal antibody, dilution 1:2,000; cat. no. 2920;
Cell Signaling Technology, Inc.), phosphorylated ERK1/2 (rabbit
anti-human monoclonal antibody, dilution 1:2,000; cat. no. 4370;
Cell Signaling Technology, Inc.), or ERK1/2 (rabbit anti-human
monoclonal antibody, dilution 1:1,000; cat. no. 4695; Cell
Signaling Technology, Inc.) at 4°C overnight. The membranes were
then incubated with horseradish peroxidase-linked goat
anti-rabbit/mouse secondary antibody (dilution 1:5,000, OriGene
Technologies, Inc., Beijing, China) at room temperature for 1.5 h.
β-actin (mouse anti-human monoclonal antibody, dilution 1:1,000;
cat. no. TA-09; OriGene Technologies, Inc.) was used as a loading
control. Bands were visualized using a chemiluminescence detection
system, and was analyzed using the Tanon Gis software (Tanon,
China).
Flow cytometry
Cell cycle analyses were performed by flow
cytometry. Briefly, after transfection with pcDNA3.1-EZH2 or
control pcDNA3.1 as well as siEZH2 or control siRNAs for 72 h,
MCF-7 TamR and parental cells were seeded into six-well plates and
treated with 4-OH TAM (8 µM) for 48 h. Then, cells were collected,
washed, fixed with 75% ethanol at −20°C for 24 h. Cells stained
with the propidium iodide (PI) at a final concentration of 10 µl/ml
for 30 min in the dark. Data acquisitions were performed using a
flow cytometer (BD FACSCalibur™; BD Biosciences, Franklin Lakes,
NJ, USA). The percentage of cells in the G1, S, and G2 phases was
analyzed. All experiments were repeated at least three times.
Methylation analysis by
methylation-specific PCR (MSP)
After transfection with pcDNA3.1-EZH2 or control
pcDNA3.1 as well as siEZH2 or control siRNAs for 72 h, MCF-7 TamR
and parental cells were treated with 4-OH TAM (8 µM) for 48 h.
Total DNA was extracted from cells using the cell/tissue genomic
DNA extraction kit (Beijing Transgen Biotech Co., Ltd., Beijing,
China) according to the manufacturer's instructions. DNA (1 µg) was
bisulfite-treated with the CpGenome™ DNA modification
kit (EMD Millipore, Billerica, MA, USA) according to the
manufacturer's protocol. Methylation-specific PCR was carried out
to investigate the methylation status of the p16 gene. PCR primers
specific to unmethylated and methylated bisulfite-modified DNA
(22) are shown in Table I. PCR reactions were performed as
follows: 95°C for 10 min, 94°C for 30 sec, annealing at 62°C for 30
sec, and extension at 72°C for 30 sec; a total of 40 cycles;
followed by a final extension at 72°C for 10 min. PCR products were
separated by 2% gel electrophoresis, and the density of the
methylated band (M) or the unmethylated (U) bands were used to
assess the methylation levels of p16. Results from triplicate
experiments were used to determine methylation status.
Statistical analysis
Data analyses were performed using the SPSS 16.0
software package (SPSS, Inc., Chicago, IL, USA). Quantitative data
are expressed as mean ± SEM. Student's t test was used to compare
differences among groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
EZH2 is overexpressed in MCF-7 TamR
cells
We generated MCF-7 TamR cells by treating MCF-7
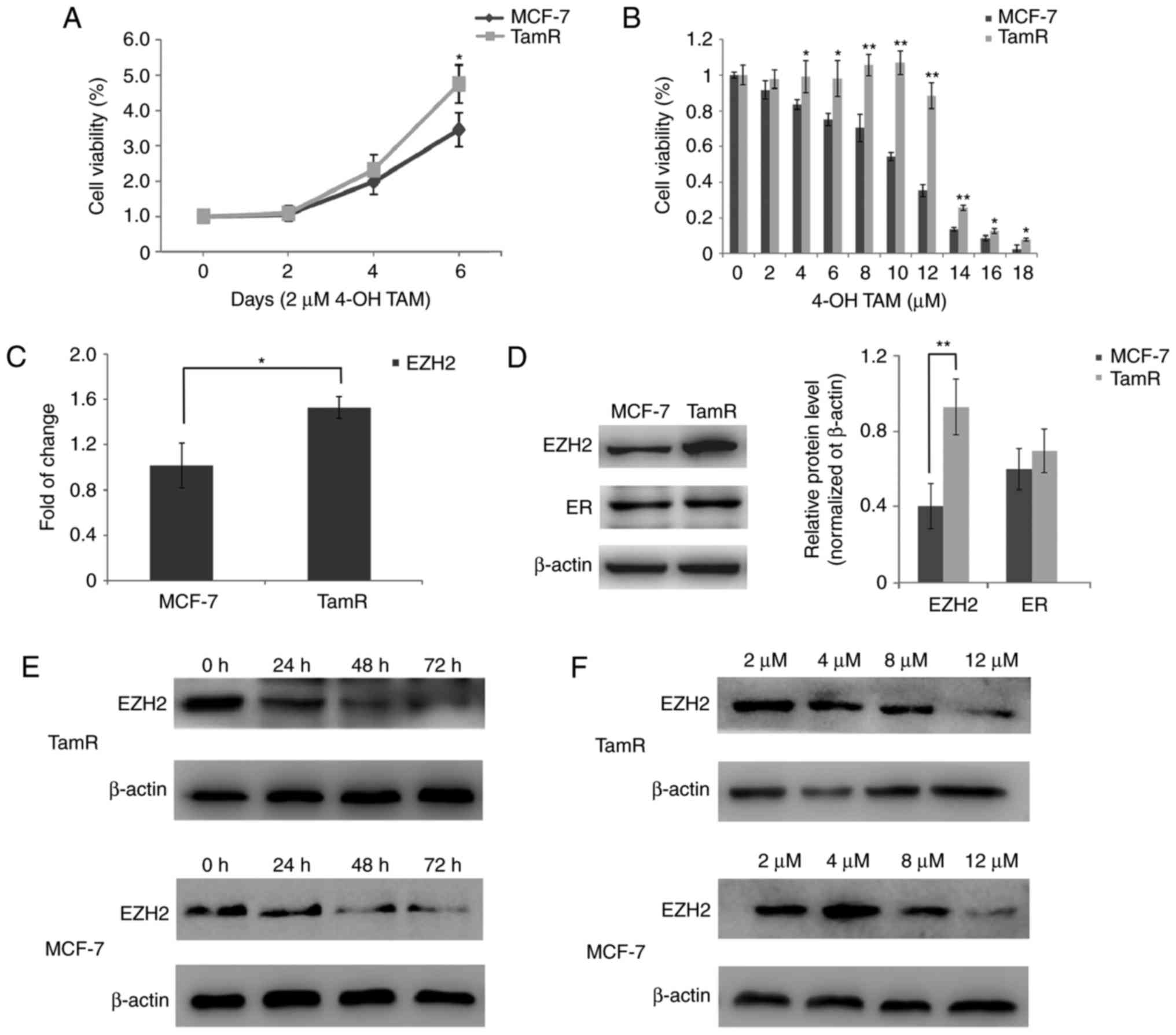
parental cells with 4-OH TAM for 10 months. MTT assays showed that
in the presence of 2 µM 4-OH TAM, cell viability of both MCF-7
parental and TamR cells was increased in a time-dependent manner,
and the growth was faster for MCF-7 TamR cells compared with their
parental cells. At 6 days after 2 µM 4-OH TAM treatment, cell
viability of MCF-7 parental cells was significantly lower compared
with MCF-7 TamR cells (P<0.05; Fig.
1A). In addition, in the presence of 4-OH TAM, cell viability
of MCF-7 parental cells was decreased in a concentration-dependent
manner, and cell viability of MCF-7 TamR cells was significantly
higher than that of MCF-7 parental cells at concentrations ≥4 µM
4-OH TAM for 2 days (P<0.05; Fig.
1B). These results indicated that MCF-7 TamR cells were
resistant to tamoxifen.
We further investigated EZH2 expression in MCF-7
TamR cells. Quantitative RT-PCR results showed that the EZH2 mRNA
expression was significantly higher in MCF-7 TamR cells than in
MCF-7 parental cells (P<0.05; Fig.
1C). Western blot analysis showed that EZH2 expression was
significantly increased in MCF-7 TamR cells compared with MCF-7
parental cells (P<0.01; Fig.
1D). The treatment of 4-OH TAM decreased the EZH2 expression
levels in a dose- and time-dependent manner (Fig. 1E and F). We also examined the ER
expression levels in MCF-7 TamR cells and their parental cells, and
found that there was not significant different from these two cell
lines (Fig. 1D).
EZH2 overexpression decreases
tamoxifen sensitivity of MCF-7 cells
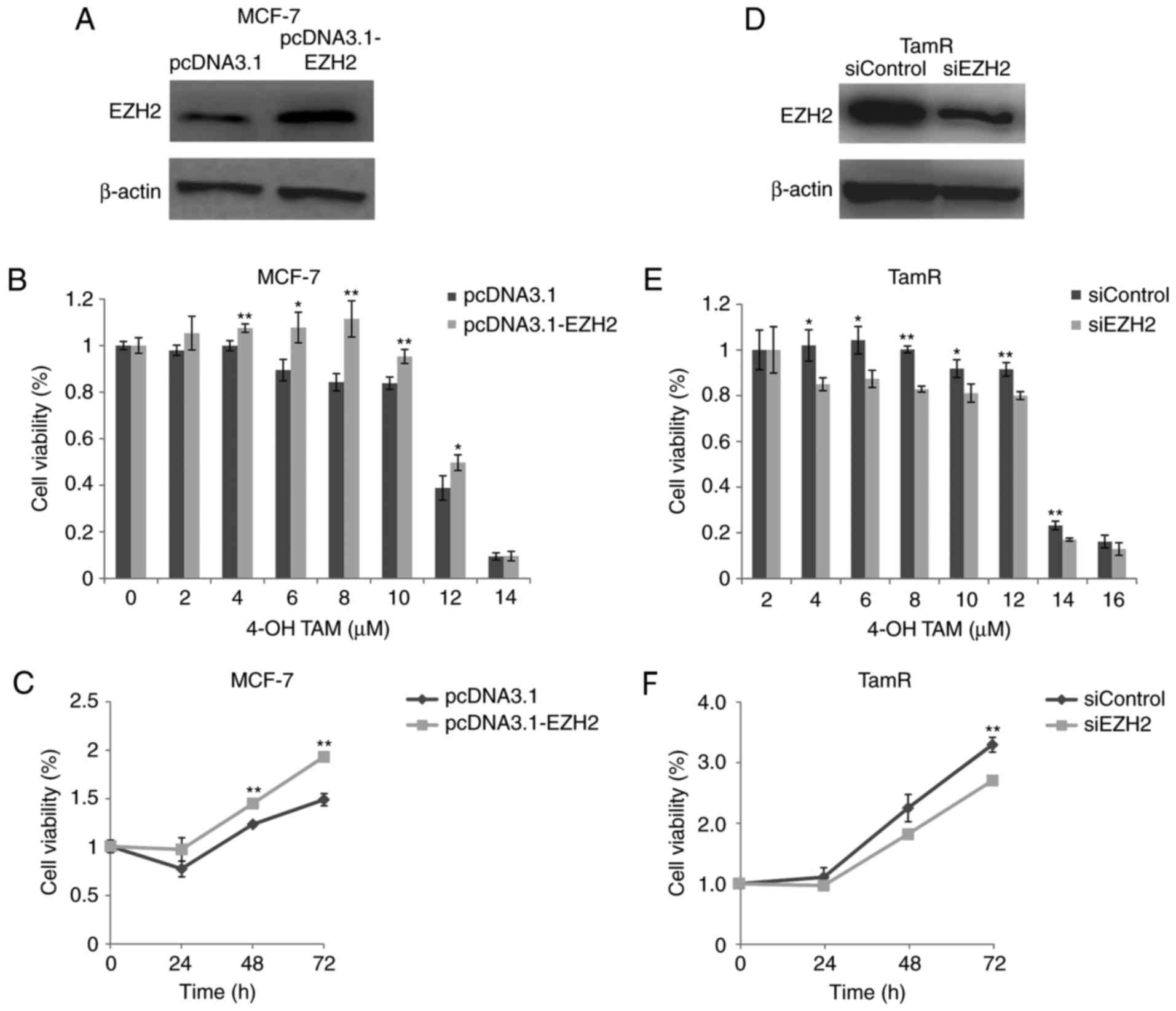
We then investigated the role of EZH2 in tamoxifen
sensitivity by transfecting MCF-7 parental cells with pcDNA3.1-EZH2
expression vectors. Western blot analysis showed that EZH2
expression was significantly increased in MCF-7 parental cells
transfected with pcDNA3.1-EZH2 vectors (Fig. 2A). MTT assays showed that cell
viability of EZH2-overexpressing MCF-7 cells was significantly
increased in the presence of 4-OH TAM (≥4 µM) compared with control
cells (P<0.05; Fig. 2B). In
addition, cell viability of EZH2-overexpressing MCF-7 cells was
significantly increased after 8 µM 4-OH TAM treatment for 48–72 h
compared with control cells (P<0.01; Fig. 2C). These results suggested that
EZH2 overexpression decreased tamoxifen sensitivity of MCF-7
cells.
EZH2 knockdown increases tamoxifen
sensitivity of MCF-7 TamR cells
We further investigated whether EZH2 inhibition
increased the sensitivity of MCF-7 TamR cells to tamoxifen, using
siRNAs against EZH2. Western blot analysis showed that EZH2
expression was decreased in MCF-7 TamR cells transfected with
EZH2-siRNAs (Fig. 2D). MTT assays
showed that cell viability of MCF-7 TamR cells treated with
EZH2-siRNAs was significantly decreased in the presence of 4-OH TAM
compared with control cells (P<0.05; Fig. 2E). Cell viability of MCF-7 TamR
cells treated with EZH2-siRNAs was significantly decreased after 8
µM 4-OH TAM treatment for 72 h compared with control cells
(P<0.01; Fig. 2F). These
findings suggested that EZH2 knockdown increased the sensitivity of
MCF-7 TamR cells to 4-OH TAM.
EZH2 knockdown induces cell cycle
arrest in MCF-7 TamR cells
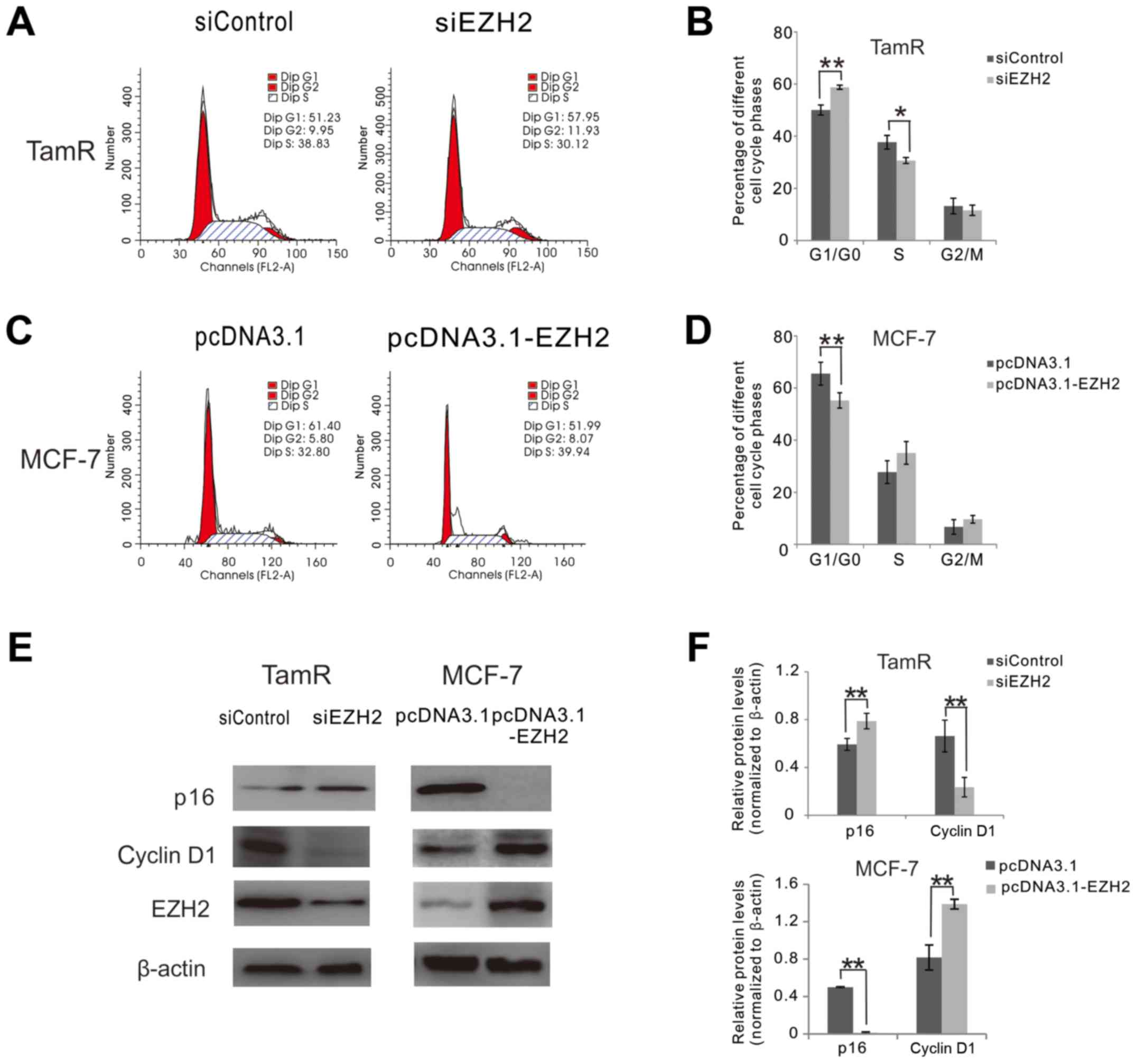
To understand the role of EZH2 in cell growth of
MCF-7 TamR cells, we performed cell-cycle analysis in MCF-7 TamR
cells treated with EZH2-siRNAs. Flow cytometry results showed that
the percentage of cells in the G0/G1 phase was significantly
increased and the percentage of cells in the S phase was
significantly decreased in MCF-7 TamR cells treated with
EZH2-siRNAs compared with control cells (P<0.05; Fig. 3A and B), suggesting that EZH2
knockdown induced cell cycle arrest in MCF-7 TamR cells.
We then examined the role of EZH2 in cell cycle of
MCF-7 parental cells by overexpressing pcDNA3.1-EZH2 vectors. Flow
cytometry results showed that the percentage of cells in the G0/G1
phase was significantly decreased in EZH2-overexpressing MCF-7
cells compared with control cells (P<0.01; Fig. 3C and D). These results further
suggested that EZH2 overexpression promoted cell cycle progression
in MCF-7 cells.
Since cell cycle progression is promoted by
cyclin-dependent kinases (CDKs) such as cyclin D1 and inhibited by
CDK inhibitor p16, we then investigated the role of EZH2 in the
expression of cyclin D1 and p16 in MCF-7 TamR cells. Western blot
analysis showed that EZH2 knockdown significantly reduced cyclin D1
expression and increased p16 expression in MCF-7 TamR cells
(P<0.01; Fig. 3E and F). In
contrast, EZH2 overexpression increased cyclin D1 expression and
decreased p16 expression in MCF-7 parental cells (P<0.01;
Fig. 3E and F).
EZH2 knockdown decreases p16 gene
methylation in MCF-7 TamR cells
We further investigated the role of EZH2 in p16
methylation in MCF-7 TamR cells and their parental cells, using
methylation-specific PCR. As shown in Fig. 4A, EZH2 knockdown significantly
decreased the DNA methylation level, but increased the
unmethylation level of p16 in MCF-7 TamR cells (P<0.05; Fig. 4A). In contrast, EZH2 overexpression
significantly increased the DNA methylation level, but decreased
unmethylation level of p16 in MCF-7 cells (P<0.05; Fig. 4B).
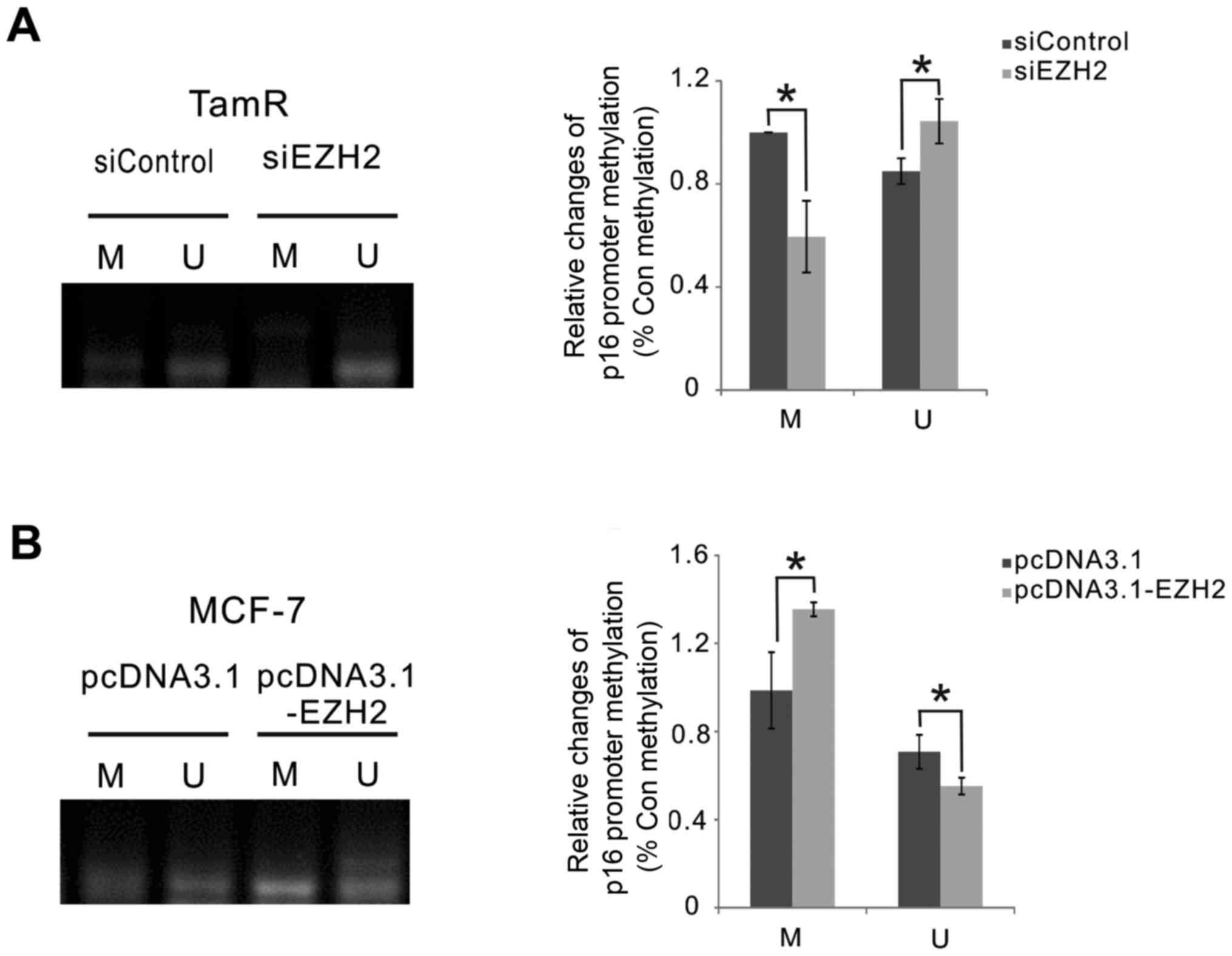 | Figure 4.EZH2 affected the methylation of p16
in MCF-7 TamR and their parental cells. (A) MCF-7 TamR cells were
transfected with siRNAs against EZH2 or control siRNAs for 72 h,
and were then treated with 8 µM 4-OH TAM for 48 h. MSP analysis was
performed to detect the p16 promoter methylation. (B) MCF-7 cells
were transfected with pcDNA3.1-EZH2 expression vector or pcDNA3.1
vector for 72 h, and were then treated with 8 µM 4-OH TAM for 48 h.
MSP analysis was performed to detect the p16 promoter methylation
(n=3). *P<0.05, as indicated. EZH2, enhancer of zeste homologue
2; 4-OH TAM, 4-hydroxytamoxifen; TamR, tamoxifen resistant; siRNA,
small interfering RNA; MSP, methylation-specific polymerase chain
reaction; U, unmethylated; M, methylated. |
The AKT and ERK signaling pathways are
involved in EZH2 expression in MCF-7 TamR cells
It has been reported that AKT and ERK signaling
pathway regulates EZH2 expression in breast cancer (12,23,24).
We then examined the expression of phosphorylated AKT, AKT,
phosphorylated ERK1/2 and ERK1/2 in MCF-7 parental and TamR cells.
Western blot analysis showed that the expression level of
phosphorylated AKT, AKT, phosphorylated ERK1/2 and ERK1/2 was
significantly lower in MCF-7 TamR cells compared with MCF-7
parental cells (P<0.01; Fig. 5A and
B). These results suggested that the AKT and ERK signaling
pathways are involved in EZH2 expression in MCF-7 TamR cells.
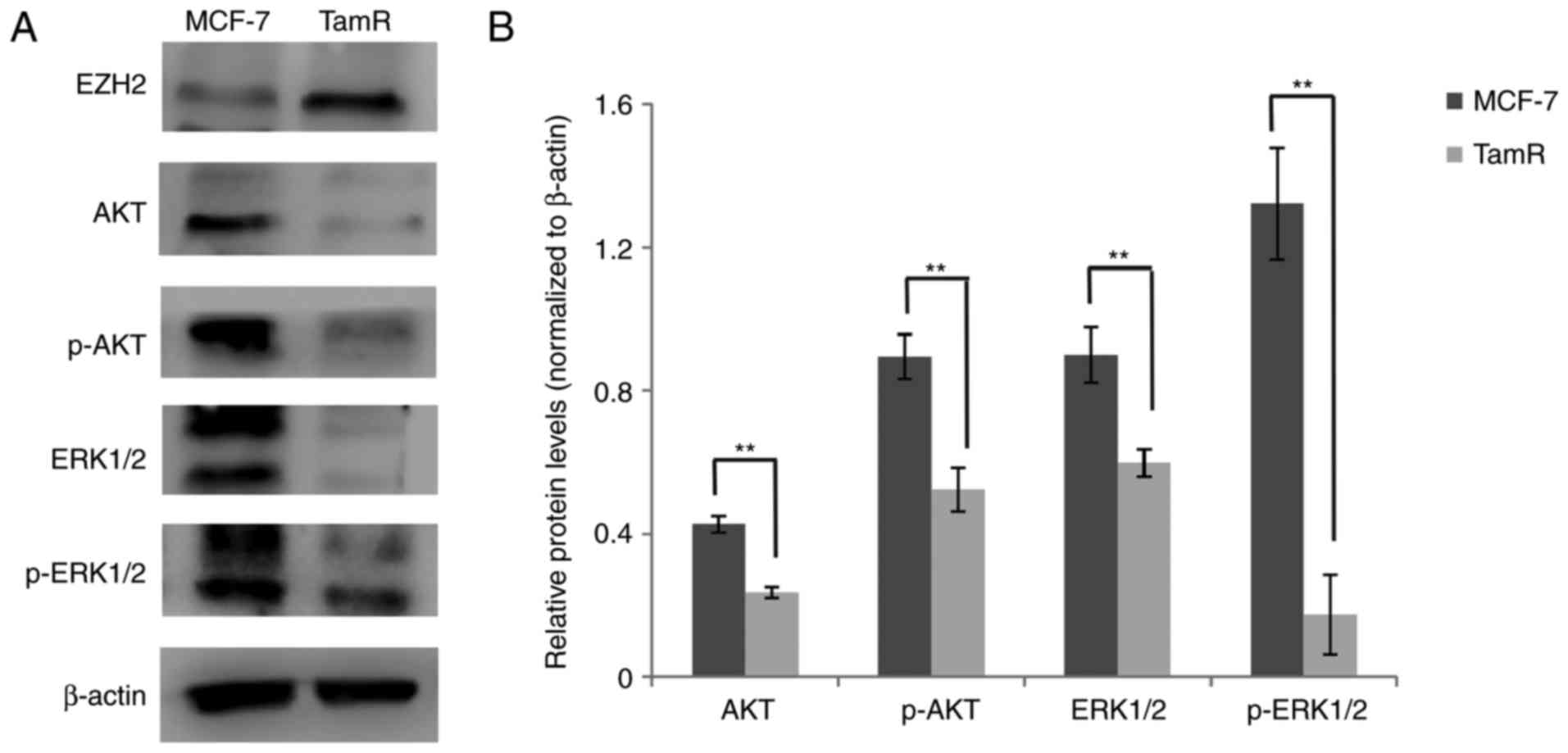 | Figure 5.The expression of p-AKT, AKT,
p-ERK1/2 and ERK1/2 in MCF-7 parental and TamR cells. (A) Western
blot analysis revealing the protein expression of EZH2, AKT, p-AKT,
ERK1/2 and p-ERK1/2 in MCF-7 TamR and their parental cells. β-actin
was used as the loading control. (B) Quantification of AKT, p-AKT,
ERK1/2 and p-ERK1/2 expression (n=3). **P<0.01, as indicated.
EZH2, enhancer of zeste homologue 2; TamR, tamoxifen resistant; p-,
phosphorylated; AKT, protein kinase B; ERK, extracellular
signal-regulated kinase. |
Discussion
In the present study, we examined EZH2 expression in
MCF-7 TamR cells, and found that EZH2 expression was significantly
increased in MCF-7 TamR cells compared with parental control cells.
We further found that EZH2 overexpression decreased the sensitivity
of MCF-7 cells to tamoxifen, and EZH2 knockdown increased the
sensitivity of MCF-7 TamR cells. Furthermore, we found that EZH2
knockdown induced cell cycle arrest in MCF-7 TamR cells by
decreasing Cyclin D1 expression and increasing p16 expression.
Moreover, EZH2 knockdown reduced p16 gene methylation in MCF-7 TamR
cells. Our findings suggest that EZH2 overexpression contributes to
tamoxifen resistance in breast cancer, and EZH2 inhibition may
reverse tamoxifen resistance in breast cancer by regulating cell
cycle via the demethylation of the p16 gene.
EZH2 overexpression has been found in many tumors
including breast cancer (12).
However, the mechanisms underlying EZH2 overexpression in breast
cancer are not fully understood. It has been reported that the AKT
and MEK/ERK signaling pathways promoted EZH2 overexpression in
breast cancer (12,23,24).
In the present study, we found that EZH2 was overexpressed in MCF-7
TamR cells. However, the mechanisms underlying EZH2 overexpression
in TamR cells remain unclear. It has been known that AKT and
MEK/ERK signaling activation contributes to tamoxifen resistance in
breast cancer (25,26). We found that the expression of AKT,
p-AKT, ERK1/2, p-ERK1/2 significantly reduced in MCF-7 TamR cells
than their parental cells. Therefore, it appears that AKT and
MEK/ERK signaling may be responsible for EZH2 overexpression in
TamR cells. Further studies are warranted to investigate the
signaling mechanisms underlying EZH2 overexpression in TamR breast
cancer cells.
Cell cycle regulators such as cyclins, CDKs and CDK
inhibitors play an important role in regulation of cell cycle
progression and have been demonstrated to be associated with
tamoxifen resistance (26). Cyclin
D1 promotes cell cycle progression to the S phase, and has been
found to be upregulated in tamoxifen-resistant breast cancer cells
(27). Cyclin D1 overexpression is
associated with poor clinical outcomes in breast cancer patients
following tamoxifen treatment (28,29).
It has been reported that cyclin D1 expression is necessary for
proliferation of tamoxifen-resistant breast cancer cells (30). In the present study, we found that
EZH2 knockdown increased the sensitivity of MCF-7 TamR cells to
tamoxifen, and induced cell cycle arrest in MCF-7 TamR cells by
inhibiting cyclin D1 expression. In addition, EZH2 overexpression
increased cyclin D1 expression in MCF-7 parental cells, accompanied
by a decrease in tamoxifen sensitivity. These findings suggest that
EZH2 overexpression contributes to tamoxifen resistance in breast
cancer cells by upregulating cyclin D1.
As a tumor suppressor, p16 is a cyclin-dependent
kinase inhibitor that binds to CDK4/6, and subsequently prevents
the interaction of CDK4/6 with cyclin D1, resulting in inhibition
of cell cycle progression (31).
In this study, we found that EZH2 knockdown induced cell cycle
arrest and increased p16 expression in MCF-7 TamR cells, and EZH2
overexpression decreased p16 expression in MCF-7 parental cells,
suggesting that EZH2 knockdown inhibits cell cycle progression by
upregulation of p16. Hypermethylation of the p16 gene is one of the
major mechanisms responsible for downregulation of p16 expression
in many cancers including breast cancer, leading to cell cycle
progression (32–34). In this study, we found that EZH2
knockdown reduced the DNA methylation level of the p16 promoter in
MCF-7 TamR cells, suggesting that EZH2 knockdown upregulates p16
expression via inhibition of p16 promoter methylation. It has been
reported that EZH2 controls DNA methylation by directly interacting
with the DNA methyltransferases (35). Therefore, EZH2 may promote p16
promoter methylation via DNA methyltransferase.
It has been reported that an increase in ER
expression can re-sensitize TamR cells to tamoxifen in breast
cancer cells (36), suggesting
that downregulation of ER expression may contribute to tamoxifen
resistance. In the present study, we examined ER expression in
MCF-7 parental and TamR cells, and found that ER expression was not
significantly different between MCF-7 parental and TamR cells,
suggesting that tamoxifen resistance might be mediated by
ER-independent signaling pathways in MCF-7 TamR cells.
Several studies have shown that EZH2 overexpression
correlates with pathological types, histological grade, ER
negativity, PR negativity, and HER-2 positivity as well as poor
prognosis in breast cancer (17,37,38).
In addition, low EZH2 expression is associated with a reduced risk
of developing breast cancer (39).
It has been reported that EZH2 inhibition decreases proliferation
and promotes apoptosis in breast cancer cells in vitro, and
produces anti-tumor activity in vivo (40,41).
Therefore, EZH2 inhibition may represent a promising new
therapeutic strategy for the treatment of breast cancer. Several
small molecule inhibitors of methyl transferase have been
identified for the treatment of many cancers such as breast cancer
and leukemia, and combination therapy with hypomethylators and
chemotherapeutic agents can synergistically inhibit breast cancer
cells (42–44). Recently, Song et al
(40) reported that ZLD1039, a
highly selective small molecule inhibitor of EZH2, inhibited tumor
growth and metastasis in breast cancer xenograft mice. Furthermore,
a phase I/II clinical trial has been initiated for EZH2 inhibitor
EPZ-6438 for the treatment of advanced solid tumor (45). In this study, we found that EZH2
inhibition increased the sensitivity of MCF-7 TamR breast cancer
cells to tamoxifen, suggesting that EZH2 inhibitors may be used to
reverse tamoxifen resistance, and combination of EZH2 inhibitors
with tamoxifen may be effective for treating ER-positive breast
cancer.
In summary, we found that EZH2 was overexpressed in
MCF-7 TamR breast cancer cells, and EZH2 knockdown by siRNAs
increased the sensitivity of MCF-7 TamR cells to tamoxifen. In
addition, EZH2 knockdown induced cell cycle arrest by decreasing
cyclin D1 expression and increasing p16 expression. Inhibition of
p16 promoter methylation by EZH2 knockdown resulted in upregulation
of p16 expression. Our findings suggest that EZH2 inhibition may be
used for reversing tamoxifen resistance in breast cancer.
Acknowledgements
The present study was supported by grants from the
Natural Science Foundations of Liaoning Province of China (no.
2015020723) to Yue Fang, and (no. 2015020501) to Fan Yao. This
study was also supported by Research Fund for the Doctoral Program
of Higher Education of Liaoning Province, China (no. 20091110) to
Yue Fang.
References
|
1
|
Global Burden of Disease Cancer
Collaboration, ; Fitzmaurice C, Dicker D, Pain A, Hamavid H,
Moradi-Lakeh M, MacIntyre MF, Allen C, Hansen G, Woodbrook R, et
al: The Global burden of cancer 2013. JAMA Oncol. 1:505–527. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li F, Dou J, Wei L, Li S and Liu J: The
selective estrogen receptor modulators in breast cancer prevention.
Cancer Chemother Pharmacol. 77:895–903. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rondón-Lagos M, Villegas VE, Rangel N,
Sanchez MC and Zaphiropoulos PG: Tamoxifen resistance: Emerging
molecular targets. Int J Mol Sci. 17:E13572016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wrobel K, Zhao YC, Kulkoyluoglu E, Chen
KL, Hieronymi K, Holloway J, Li S, Ray T, Ray PS, Landesman Y, et
al: ERα-XPO1 cross talk controls tamoxifen sensitivity in tumors by
altering ERK5 cellular localization. Mol Endocrinol. 30:1029–1045.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Luqmani YA and Alam-Eldin N: Overcoming
resistance to endocrine therapy in breast cancer: New approaches to
a nagging problem. Med Princ Pract. 25 Suppl 2:S28–S40. 2016.
View Article : Google Scholar
|
|
6
|
Mahara S, Lee PL, Feng M, Tergaonkar V,
Chng WJ and Yu Q: HIFI-α activation underlies a functional switch
in the paradoxical role of Ezh2/PRC2 in breast cancer. Proc Natl
Acad Sci USA. 113:pp. E3735–E3744. 2016; View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shenoy N, Vallumsetla N, Zou Y, Galeas JN,
Shrivastava M, Hu C, Susztak K and Verma A: Role of DNA methylation
in renal cell carcinoma. J Hematol Oncol. 8:882015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Qi W, Chan H, Teng L, Li L, Chuai S, Zhang
R, Zeng J, Li M, Fan H, Lin Y, et al: Selective inhibition of Ezh2
by a small molecule inhibitor blocks tumor cells proliferation.
Proc Natl Acad Sci USA. 109:pp. 21360–21365. 2012; View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hubaux R, Thu KL, Coe BP, MacAulay C, Lam
S and Lam WL: EZH2 promotes E2F-driven SCLC tumorigenesis through
modulation of apoptosis and cell-cycle regulation. J Thorac Oncol.
8:1102–1106. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cai L, Wang Z and Liu D: Interference with
endogenous EZH2 reverses the chemotherapy drug resistance in
cervical cancer cells partly by up-regulating dicer expression.
Tumour Biol. 37:6359–6369. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sun R, Shen J, Gao Y, Zhou Y, Yu Z,
Hornicek F, Kan Q and Duan Z: Overexpression of EZH2 is associated
with the poor prognosis in osteosarcoma and function analysis
indicates a therapeutic potential. Oncotarget. 7:38333–38346. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fujii S, Tokita K, Wada N, Ito K, Yamauchi
C, Ito Y and Ochiai A: MEK-ERK pathway regulates EZH2
overexpression in association with aggressive breast cancer
subtypes. Oncogene. 30:4118–4128. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Varambally S, Dhanasekaran SM, Zhou M,
Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt
RG, Otte AP, et al: The polycomb group protein EZH2 is involved in
progression of prostate cancer. Nature. 419:624–629. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim KH and Roberts CW: Targeting EZH2 in
cancer. Nat Med. 22:128–134. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wassef M, Michaud A and Margueron R:
Association between EZH2 expression, silencing of tumor suppressors
and disease outcome in solid tumors. Cell Cycle. 15:2256–2262.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Italiano A: Role of the EZH2 histone
methyltransferase as a therapeutic target in cancer. Pharmacol
Ther. 165:26–31. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jang SH, Lee JE, Oh MH, Lee JH, Cho HD,
Kim KJ, Kim SY, Han SW, Kim HJ, Bae SB and Lee HJ: High EZH2
protein expression is associated with poor overall survival in
patients with luminal a breast cancer. J Breast Cancer. 19:53–60.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Reijm EA, Timmermans AM, Look MP,
Meijer-van Gelder ME, Stobbe CK, van Deurzen CH, Martens JW,
Sleijfer S, Foekens JA, Berns PM and Jansen MP: High protein
expression of EZH2 is related to unfavorable outcome to tamoxifen
in metastatic breast cancer. Ann Oncol. 25:2185–2190. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Knowlden JM, Hutcheson IR, Jones HE,
Madden T, Gee JM, Harper ME, Barrow D, Wakeling AE and Nicholson
RI: Elevated levels of epidermal growth factor receptor/c-erbB2
heterodimers mediate an autocrine growth regulatory pathway in
tamoxifen-resistant MCF-7 cells. Endocrinology. 144:1032–1044.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Coward WR, Feghali-Bostwick CA, Jenkins G,
Knox AJ and Pang L: A central role for G9a and EZH2 in the
epigenetic silencing of cyclooxygenase-2 in idiopathic pulmonary
fibrosis. FASEB J. 28:3183–3196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Velagapudi SP, Gallo SM and Disney MD:
Sequence-based design of bioactive small molecules that target
precursor microRNAs. Nat Chem Biol. 10:291–297. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pan FP, Zhou HK, Bu HQ, Chen ZQ, Zhang H,
Xu LP, Tang J, Yu QJ, Chu YQ, Pan J, et al: Emodin enhances the
demethylation by 5-Aza-CdR of pancreatic cancer cell
tumor-suppressor genes P16, RASSF1A and ppENK. Oncol Rep.
35:1941–1949. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chang LC, Lin HY, Tsai MT, Chou RH, Lee
FY, Teng CM, Hsieh MT, Hung HY, Huang LJ, Yu YL and Kuo SC: YC-1
inhibits proliferation of breast cancer cells by down-regulating
EZH2 expression via activation of c-Cbl and ERK. Br J Pharmacol.
171:4010–4025. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cha TL, Zhou BP, Xia W, Wu Y, Yang CC,
Chen CT, Ping B, Otte AP and Hung MC: Akt-mediated phosphorylation
of EZH2 suppresses methylation of lysine 27 in histone H3. Science.
310:306–310. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Riggins RB, Schrecengost RS, Guerrero MS
and Bouton AH: Pathways to tamoxifen resistance. Cancer Lett.
256:1–24. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Viedma-Rodriguez R, Baiza-Gutman L,
Salamanca-Gómez F, Diaz-Zaragoza M, Martínez-Hernández G, Ruiz
Esparza-Garrido R, Velázquez-Flores MA and Arenas-Aranda D:
Mechanisms associated with resistance to tamoxifen in estrogen
receptor-positive breast cancer (Review). Oncol Rep. 32:3–15. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kilker RL, Hartl MW, Rutherford TM and
Planas-Silva MD: Cyclin D1 expression is dependent on estrogen
receptor function in tamoxifen-resistant breast cancer cells. J
Steroid Biochem Mol Biol. 92:63–71. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Stendahl M, Kronblad A, Rydén L, Emdin S,
Bengtsson NO and Landberg G: Cyclin D1 overexpression is a negative
predictive factor for tamoxifen response in postmenopausal breast
cancer patients. Br J Cancer. 90:1942–1948. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rudas M, Lehnert M, Huynh A, Jakesz R,
Singer C, Lax S, Schippinger W, Dietze O, Greil R, Stiglbauer W, et
al: Cyclin D1 expression in breast cancer patients receiving
adjuvant tamoxifen-based therapy. Clin Cancer Res. 14:1767–1774.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kilker RL and Planas-Silva MD: Cyclin D1
is necessary for tamoxifen-induced cell cycle progression in human
breast cancer cells. Cancer Res. 66:11478–11484. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hara E, Smith R, Parry D, Tahara H, Stone
S and Peters G: Regulation of p16CDKN2 expression and its
implications for cell immortalization and senescence. Mol Cell
Biol. 16:859–867. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shim YH, Park HJ, Choi MS, Kim JS, Kim H,
Kim JJ, Jang JJ and Yu E: Hypermethylation of the p16 gene and lack
of p16 expression in hepatoblastoma. Mod Pathol. 16:430–436. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lee JJ, Ko E, Cho J, Park HY, Lee JE, Nam
SJ, Kim DH and Cho EY: Methylation and immunoexpression of p16
(INK4a) tumor suppressor gene in primary breast cancer tissue and
their quantitative p16 (INK4a) hypermethylation in plasma by
real-time PCR. Korean J Pathol. 46:554–561. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang L, Tang L, Xie R, Nie W, Chen L and
Guan X: p16 promoter hypermethylation is associated with increased
breast cancer risk. Mol Med Rep. 6:904–908. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Vire E, Brenner C, Deplus R, Blanchon L,
Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden
JM, et al: The Polycomb group protein EZH2 directly controls DNA
methylation. Nature. 439:871–874. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li Y, Meeran SM and Tollefsbol TO:
Combinatorial bioactive botanicals re-sensitize tamoxifen treatment
in ER-negative breast cancer via epigenetic reactivation of ERα
expression. Sci Rep. 7:93452017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang X, Hu B, Shen H, Zhou H, Xue X, Chen
Y, Chen S, Han Y, Yuan B, Zhao H, et al: Clinical and prognostic
relevance of EZH2 in breast cancer: A meta-analysis. Biomed
Pharmacother. 75:218–225. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Guo S, Li X, Rohr J, Wang Y, Ma S, Chen P
and Wang Z: EZH2 overexpression in different immunophenotypes of
breast carcinoma and association with clinicopathologic features.
Diagn Pathol. 11:412016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Beca F, Kensler K, Glass B, Schnitt SJ,
Tamimi RM and Beck AH: EZH2 protein expression in normal breast
epithelium and risk of breast cancer: Results from the nurses'
health studies. Breast Cancer Res. 19:212017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Song X, Gao T, Wang N, Feng Q, You X, Ye
T, Lei Q, Zhu Y, Xiong M, Xia Y, et al: Selective inhibition of
EZH2 by ZLD1039 blocks H3K27 methylation and leads to potent
anti-tumor activity in breast cancer. Sci Rep. 6:208642016.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang L, Deng L, Chen F, Yao Y, Wu B, Wei
L, Mo Q and Song Y: Inhibition of histone H3K79 methylation
selectively inhibits proliferation, self-renewal and metastatic
potential of breast cancer. Oncotarget. 5:10665–10677. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Feng Z, Yao Y, Zhou C, Chen F, Wu F, Wei
L, Liu W, Dong S, Redell M, Mo Q and Song Y: Pharmacological
inhibition of LSD1 for the treatment of MLL-rearranged leukemia. J
Hematol Oncol. 9:242016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Song Y, Wu F and Wu J: Targeting histone
methylation for cancer therapy: Enzymes, inhibitors, biological
activity and perspectives. J Hematol Oncol. 9:492016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cang S, Ma Y, Chiao JW and Liu D:
Phenethyl isothiocyanate and paclitaxel synergistically enhanced
apoptosis and alpha-tubulin hyperacetylation in breast cancer
cells. Exp Hematol Oncol. 3:52014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Huang T, Lin C, Zhong LL, Zhao L, Zhang G,
Lu A, Wu J and Bian Z: Targeting histone methylation for colorectal
cancer. Therap Adv Gastroenterol. 10:114–131. 2017. View Article : Google Scholar : PubMed/NCBI
|