Introduction
Intestinal epithelial cells are crucial components
of the intestinal mucosal barrier, and damage to these cells
increases the permeability of this barrier, which may lead to
increased translocation of gut-derived bacterial endotoxins
(1). Previous studies on the
effects of heat stress on these cells have reported a significant
level of apoptosis in the rat small intestine (2,3).
Indeed, apoptosis in the small intestine was demonstrated to serve
a major role in the pathogenesis of heat stroke (2,3).
Heat stress may induce the production of reactive
oxygen species (ROS), which may lead to cellular dysfunction and
cell death (3–5). Nuclear factor (NF)-κB was previously
reported to be induced by a multitude of stimuli, such as
cytokines, oxidative stress or thermal stress (6). A previous study demonstrated that
NF-κB is activated in response to heat stress in HeLa human
cervical cancer cells (6). Many
inducers of NF-κB expression may be inhibited by antioxidants,
which suggested that ROS may modulate the signal transduction
pathway leading to NF-κB activation (7,8).
However, the role of ROS heat stress-induced NF-κB activation in
IEC-6 rat small intestinal epithelial cells remains unclear.
Cultured IEC-6 cells are similar to the mature intestinal
epithelium (3), and were used as a
model to investigate the mechanisms of intestinal epithelial cell
survival and apoptosis.
The heterodimeric NF-κB complex comprises two DNA
binding subunits, p50 and p65, which may form either homo- or
heterodimers (9). NF-κB signaling
is a transcriptional regulator of several genes that are involved
the inflammatory response, cell growth, cell survival and apoptosis
(9,10). A number of studies have reported
that NF-κB activation may downregulate proapoptotic signaling and
thus prevent apoptosis in many cells types (10,11).
By contrast, other studies have demonstrated that NF-κB activation
may by an inducer of apoptosis (12,13).
However, the mechanisms underlying heat stress-induced apoptosis in
IEC-6 cells and the involvement of NF-κB activation are still
unknown.
Heat shock transcription factor 1 (HSF1) is a master
regulator of the genes that encode molecular chaperones and serves
a role in the attenuation of apoptosis induced by multiple factors
(14,15). Upon heat shock, HSF1 rapidly
translocates into the nucleus and exhibits the properties of a
stable trimer which correlates with the acquisition of DNA binding
activity; furthermore, the transcriptionally active form of HSF1
becomes inducibly phosphorylated (16). Previous studies have reported
interactions between HSF1 and NF-κB, which serve opposite roles in
cytoprotection and cell injury (17).
The signal-transducing transcription factor c-Jun,
also known as activating protein 1 (AP1), has previously been
reported to serve a role in cell cycle progression, differentiation
and transformation, as well as apoptosis (18,19).
c-Jun protein activity is regulated by phosphorylation at specific
sites; for example, Ser63 phosphorylation in the transactivation
domain leads to an increased ability of c-Jun to activate the
transcription of target genes (20). Previous studies have demonstrated
that c-Jun is able to physically interact with NF-κB p65 through
the Rel homology domain (21).
Nevertheless, whether NF-κB can interact with HSF1 and c-Jun, and
thereby influence IEC-6 cell apoptosis, still remains unknown.
Results from the present study demonstrated that
heat stress-induced increases of ROS levels may lead to NF-κB
activation, which in turn may activate caspase-3 and, thus,
apoptosis in IEC-6 cells. In addition, a putative role for NF-κB in
the regulation of HSF1 and c-Jun activation induced by heat stress
treatment was investigated. This study also aimed to examine
whether HSF1 might prevent apoptosis and c-Jun activation-induced
apoptosis in the same experimental settings.
Materials and methods
Cell culture and treatments
Approximately 2×106 IEC-6 cells (ATCC,
Manassas, VA, USA) were grown as a monolayer in Dulbecco's Modified
Eagle's Medium (DMEM) supplemented with 10% heat inactivated fetal
bovine serum (FBS) (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), 100 U/ml of penicillin, and 100 µg/ml of
streptomycin (Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C
in a humidified atmosphere of 5% CO2 and 95% air. To
induce heat stress, culture dishes were placed into a circulating
water bath at 43±0.5°C for indicated times; control cells were held
at 37±0.5°C. Following heat-stress culture, the media was replaced
and cells were further incubated at 37°C for 0, 2, 6 and 12 h.
Measurement of ROS levels
Levels of intracellular ROS were assessed using a
ROS assay kit (Beyotime Institute of Biotechnology, Haimen, China).
Dichlorofluorescein diacetate (DCFH-DA; Molecular Probes; Thermo
Fisher Scientific, Inc.) enters the cells and reacts with ROS,
producing the fluorophore DCF. Briefly, cells were either kept
untreated or incubated at 43°C for 20 (short-term heat stress), 40,
60 (moderate-term heat stress) and 80 min (long-term heat stress),
followed by an additional incubation at 37°C for 6 h. IEC-6 cells
(3×105) with the antioxidant glutathione (GSH; 100
µmol/l) for 1 h, followed by exposure to heat stress at 43°C for 60
min. Control cells were always incubated at 37°C. Cells
(3×105) were harvested, washed with serum-free DMEM
culture medium, and stained with 10 µM DCFH-DA for 30 min at 37°C
in the dark. Following this, the cells were harvested by trypsin,
the supernatants was removed by centrifugation (1,000 × g for 3 min
at room temperature) and resuspended in serum-free DMEM culture
medium three times. The fluorescence intensity was determined using
a flow cytometer (FACSCanto™ II; BD Biosciences, San Jose, CA, USA)
and analyzed using FlowJo software version 9.0 (FlowJo LLC,
Ashland, OR, USA).
Flow cytometric analysis of cell
apoptosis
Cell apoptosis was analyzed by flow cytometry with
annexin V-fluorescein isothiocyanate (FITC) Apoptosis Detection kit
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. Briefly, cells were either kept untreated
or incubated at 43°C for 20, 40, 60, 80 min, followed by an
additional incubation at 37°C for 6 h. Control cells were always
incubated at 37°C. IEC-6 cells (1×106) were collected,
washed in ice cold PBS and resuspended in the binding buffer
containing 5 µl annexin V-FITC (5 µg/ml). Following incubation at
room temperature for 10 min, the buffer was removed by
centrifugation (1,000 × g, 3 min, room temperature) and cells were
resuspended in reaction buffer containing 10 µl propidium iodide
(PI; 5 µg/ml) for 10 min at room temperature. Flow cytometric
analysis was immediately performed to detect apoptosis (FACSCanto™
II; BD Biosciences), The combination of Annexin V-FITC and
propidium iodide allows for the distinction between early apoptotic
cells (Annexin V-FITC positive), late apoptotic and/or necrotic
cells (Annexin V-FITC and propidium iodide positive), and viable
cells (unstained). The fluorescence intensity was analyzed using
FlowJo software version 9.0 (FlowJo LLC, Ashland, OR, USA).
Small interfering (si)RNA
transfection
siRNAs for NF-κB p65 and HSF1 were designed and
synthesized by Shanghai GenePharma Co. Ltd. (Shanghai, China). The
sequence of each siRNA and the negative control (non-targeting
siRNA) are shown in Table I. Prior
to transfection, 1×105 IEC-6 cells were plated onto a
6-well plate (Nest Biotechnology Co., Ltd., Wuxi, China) and
incubated for 24 h to 30–50% confluence at 37°C. Cells were
transfected with 1 µM siRNA (all siRNAs were used at this
concentration) using siRNAMate Transfection Reagent (Shanghai
GenePharma Co. Ltd.) and incubated for 12 h at 37°C, according to
the manufacturer's protocol. Cells were incubated following 48–72 h
at 37°C for further experiments.
 | Table I.Small interfering RNA oligonucleotide
sequences. |
Table I.
Small interfering RNA oligonucleotide
sequences.
| Gene | Sequence (5′→3′) |
|---|
| p65 | Sense:
GCCCUAUCCCUUUACGUCATT |
|
| Antisense:
UGACGUAAAGGGAUAGGGCTT |
| HSF1 | Sense:
GGAAAGUGGUCCACAUCGATT |
|
| Antisense:
UCGAUGUGGACCACUUUCCTT |
| Negative control | Sense:
UUCUCCGAACGUGUCACGUTT |
|
| Antisense:
ACGUGACACGUUCGGAGAATT |
Adenoviral infection
Adenoviruses (Ad) that constitutively overexpressed
p65 (Ad-p65) or empty construct (Ad-empty) were constructed by
Vigene Biosciences (Jinan, China). Cells were infected with the
adenoviruses in serum-free DMEM for 6 h and then the media was
replaced with DMEM supplemented with 10% FBS. Cells
(1×105/well) were infected with 100 MOI Ad in serum-free
DMEM for 6 h at 37°C, according to the manufacturer's protocol,
following which the media was replaced with DMEM supplemented with
10% FBS, cells were incubated following 48–72 h at 37°C for further
experiments.
Western blot analysis
IEC-6 cells (~1×106) were kept at 37°C or
43°C for 60 min, and further incubated for 2, 6 or 12 h at 37°C.
For cytoplasmic and nuclear protein of p65 extraction, cells were
lysed in NE-PER Nuclear and Cytoplasmic Extraction Reagents
(Pierce; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. The IEC-6 cells were homogenized in
radioimmunoprecipitation buffer with phenylmethylsulfonyl fluoride
(Sigma-Aldrich; Merck KGaA). Following centrifugation at 14,000 × g
at 4°C for 10 min, the supernatants were used for western blot
analysis. Protein concentration was determined using a
Bicinchoninic Acid Protein assay kit (Thermo Fisher Scientific,
Inc.). Proteins (20 µg/well) were separated by SDS-PAGE using 10%
SDS polyacrylamide gels and transferred onto polyvinylidene
difluoride membranes. Membranes were blocked with blocking solution
(5% skimmed milk diluted with PBS) at room temperature for 2 h,
followed by incubation with primary antibodies overnight at 4°C.
The following rabbit primary antibodies were used at a 1:2,000
dilution: p65 (cat. no. Ab16502; Abcam, Cambridge, MA, USA), HSF1
(cat. no. Ab59963; Abcam), phosphorylated (p)-HSF1 (cat. no.
Ab52757; Abcam), Lamin-B1 (cat. no. Ab16048; Abcam), c-Jun (rabbit
antibodies; cat. no. 9165p; Cell Signaling Technology, Inc.,
Danvers, MA, USA), phosphorylated (p)-c-Jun (rabbit antibodies;
cat. no. 8222S; Cell Signaling Technology, Inc.), caspase-3 (rabbit
antibodies; cat. no. 14220S; Cell Signaling Technology, Inc.),
cleaved caspase-3 (rabbit antibodies; cat. no. 9654S; Cell
Signaling Technology, Inc.) and GAPDH (rabbit antibodies; cat. no.
ab70699; Abcam). An anti-rabbit horseradish peroxidase-conjugated
immunoglobulin G antibody (cat. no. TA130023; 1:5,000; OriGene
Technologies, Inc., Beijing, China) was used as the secondary
antibody for incubation for 2 h at room temperature. Antibodies
were detected with Enhanced Chemiluminescence Western Blot
Detection reagent (Pierce; Thermo Fisher Scientific, Inc.).
Membranes were exposed to light-sensitive film and quantified using
ImageJ software (version 1.3.4.67; National Institutes of Health,
Bethesda, MD, USA).
Measurement of p65 and HSF1
DNA-binding capacity by ELISA
Nuclear extracts were prepared from treated and
control cells (~5×106 cells) using a Nuclear Extract kit
(Active Motif, Shanghai, China) according to the manufacturer's
protocol. The ability of p65 and HSF1 to bind the DNA consensus
sequence was assessed using an ELISA-based TransAM NF-κB kit (cat.
no. 40096; Active Motif, Carlsbad, CA, USA) and an ELISA-based
TransAM HSF1 kit (cat. no. 47096; Active Motif), according to the
manufacturer's protocol, quantitative analysis was performed by
spectrophotometry at 450 nm using an automatic microplate reader
(SpectraMax® M5; Molecular Devices, LLC, Sunnyvale, CA,
USA).
Statistical analysis
All data were analyzed for statistical significance
using SPSS 13.0 software (SPSS, Chicago, IL, USA). Data were
expressed as the mean ± standard deviation from at least three
independent experiments performed in duplicate. One-way analysis of
variance was performed followed by Fisher's least significant
difference post hoc test for multiple comparisons. P<0.05 was
considered to indicate a statistically significant difference.
Results
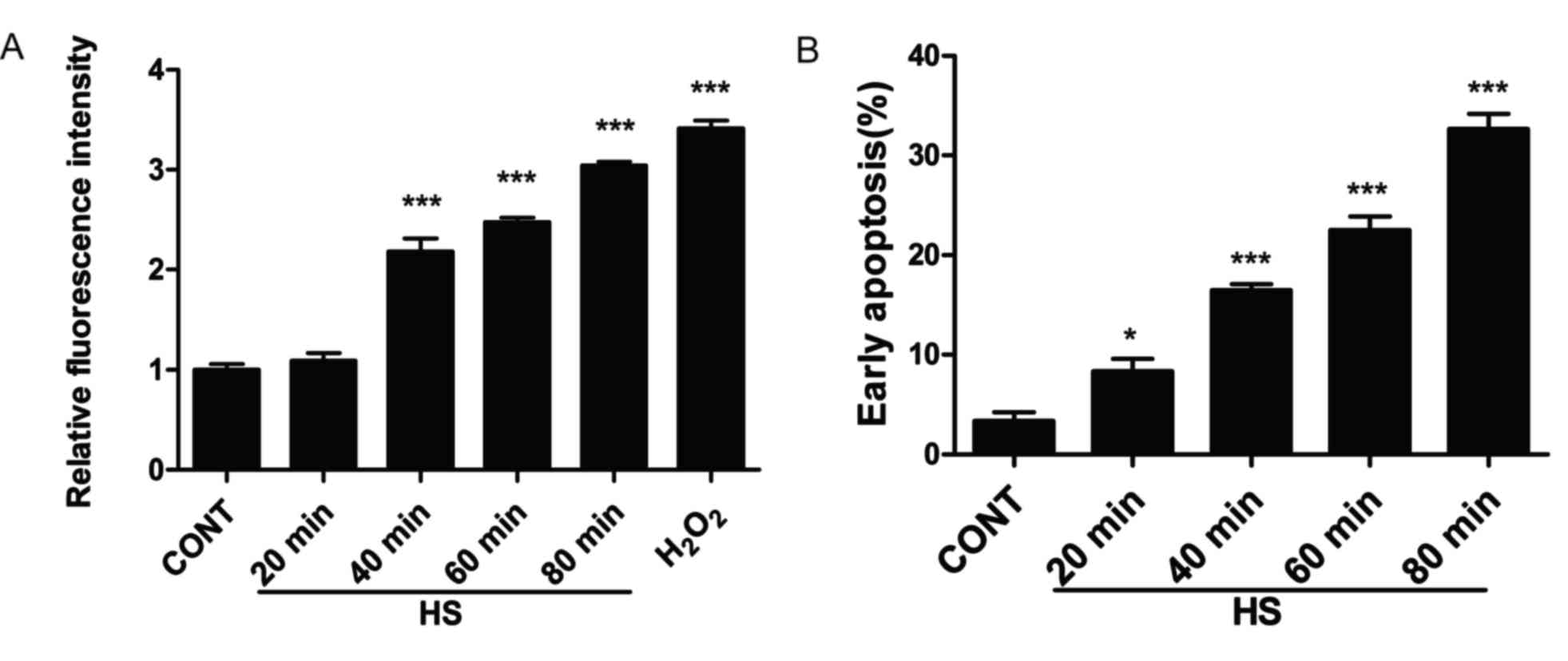
Heat stress increases ROS generation
and apoptosis in IEC-6 cells
As ROS generation serves an important role in heat
stress (3–5), the effects of heat stress on ROS
accumulation was examined in IEC-6 cells. IEC-6 cells were exposed
to heat stress (43°C) for different time intervals (20, 40, 60 and
80 min), followed by an additional incubation at 37°C for 6 h in
fresh media. Intracellular ROS levels increased in a time-dependent
manner (Fig. 1A). In addition,
apoptotic rates were quantitative using flow cytometry. Cells were
exposed to different durations of heat stress, and the results
demonstrate that the number of early apoptotic cells gradually
increased and reached a peak of 32.37% in IEC-6 cells exposed
incubated at 43°C for 80 min (Fig.
1B).
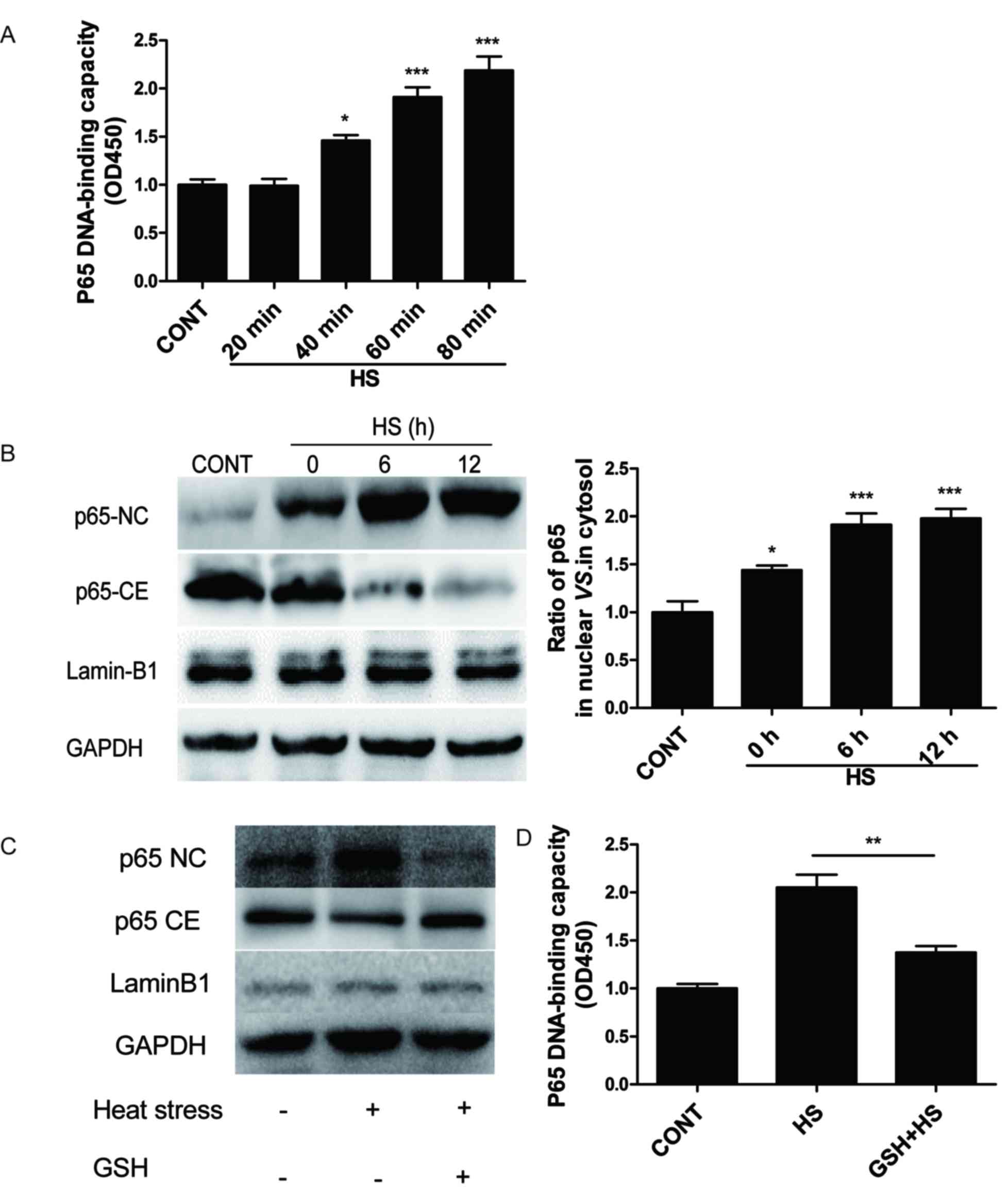
Role of ROS in activating p65 in heat
stress-treated IEC-6 cells
Compared with the untreated control cells, the
capacity of p65 to bind to DNA in IEC-6 cells was not modified at
20 min of heat stress at 43°C (Fig.
2A). By contrast, at 40 min heat stress, the DNA-binding
capacity of p65 increased and continued to increase significantly
at 60 and 80 min (Fig. 2A).
Additionally, a time course of p65 activation was analyzed in heat
stressed IEC-6 cells at 43°C for 60 min followed by a recovery
period at 37°C for 0, 6, or 12 h. When nuclear and cytoplasmic
extracts of these timepoints were collected and analyzed by western
blot, nuclear levels of p65 increased as the recovery time
increased (Fig. 2B). Collectively,
these data indicated that heat stress may induce the activation of
NF-κB p65, but this event seems not to occur during 20 min of heat
stress at 43°C (short-term heat stress).
The role of heat stress-induced ROS generation in
p65 activation was examined by pretreating IEC-6 cells with the
antioxidant glutathione (GSH; 100 µmol/l) for 1 h, followed by
exposure to heat stress at 43°C for 60 min. Cells pretreated with
GSH significantly decreased the heat stress-mediated translocation
of p65 to nucleus and the binding capacity of p65 to DNA (Fig. 2C and D, respectively). These data
suggested that heat stress treatment of IEC-6 cells activated the
NF-κB p65 pathway through the release of ROS.
p65 and HSF1 activation is involved in
heat stress-induced apoptosis in IEC-6 cells
To clarify the role of NF-κB p65 and HSF1 in heat
stress-induced apoptosis in IEC-6 cells, the effects of p65-siRNA
and HSF1-siRNA knockdown, as well as Ad-p65 overexpression were
examined (Fig. 3A-C). Following
p65-siRNA mediated knockdown, apoptosis was significantly reduced
in heat stress-treated IEC-6 cells (Fig. 3D). By contrast, apoptosis was
markedly increased in heat stress-treated cells that were
co-treated with Ad-p65 compared with the same cells transfected
with Ad-empty control (Fig. 3E).
These observations implied that activated p65 may be able to
mediate heat stress-induced apoptosis in IEC-6 cells. In addition,
heat stress-induced IEC-6 cells co-treated with HSF1-siRNA
exhibited a significant increase in apoptosis, which demonstrated
that HSF1 may serve an antiapoptotic role in IEC-6 cells (Fig. 3C and D).
 | Figure 3.Role of p65 and HSF1 in heat
stress-induced cell apoptosis in IEC-6 cells. Cells were
transfected with NC-siRNA, p65-siRNA or HSF1-siRNA, as well as
Ad-empty or Ad-p65 for 48 h. Western blot analysis was used to
detect the effectiveness of transfection for (A-C) p65-siRNA
knockdown, Ad-p65 overexpression and HSF1-siRNA knockdown
expression. (B and C) Cells were exposed to heat stress for 60,
followed by a 6 h incubation at 37°C, and apoptosis was analyzed by
flow cytometry using Annexin V-fluorescein isothiocyanate/propidium
iodide staining. Data are presented as the mean ± standard
deviation of three separate experiments; *P<0.05, **P<0.01
and ***P<0.001. Ad, adenovirus; CONT, untreated control; HSF1,
heat shock transcription factor 1; NC, negative control; ns, not
significant; siRNA, small interfering RNA. |
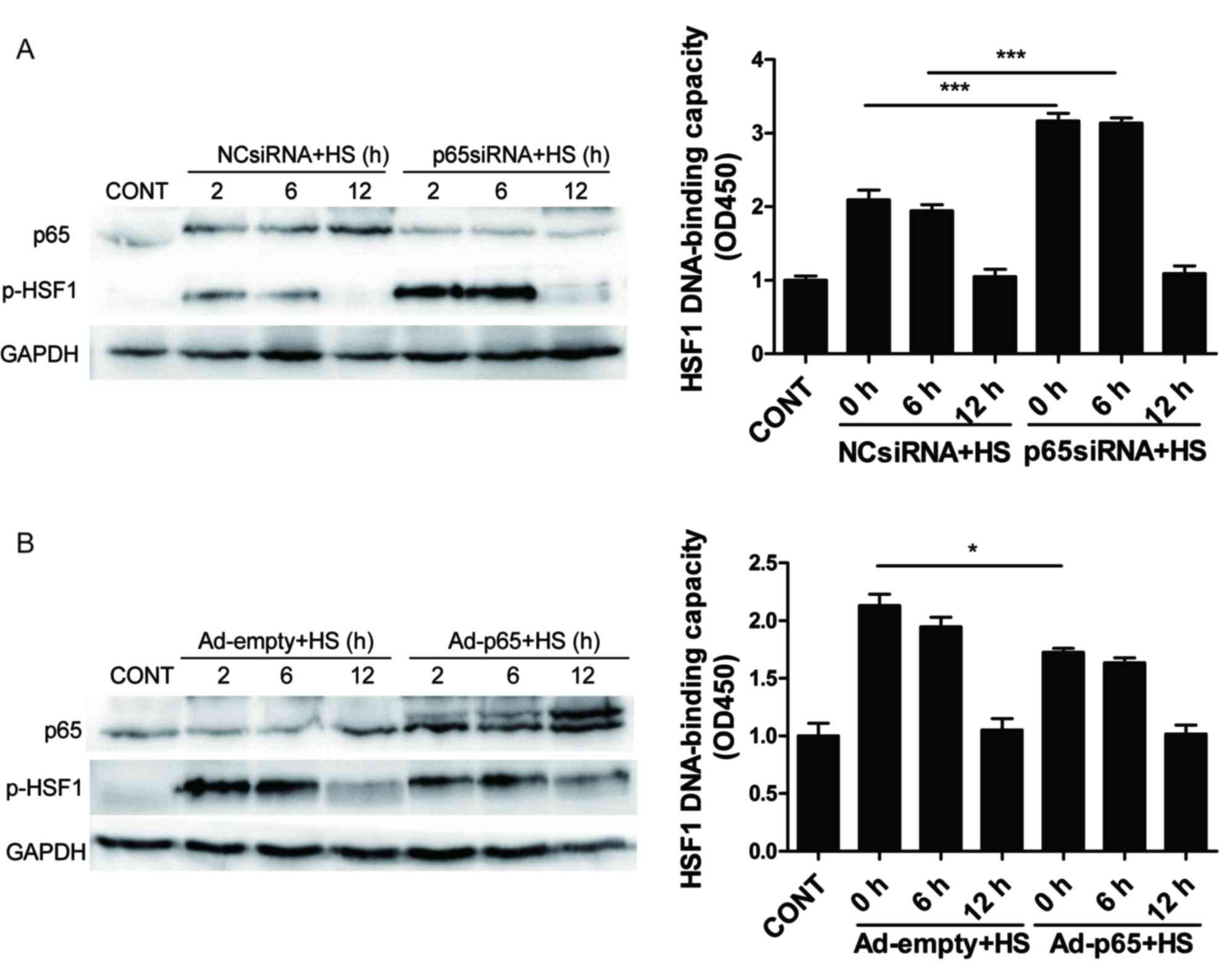
Role of p65 in HSF1 activation in heat
stress-induced apoptosis in IEC-6 cells
Whether HSF1 activation was linked with NF-κB
signaling in the experimental settings was investigated.
p65-siRNA-mediated knockdown significantly increased the heat
stress-induced phosphorylation of HSF1 as well as the HSF1
DNA-binding capacity at 2 and 6 h post-heat stress exposure
compared with the heat stress-treated cells that were co-treated
with the NC-siRNA control, no significant change was observed at 12
h post-heat stress. HSF1 phosphorylation occurred early (0 and 6 h)
following heat stress, and HSF1 phosphorylation level returned to
normal at 12 h following heat stress, which demonstrated that HSF1
is not constitutively phosphorylated after heat stress. (Fig. 4A and B, respectively). Ad-p65
overexpression led to a slight decrease in HSF1 phosphorylation in
heat stress-treated cells, compared with Ad-empty treated IEC-6
cells at 2 h post-heat stress, no significant change was observed
at 6 and 12 h post-heat stress (Fig.
4C). Similar results were observed for HSF1 DNA-binding
activity (Fig. 4D). These results
indicated that p65 activation may have potential inhibitory effects
on heat stress-induced HSF1 activation.
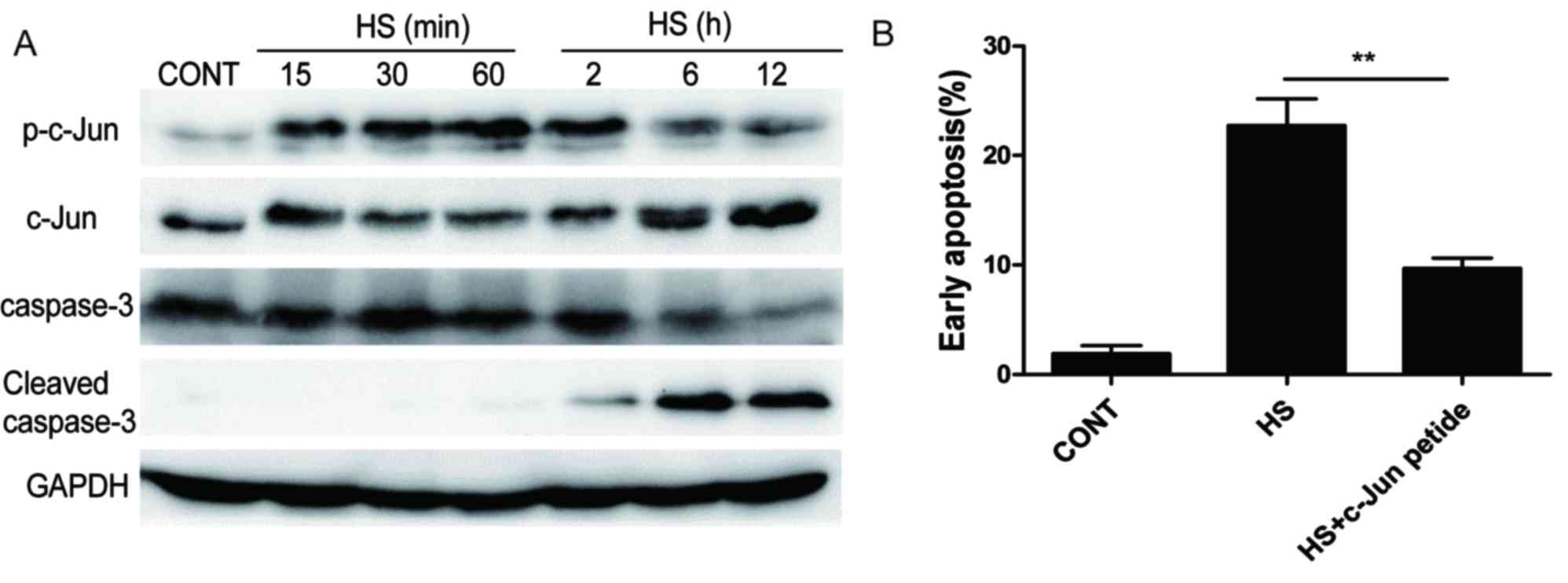
Phosphorylation of c-Jun is necessary
for apoptosis in heat-stressed IEC-6 cells
To examine the effects of heat stress on c-Jun
activation and expression, IEC-6 cells were exposed to 43°C heat
stress for the indicated times. Heat stress resulted in an increase
in the phosphorylation level of c-Jun within 15 min of exposure,
reached a peak at 2 h and subsequently began to decline (Fig. 5A); the rapid increase of c-Jun
phosphorylation on Ser63 and subsequent accumulation of c-Jun
protein appeared to occur in a time-dependent manner. In addition,
heat stress treatment resulted in the increased cleavage of
caspase-3 at 2 h of exposure and was expressed at high levels for
12 h, which indicated the activation of apoptosis.
To determine whether c-Jun phosphorylation is
necessary for apoptosis, IEC-6 cells were exposed to heat stress in
the presence or absence of c-Jun peptide, which effectively acts as
c-Jun inhibitor. The inhibition of c-Jun phosphorylation resulted
in a significant decrease in the number of apoptotic IEC-6 cells
(Fig. 5B). These results suggested
that c-Jun phosphorylation may be involved in heat stress induced
apoptosis in IEC-6 cells.
Influence of NF-κB signaling on c-Jun
phosphorylation and caspase-3 activation
The effects of p65 on c-Jun phosphorylation in heat
stressed IEC-6 cells were also examined. Western blotting results
demonstrated that heat stress-induced c-Jun phosphorylation was
reduced by knockdown of p65 at 6 and 12 h post-heat stress
exposure, whereas it was increased by in cells overexpressing p65
(Fig. 6A and B, respectively). In
addition, p65 knockdown caused a decrease in the cleavage of
caspase-3, whereas p65 overexpression led to an increase in caspase
3 cleavage (Fig. 6C and D).
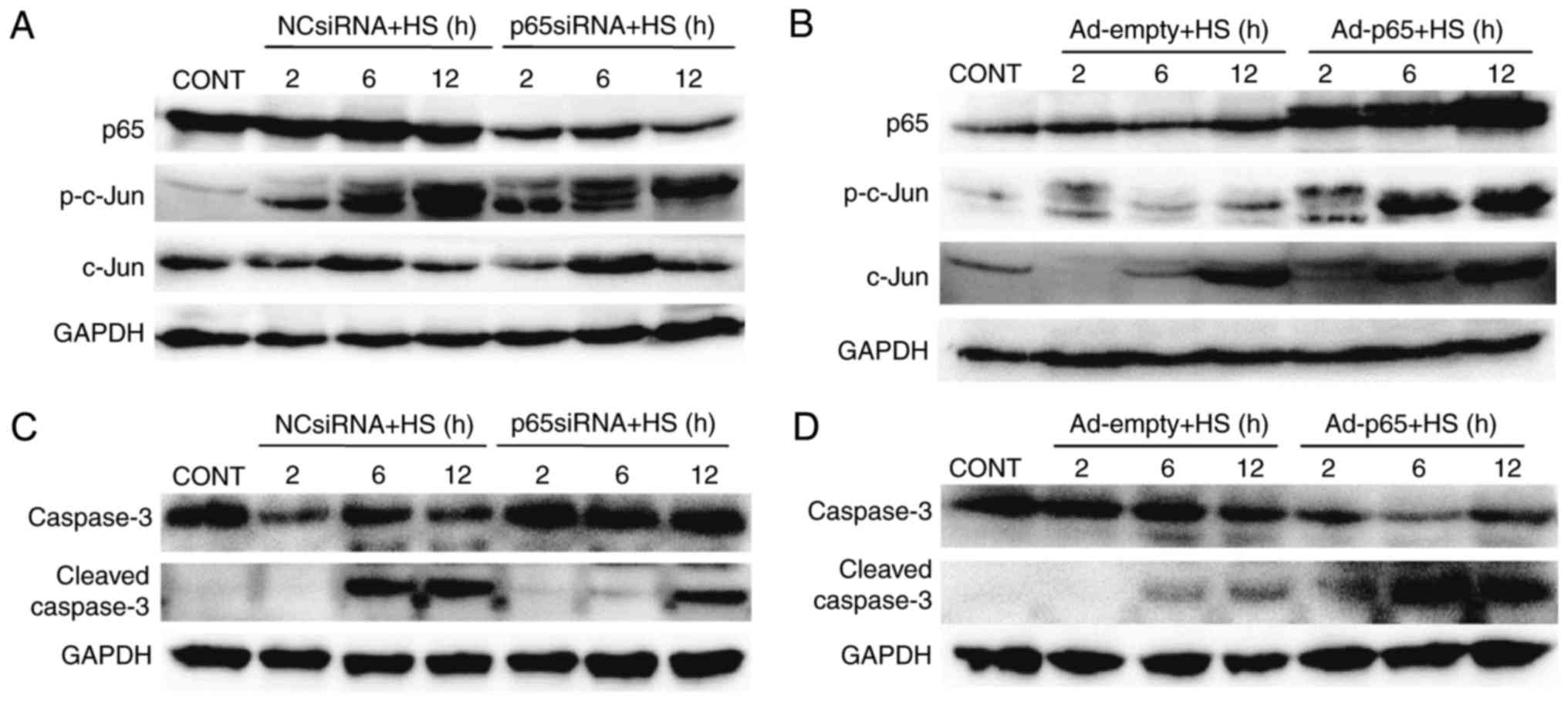 | Figure 6.p65 is involved in the c-Jun
activation in IEC-6 cells. Cells were transfected with NC-siRNA,
p65-siRNA, Ad-empty or Ad-p65, exposed to heat stress for 60 min,
and further incubated for indicated times. (A and B) Expression of
p-c-Jun and c-Jun were detected by western blotting following
treatment with (A) p65-siRNA or (B) Ad-p65. (C and D) Expression of
caspase-3 and cleaved caspase-3 was detected by western blotting
following treatment with (C) p65-siRNA or (D) Ad-p65. The images
are representative of three independent experiments. Ad,
adenovirus; CONT, untreated control; HS, heat stress; NC, negative
control; p, phosphorylated; siRNA, small interfering RNA. |
Discussion
Heat stroke is a life-threatening condition and is
the leading cause of morbidity and mortality during heat waves; it
is characterized by a rapidly increasing core temperature to
>40°C and multiple organ dysfunction syndrome (1). As global temperatures continue to
rise, heat stroke morbidity and mortality rates may also continue
to increase (1,22,23).
Heat stroke is a condition of complex pathogenesis in which the
intestine appears to serve a key role (1,22);
for example, gut-derived endotoxinemia has been implicated in the
pathophysiology of heat stroke (1–3). In
addition, studies performed in animal models demonstrated
widespread apoptosis in the intestine of animals affected by this
type of injury (22,23). Although apoptosis is a normal
physiological process, the disruption of intestinal mucosa by
apoptosis allows bacterial components or complete microorganisms to
enter the bloodstream, which further complicates the already
complex pathophysiology of heat stroke (23). Therefore, an examination of the
mechanisms of heat stroke-induced apoptosis in intestinal cell
models, such as the IEC-6 cell line used in the present study, is
required for a better understanding of heat stroke
pathogenesis.
Oxidative stress generated during heat stress, as
well as ROS modulation of the NF-κB pathway, have been previously
demonstrated in a number of cell types (3–7).
Therefore, the present study examined whether ROS was involved in
the activation of NF-κB during heat stroke. The biological effects
of heat stress on IEC-6 cell apoptosis was investigated, and the
results demonstrated that heat stress was able to induce ROS
accumulation, which might subsequently lead to NF-κB activation. In
addition, the DNA-binding capacity of NF-κB p65 was not modified in
response to short-term heat stress exposure. By contrast, during
moderate- and long-term heat stress treatments, the activity of
NF-κB increased. These results indicated that short-term heat
treatment may not induce notable ROS accumulation, which is
necessary for NF-κB activation.
Previous studies have reported that NF-κB binds to
DNA in heat-stressed HeLa cells (6). The results of the present study are
in line with these previous results; however, the present results,
to the best of our knowledge, are the first to establish a link
between apoptosis and NF-κB signaling in IEC-6 cells. An increasing
number of studies have suggested that NF-κB serves an important
role in preventing apoptosis (10,11).
By contrast, NF-κB activation has also been reported to induce
apoptosis in certain cell types, as demonstrated by the induction
of Fas ligand expression in Jurkat T lymphocyte cells treated with
the drugs etoposide or teniposide (12). The promotion or inhibition of
apoptosis by NF-κB signaling may depend on the cell type and the
type of inducer (24). The present
study demonstrated that heat stress-induced NF-κB activity may
promote cell apoptosis in IEC-6 cells and that NF-κB p65 knockdown
increased the heat stress-induced phosphorylation of HSF1 and HSF1
DNA-binding activity. HSF1 is able to block apoptosis at different
stages owing to its regulatory role on genes encoding molecular
chaperones that are involved in the apoptotic pathway (15). The present study demonstrated that
heat stress-induced HSF1 activity may inhibit apoptosis in IEC-6
cells, and that NF-κB p65 activity may be linked to the suppression
of HSF1 activation in these cells. A previous study reported that
inhibition of NF-κB by prostaglandin A1 may be associated with HSF
activation in Jurkat T cells, CEM-SS T lymphoid cells and HeLa
cells, the possibility then exists that NF-κB may be affected by
the state of transcriptional activation of HSF1, or that
simultaneous activation of these transcription factors may be
incompatible (25). The inhibition
of NF-κB signaling by aspirin has also been associated with HSF
activation (26). In addition, it
was reported that the inhibition of NF-κB activation was common to
several HSF inducers, including heat shock, which suggested that
the regulatory pathways of NF-κB and HSF may be linked and offers
new avenues in the search for effective NF-κB inhibitors as useful
immunosuppressive, anti-inflammatory and anti-apoptotic drugs
(25).
A previous study reported that NF-κB interacts
directly with c-Jun and enhances the transcription of
AP1-responsive genes in HeLa cells (21). In the present study, NF-κB was
demonstrated to be involved in the regulation of c-Jun
phosphorylation. Previous studies have indicated that c-Jun, a
signal-transducing transcription factor of the AP-1 family, is
associated with apoptosis (18,19).
The present study used c-Jun peptide to inhibit c-Jun
phosphorylation and this treatment resulted in a substantially
decreased number of IEC-6 cells in early apoptosis after heat
stress. These data suggested that a pro-apoptotic pathway may be
induced by NF-κB via enhancement of c-Jun phosphorylation and
caspase-3 activation. However, additional studies are required to
determine the potential mechanism by which NF-κB modulates c-Jun
phosphorylation.
In the present study, the effects of three different
transcription factors, NF-κB, HSF1 and c-Jun, as well as NF-κB
p65-mediated regulation of HSF1 and c-Jun in heat stress-induced
IEC-6 apoptosis was examined. The putative interactions between
NF-κB, HSF1 and c-Jun appeared to be important for apoptosis in
IEC-6 cells, and the interactive mechanism of these three
transcription factors merits further study. In conclusion, an
understanding of NF-κB regulation and the mechanism by which NF-κB
induces cell apoptosis during heat stroke may lead to the
development of novel strategies for treating heat-induced illnesses
related to the intestinal mucosa, in which intestinal epithelial
cell apoptosis serves a major etiological role.
Acknowledgements
The present study was supported by the project team
of The Natural Science Foundation of Guangdong Province (grant no.
s2013030013217).
References
|
1
|
Bouchama A and Knochel JP: Heat stroke. N
Engl J Med. 346:1978–1988. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gao Z, Liu F, Yin P, Wan C, He S, Liu X,
Zhao H, Liu T, Xu J and Guo S: Inhibition of heat-induced apoptosis
in rat small intestine and IEC-6 cells through the AKT signaling
pathway. BMC Vet Res. 9:2412013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yu J, Liu F, Yin P, Zhao H, Luan W, Hou X,
Zhong Y, Jia D, Zan J, Ma W, et al: Involvement of oxidative stress
and mitogen-activated protein kinase signaling pathways in heat
stress-induced injury in the rat small intestine. Stress.
16:99–113. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hsu YL, Yu HS, Lin HC, Wu KY, Yang RC and
Kuo PL: Heat shock induces apoptosis through reactive oxygen
species involving mitochondrial and death receptor pathways in
corneal cells. Exp Eye Res. 93:405–412. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee SJ, Yang ES, Kim SY, Kim SY, Shin SW
and Park JW: Regulation of heat shock-induced apoptosis by
sensitive to apoptosis gene protein. Free Radic Biol Med.
45:167–176. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kretz-Remy C1, Munsch B and Arrigo AP:
NFkappa B-dependent transcriptional activation during heat shock
recovery. Thermolability of the NF-kappaB. Ikappa B complex. J Biol
Chem. 276:43723–43733. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kretz-Remy C, Mehlen P, Mirault ME and
Arrigo AP: Inhibition of I kappa B-alpha phosphorylation and
degradation and subsequent NF-kappa B activation by glutathione
peroxidase overexpression. J Cell Biol. 133:1083–1093. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xu D, Chen M, Ren X, Ren X and Wu Y:
Leonurine ameliorates LPS-induced acute kidney injury via
suppressing ROS-mediated NF-κB signaling pathway. Fitoterapia.
97:148–155. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Baldwin AS: Control of oncogenesis and
cancer therapy resistance by the transcription factor NF-kappaB. J
Clin Invest. 107:241–246. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pahl HL: Activators and target genes of
Rel/NF-kappaB transcription factors. Oncogene. 18:6853–6866. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Monks NR, Biswas DK and Pardee AB:
Blocking anti-apoptosis as a strategy for cancer chemotherapy:
NF-kappaB as a target. J Cell Biochem. 92:646–650. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kasibhatla S, Brunner T, Genestier L,
Echeverri F, Mahboubi A and Green DR: DNA damaging agents induce
expression of Fas ligand and subsequent apoptosis in T lymphocytes
via the activation of NF-kappa B and AP-1. Mol Cell. 1:543–551.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Collins T: Endothelial nuclear
factor-kappa B and the initiation of the atherosclerotic lesion.
Lab Invest. 68:499–508. 1993.PubMed/NCBI
|
|
14
|
Choi YJ, Om JY, Kim NH, Chang JE, Park JH,
Kim JY, Lee HJ, Kim SS and Chun W: Heat shock transcription
factor-1 suppresses apoptotic cell death and ROS generation in
3-nitropropionic acid-stimulated striatal cells. Mol Cell Biochem.
375:59–67. 2013.PubMed/NCBI
|
|
15
|
Verma P, Pfister JA, Mallick S and D'Mello
SR: HSF1 protects neurons through a novel trimerization- and
HSP-independent mechanism. J Neurosci. 34:1599–1612. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cotto JJ, Kline M and Morimoto RI:
Activation of heat shock factor 1 DNA binding precedes
stress-induced serine phosphorylation. Evidence for a multistep
pathway of regulation. J Biol Chem. 271:3355–3358. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu L, Hu C, Huang M, Jiang M, Lu L and
Tang J: Heat shock transcription factor 1 attenuates TNFα-induced
cardiomyocyte death through suppression of NFκB pathway. Gene.
527:89–94. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bossy-Wetzel E, Bakiri L and Yaniv M:
Induction of apoptosis by the transcription factor c-Jun. EMBO J.
16:1695–1709. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Watson A, Eilers A, Lallemand D, Kyriakis
J, Rubin LL and Ham J: Phosphorylation of c-Jun is necessary for
apoptosis induced by survival signal withdrawal in cerebellar
granule neurons. J Neurosci. 18:751–762. 1998.PubMed/NCBI
|
|
20
|
Zhu J, Zhang J, Huang H, Li J, Yu Y, Jin
H, Li Y, Deng X, Gao J, Zhao Q and Huang C: Crucial role of c-Jun
phosphorylation at Ser63/73 mediated by PHLPP protein degradation
in the cheliensisin a inhibition of cell transformation. Cancer
Prev Res (Phila). 7:1270–1281. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stein B, Baldwin AS Jr, Ballard DW, Greene
WC, Angel P and Herrlich P: Cross-coupling of the NF-kappa B p65
and Fos/Jun transcription factors produces potentiated biological
function. EMBO J. 12:3879–3891. 1993.PubMed/NCBI
|
|
22
|
Liu Z, Sun X, Tang J, Tang Y, Tong H, Wen
Q, Liu Y and Su L: Intestinal inflammation and tissue injury in
response to heat stress and cooling treatment in mice. Mol Med Rep.
4:437–443. 2011.PubMed/NCBI
|
|
23
|
Boberts GT, Ghebeh H, Chishti MA,
Al-Mohanna F, El-Sayed R, Al-Mohanna F and Bouchama A:
Microvascular injury, thrombosis, inflammation, and apoptosis in
the pathogenesis of heatstroke: A study in baboon model.
Arterioscler Thromb Vasc Biol. 28:1130–1136. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lawrence T and Fong C: The resolution of
inflammation: Anti-inflammatory roles for NF-kappaB. Int J Biochem
Cell Biol. 42:519–523. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rossi A, Elia G and Santoro MG: Inhibition
of nuclear factor kappa B by prostaglandin A1: An effect associated
with heat shock transcription factor activation. Proc Natl Acad Sci
USA. 94:pp. 746–750. 1997; View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kopp E and Ghosh S: Inhibition of NF-kappa
B by sodium salicylate and aspirin. Science. 265:956–959. 1994.
View Article : Google Scholar : PubMed/NCBI
|