Introduction
Pulmonary artery hypertension (PAH) is a disease
caused by a variety of elements, and is characterized by a
progressive increase in pulmonary circulatory resistance and
pressure in the pulmonary artery (PA), which results in mortality
owing to right-sided heart failure (1). Although the mechanism of PAH remains
unclear, hypoxia is a known trigger factor. Hypoxia-induced PAH and
sustained pulmonary vascular contraction have anti-apoptotic
effects, which lead to imbalanced pulmonary vascular remodeling
(2). Regeneration and extended
survival time of PA smooth muscle cells (PASMCs) may lead to
hypertrophy, vascular wall thickening, vascular diameter narrowing
and increased resistance to blood flow and perfusion (3). Therefore, an imbalance in the
proliferation and apoptosis in PASMCs may be associated with the
development and progression of pulmonary vascular remodeling and
targeting the progression of vascular remodeling may serve as a
novel treatment strategy.
MicroRNAs (miRNAs) are small endogenous non-coding
molecules ~22 nucleotides in length that act on the 3′-untranslated
region (3′-UTR) of target mRNAs and negatively regulate gene
expression. Post-transcriptional regulation by miRNAs affects a
number of physiological and pathological processes, including cell
proliferation and apoptosis, tumor development and various
cardiovascular diseases (4–7). In
previous studies involving exposure to hypoxia, a variety of miRNAs
were revealed to be dysregulated in PAs, including miRNA (miR)-138,
miR-204, miR-20a, miR-145 and miR-190, in which miR-138 expression
was revealed to be increased and therefore may serve an important
role in hypoxic pulmonary vascular remodeling (8–12).
It was demonstrated that miR-138 was expressed in the PASMCs and
was regulated by hypoxia. Exogenous miR-138 expression inhibited
apoptosis and caspase activation in PASMCs and also inhibited the
Bcl-2 expression (9). In hypoxic
conditions, overexpression of miR-138 enhanced the expression
levels of anti-apoptotic protein Bcl-2 and cleaved caspase-3
(13). Previous studies have also
demonstrated that lowering of oxygen from 21 to 2.5% induced the
phosphorylation of extracellular signal regulated kinase (ERK)1/2
in PASMCs and also suggested that the ERK1/2 signaling pathway is
associated with hypoxia-induced proliferation of PASMCs (14). These results suggest that Bcl-2,
caspase-3 and the ERK1/2 signaling pathway may be associated with
miR-138-induced hypoxia in PAH.
Potassium channel subfamily K member 3 (TASK-1) is a
member of the two-pore domain potassium channel family that has
been described in rabbit PASMCs (15) and in rat PAs (16). TASK-1 activity is dependent on
external pH and oxygen tension, which suggested that this potassium
channel may be a sensor of variations in external pH and hypoxia in
PAs. In the present study, miR-138 mimic was used to transfect
human PASMCs (HPASMCs) to establish an in vitro hypoxic PAH
model. The hypothesis that miR-138 contributes to the regulation of
proliferation, membrane potential and apoptosis of HPASMCs by
targeting TASK-1 expression was investigated. Therefore, the
results may provide an important mechanism for the hypoxic
relaxation of PAs.
Materials and methods
Cell culture
Lung tissues were obtained at lung resection from 3
patients (male/female, 2/1; ages 45–68 years) with lung cancer, but
without pulmonary vessel disease and arterial hypoxemia, from The
First Affiliated Hospital of Bengbu Medical College (Bengbu, China)
between May 2010 and February 2015. Tissues were placed on a plate
and washed with ice-cold PBS containing 100 mg/ml penicillin G and
50 µg/ml streptomycin (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). HPASMCs were isolated from distal human PAs
(<1 mm external diameter), and the smooth muscle phenotype was
confirmed immunohistochemically using an α-smooth muscle actin
(α-SMA) antibody (Fig. 1A), as
previously described (17,18). HPASMCs were cultured in RMPI-1640
medium (Thermo Fisher Scientific, Inc.) and maintained at 37°C in
5% CO2 until passage three or six. Ethical approval was
obtained from The First Affiliated Hospital of Bengbu Medical
College Ethics Committee (Bengbu, China) and written informed
consent was obtained from all patients included.
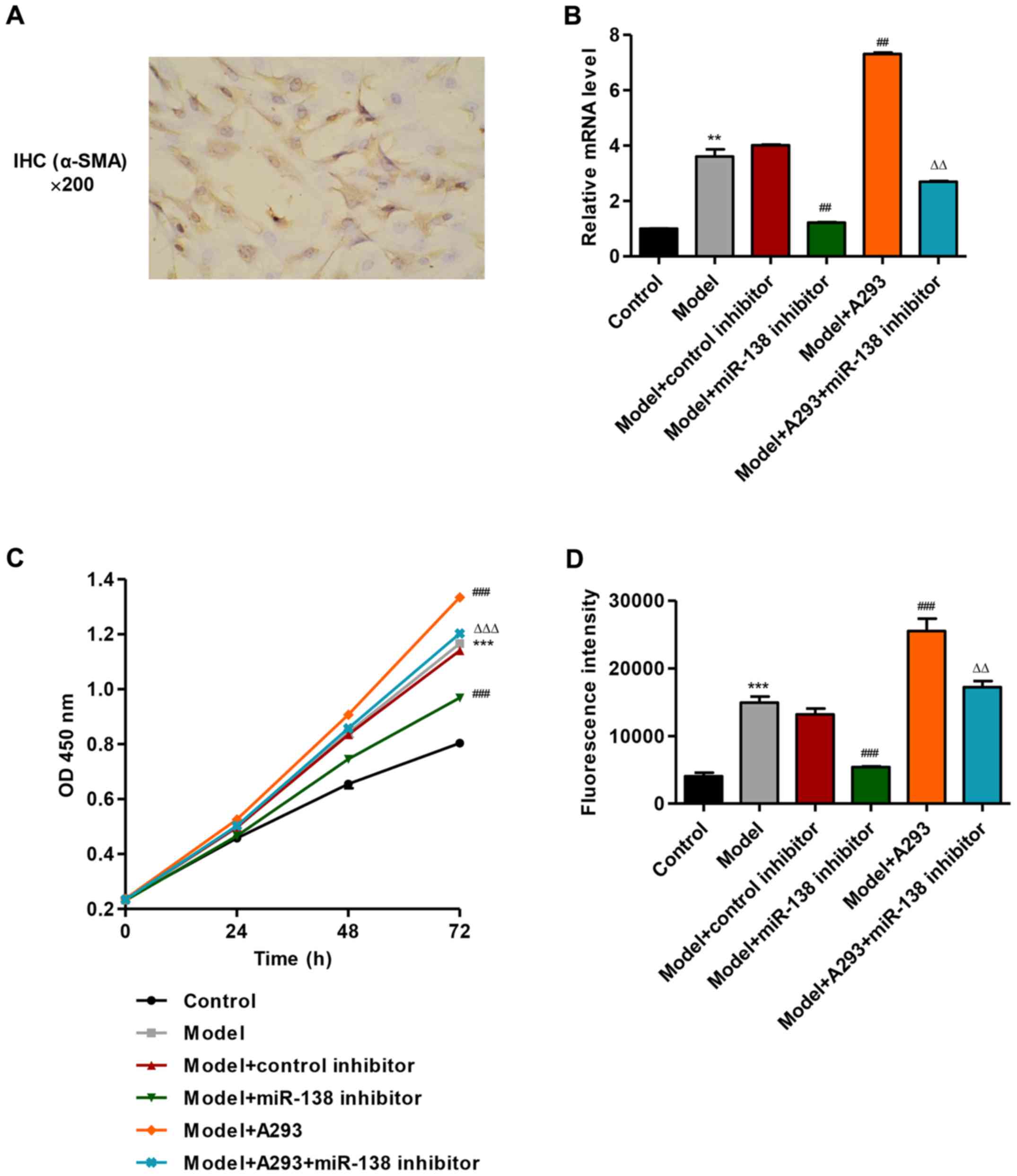 | Figure 1.Effects of miR-138 mimic or miR-138
inhibitor on proliferation and MMP of HPASMCs. HPASMCs were
transfected with miR-138 mimic (model), miR-138 inhibitor and/or
A293 TASK-1 inhibitor for 48 h. (A) Immunohistochemistry for α-SMA
confirming smooth muscle phenotype of HPASMCs. (B) Determination of
mRNA expression levels of miR-138 using RT-qPCR. (C) Cell
proliferation was measured by Cell Counting Kit-8 assay at 0, 24,
48 and 72 h. (D) MMP was measured by flow cytometry. **P<0.01,
***P<0.001 vs. control; ##P<0.01,
###P<0.001 vs. model; ΔΔP<0.01,
ΔΔΔP<0.001 vs. model + A293. α-SMA, α-smooth muscle
actin; HPASMCs, human pulmonary artery smooth muscle cells; IHC,
immunohistochemistry; miR, microRNA; MMP, mitochondrial membrane
potential; OD, optical density; TASK-1, potassium channel subfamily
K member 3. |
Cell treatment
miR-138 mimic (5′-AGCUGGUGUUGUGAAUCAGGCCG-3′) was
purchased from Shanghai GenePharma Co., Ltd. (Shanghai, China). To
establish the hypoxic PAH model, HPASMCs (3×105
cell/well) were seeded in 6-well plates and transiently transfected
with miR-138 mimic (20 µM) (19)
for 6 h at 37°C using Lipofectamine® 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.) in Opti-MEM (Gibco; Thermo Fisher
Scientific, Inc.), according to the manufacturer's instructions.
HPASMCs that were not subjected to transfection were used as a
control. Following 4–8 h transfection, the miR-138
mimic-transfected HPASMCs (model) were subsequently transfected
with an miR-138 inhibitor (20 µM; GE Healthcare Dharmacon, Inc.,
Lafayette, CO, USA) or control inhibitors (non-targeting miR-138
control inhibitor; 20 µM; GE Healthcare Dharmacon, Inc.) for 6 h at
37°C using Lipofectamine® 2000 as described above. In
addition, miR-138 mimic-transfected HPASMCs (model) were also
treated with A293 (100 µM; Sanofi S.A., Paris, France) for 4 h at
37°C in the absence or presence of miR-138 inhibitor transfection.
Following a 48 h time interval post-transfection, subsequent
analyses were performed. A total of 6 experiment groups were used
in subsequent analyses: Control group (untreated HPASMCs), model
group (miR-138 mimic - transfected HPASMCs), model + control
inhibitors group (model + non-targeting miR-138 control inhibitor),
model + miR-138 inhibitor group, model + A293 group, and the model
+ A293 + miR-138 inhibitor group.
Cell counting kit-8 (CCK-8) assay
CCK-8 (Dojindo Molecular Technologies, Rockville,
MD, USA) was used to evaluate the effects of miR-138, as previously
described (20). HPASMCs were
seeded in 96-well plates at 5×103 or 1×104
cells/well in RMPI-1640 at 37°C. CCK-8 solution was added to the
wells at 0, 24, 48 or 72 h, according to the manufacturer's
instructions. The plates were incubated for 1 h at 37°C in 5%
CO2 incubator conditions and the absorbance was read at
450 nm using a microplate reader (Shanghai Utrao Medical Instrument
Co., Ltd., Shanghai, China).
Measurement of mitochondrial membrane
potential (MMP)
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine
iodide (JC-1) probe (Molecular Probes; Thermo Fisher Scientific,
Inc.) was used to detect the alterations in MMP, which aggregates
in the intact mitochondria of non-apoptotic cells emitting
orange-red fluorescence and is widely distributed in apoptotic
cells, emitting green fluorescence in the monomeric form at 488 nm
(21). Cells (5×104
cells/well) were cultured in 24-well plate overnight at 37°C.
Following transfection with the miR-138 mimic or miR-138 inhibitor
as aforementioned, HPASMCs were washed with PBS, incubated with
JC-1 (100 nM) for 20 min at 37°C and subsequently subjected to flow
cytometry (BD Biosciences, Franklin Lakes, NJ, USA). Following
this, the results were subsequently analyzed using CellQuest
software (version 5.1; BD Biosciences).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from HPASMCs using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.), according to
the manufacturer's instructions. cDNA was synthesized from RNA with
a MMLV RT Reagent kit (Thermo Fisher Scientific, Inc.). The
specific primer (5′-AGCUGGUGUUGUGAAUCAGGCCG-3′) was used to
synthesize miR-138 cDNA. cDNA was amplified with a SYBR®
Green PCR kit (Thermo Fisher Scientific, Inc.) on the ABI-7500
Real-Time PCR platform (Invitrogen; Thermo Fisher Scientific,
Inc.). The PCR cycling conditions were as follows: 95°C for 10 min,
followed by 40 cycles at 95°C for 15 sec and 60°C for 45 sec, and a
final extension step of 95°C for 15 sec, 60°C for 1 min, 95°C for
15 sec and 60°C for 15 sec. qPCR primer sequences were as follows:
TASK-1 forward, 5′-CGAGGGAGCCACAACCAAAG-3′ and reverse,
5′-GCAGTGTGCCCAGGCATAAG-3′; Bcl-2 forward,
5′-AGCTGAGCGAGTGTCTCAAG-3′ and reverse, 5′-TGTCCAGCCCATGATGGTTC-3′;
caspase-3 forward, 5′-AACTGGACTGTGGCATTGAG-3′ and reverse,
5′-ACAAAGCGACTGGATGAACC-3′; U6 forward, 5′-CTCGCTTCGGCAGCACA-3′ and
reverse, 5′-AACGCTTCACGAATTTGCGT-3′; GAPDH forward,
5′-CACCCACTCCTCCACCTTTG-3′ and reverse, 5′-CCACCACCCTGTTGCTGTAG-3′.
Relative quantification of miR-138 expression levels was determined
using the 2−ΔΔCq method (22). U6 was used as an internal standard
for the normalization of miR-138 expression. GAPDH used to for the
normalization of the non-miRNA expression data.
Western blot analysis
Western blot analysis was performed according to
standard procedures. Briefly, total protein was isolated from
HPASMCs using radio-immunoprecipitation buffer (Amyjet Scientific,
Inc., Wuhan, China) for 10 min at 95°C, followed by centrifugation
at 400 × g at 25°C for 10 min. The protein concentration was
determined using a Bicinchoninic Acid Protein Assay kit (cat. no.
PICPI23223; Thermo Fisher Scientific, Inc.). A total of 15 µl
protein was loaded into each well and separated by 10% SDS-PAGE
(Amyjet Scientific, Inc.) and transferred to polyvinylidene
difluoride membranes (Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany). The membranes were blocked with fat-free milk for 1 h at
25°C and subsequently incubated with primary antibodies against
TASK-1 (1:800, cat. no. ab49433; Abcam, Cambridge, MA, USA), Bcl-2
(1:300, cat. no. Sc-492; Santa Cruz Biotechnology, Inc., Dallas,
TX, USA), caspase-3 (1:500, cat. no. ab44976; Abcam),
phosphorylated (p)-ERK1/2 (1:1,000, cat. no. 4376), ERK1/2
(1:1,000, cat. no. 4695) and GAPDH (1:2,000, cat. no. 5174) (all
from Cell Signaling Technology, Inc.) for 2 h at 25°C. The
membranes were then incubated with horseradish
peroxidase-conjugated goat anti-rabbit IgG (1:1,000, cat. no.
A0208), donkey anti-goat IgG (1:1,000, cat. no. A0181) and goat
anti-mouse IgG (1:1,000, cat. no. A0216) (all from Beyotime
Institute of Biotechnology, Haimen, China) secondary antibodies for
1 h at 37°C. Signals were detected using an enhanced
chemiluminescence kit (EMD Millipore, Billerica, MA, USA), and the
signal intensity was determined using ImageJ 1.46 software
(National Institutes of Health, Bethesda, MD, USA). GAPDH was used
to normalize the protein expression data.
Dual-luciferase reporter assays
TASK-1 was predicted to interact with miR-138 by
bioinformatics analysis using TargetScan, which is able to predict
biological targets of miRNAs by searching for the presence of 8, 7,
and 6mer sites that match the seed region of each miRNA (23). The mutant and wild-type 3′-UTR of
human TASK-1 were synthesized and inserted downstream of the
firefly luciferase gene in the pGL3 reporter vector (Promega
Corporation, Madison, WI, USA), yielding pGL3-mut-TASK-1 and
wild-type pGL3-TASK-1, respectively. HPASMCs (5×103
cell/well) were seeded in 96-well plates, pGL3-mut-TASK-1 (50 ng)
or wild-type pGL3-TASK-1 (50 ng) plasmids were co-transfected with
miR-138 mimic (5 ng) or miR-138 inhibitor (5 ng) using
Lipofectamine® 2000 at 37°C. Following 24 h, cells were
lysed and activities of firefly luciferase and Renilla luciferase
were examined using the Dual-Luciferase Reporter assay system
(Promega Corporation). Firefly luciferase activity was normalized
to Renilla luciferase activity.
Statistical analysis
Data are expressed as the mean ± standard deviation.
All presented data were representative of a minimum of three
independent experiments. Statistical analysis was performed using
one-way analysis of variance followed by Tukey's post hoc test.
Statistical analysis was performed using GraphPad Prism 5 software
(GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
miR-138 mimic promote proliferation
and suppress mitochondrial depolarization of HPASMCs
miR-138 mimic was transfected into HPASMCs to
establish a PAH model. As revealed in Fig. 1B, transfection of HPASMCs with the
miR-138 mimic (model) significantly increased the expression of
miR-138 by 2.61-fold compared with untreated control HPASMCs,
whereas model cells treated with the miR-138 inhibitor exhibited a
significant decrease in miR-138 expression compared with the model
group. However, the miR-138 control inhibitor did not exhibit a
marked effect on miR-138 expression compared with the model cells.
PAH model HPASMCs were also treated with 100 µM A293, a TASK-1
inhibitor, which significantly enhanced miR-138 expression compared
with the control group, and the model + A293 treated HPASMCs with
miR-138 inhibitor transfection demonstrated a significant decrease
in miR-138 expression compared with the model + A293 only group
(P<0.01; Fig. 1B).
The effects of miR-138 on cell proliferation of
HPASMCs were examined using a CCK-8 assay. Transfection of HPASMCs
with the miR-138 mimic (model) significantly increased cell
proliferation by 45.0% at 72 h compared with untransfected control
cells, whereas PAH model cells treated with the miR-138 inhibitor
exhibited a significant decrease in proliferation compared with the
model group (P<0.001; Fig. 1C).
However, the miR-138 control inhibitor did not exhibit a
significant effect on cell proliferation compared with the model
cells. PAH model HPASMCs were also treated with A293 (100 µM),
which significantly enhanced cell proliferation by 14.5% at 72 h
compared with the model group, and the model + A293 treated HPASMCs
with miR-138 inhibitor transfection significantly decreased the
proliferation compared with the model + A293 only group
(P<0.001; Fig. 1C).
The role of miR-138 on mitochondrial function in
HPASMCs was investigated by measuring the MMP by flow cytometry.
Treatment with miR-138 mimic (model group) resulted in an increase
in MMP levels by 2.70-fold compared with the control (Fig. 1D). Model cells treated with the
miR-138 inhibitor decreased the MMP level significantly compared
with model and the A293-treated model cells (P<0.01). These
results suggested that miR-138 mimic suppress mitochondrial
depolarization of HPASMCs.
miR-138 mimic regulate TASK-1 and
apoptosis-associated protein expression in HPASMCs
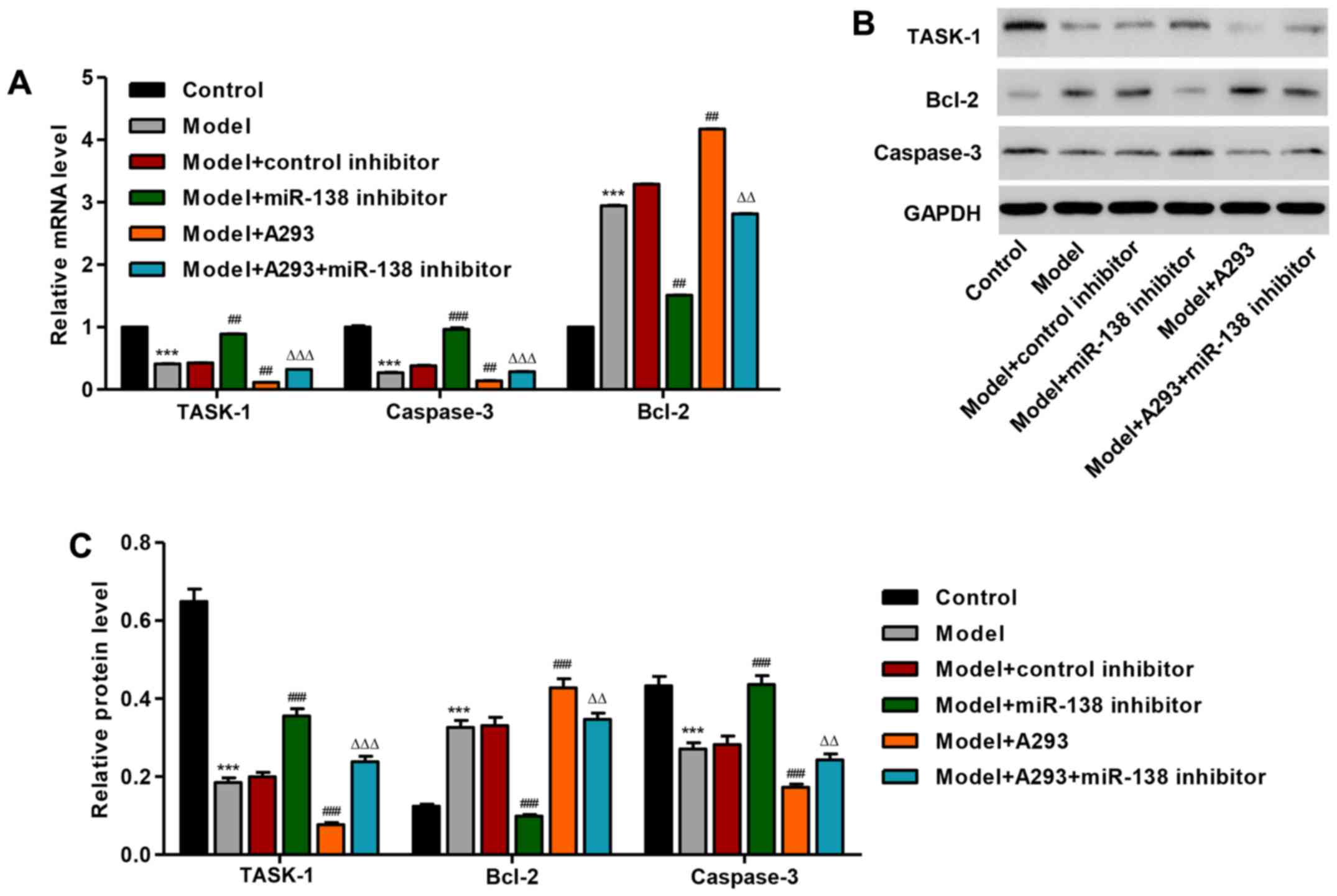
Following treatment with miR-138 mimic, the mRNA
expression of TASK-1 was significantly decreased by 58.9% in
HPASMCs compared with the control group (P<0.001; Fig. 2A). However, miR-138 inhibitor
transfected model cells demonstrated a significantly increased
expression of TASK-1 mRNA compared with the model group and the
model + A293 + miR-138 inhibitor group. Furthermore, miR-138 mimic
(model) significantly increased the mRNA expression level of Bcl-2
and decreased the mRNA expression level of caspase-3, compared with
the untreated control (P<0.001; Fig. 2A); however, co-treatment with the
miR-138 inhibitor significantly reduced the expression levels of
Bcl-2 and significantly increased the expression of caspase-3 mRNA
compared with the model group and the model + A293 + miR-138
inhibitor group, respectively. Similar results were also observed
by western blot analysis (Fig. 2B and
C).
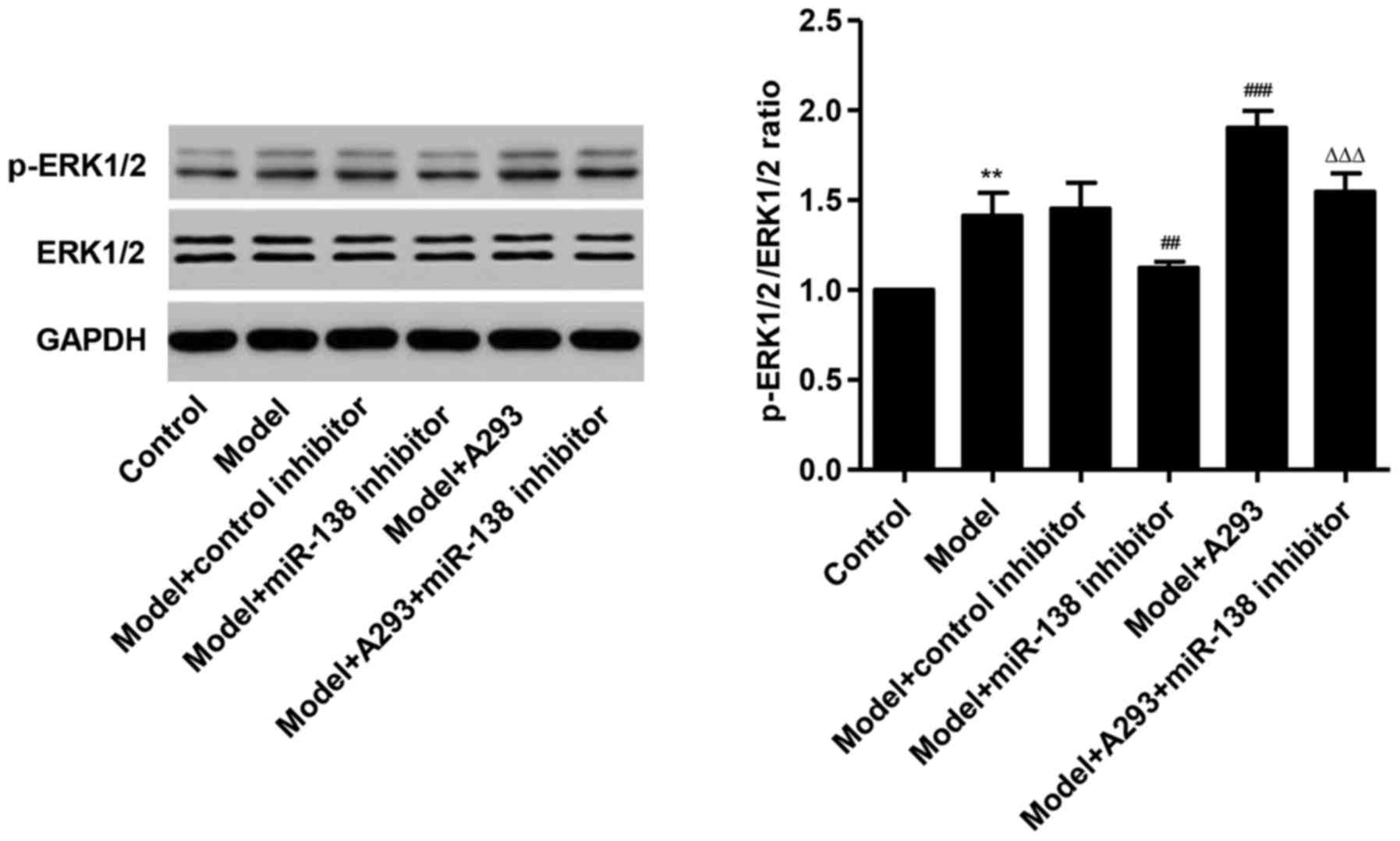
miR-138 mimic activate ERK1/2
signaling in HPASMCs
Following treatment with the miR-138 mimic, the
ratio of p-ERK1/2 to ERK1/2 was significantly increased in HPASMCs
by 40.1% compared with the control (P<0.01; Fig. 3). However, co-treatment with the
miR-138 inhibitor decreased the expression of p-ERK1/2 by 20.3 and
37.5% compared with the model group and the model + A293 + miR-138
inhibitor group, respectively. These data indicated that miR-138
mimic activated ERK1/2 signaling in HPASMCs.
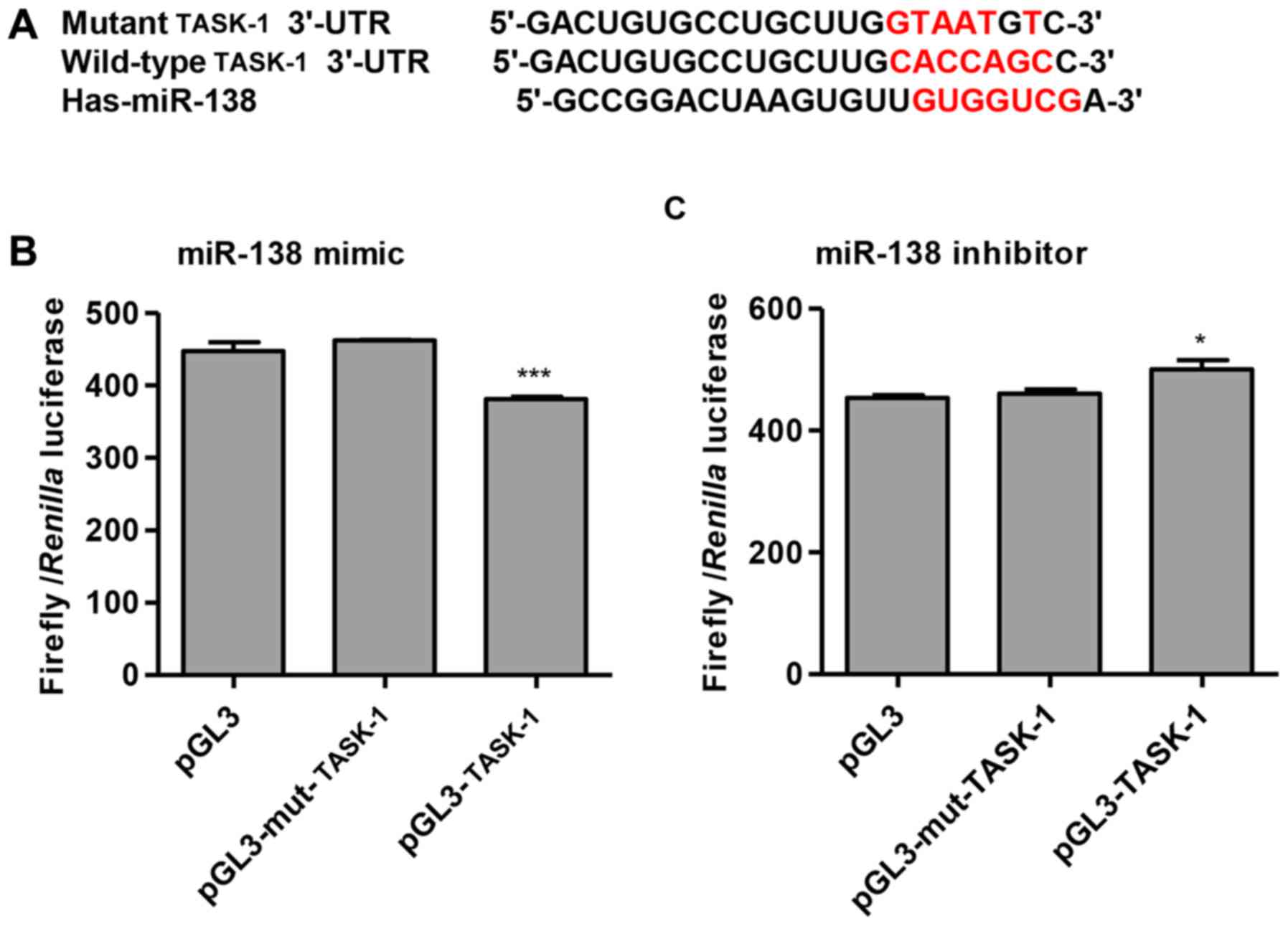
miR-138 targets TASK-1 in HPASMCs
To investigate the regulatory mechanism of miR-138,
bioinformatics analysis (TargetScan) was used, which identified
that TASK-1 mRNA contained potential miR-138 binding sites
(Fig. 4A). To confirm TASK-1 as a
miR-138-regulated target in HPASMCs, wild-type and mutant versions
of the TASK-1 3′-UTR were cloned and inserted into a pGL3
luciferase reporter vector. The luciferase assay demonstrated that
miR-138 mimic transfection significantly suppressed luciferase
activity in cells co-transfected with the wild-type TASK-1 3′-UTR
compared with cells co-transfected with the mutant TASK-1 3′-UTR
(P<0.001; Fig. 4B), and that
treatment with the miR-138 inhibitor significantly increased
luciferase activity in the presence of TASK-1 3′-UTR compared with
cells co-transfected with the mutant TASK-1 3′-UTR (P<0.05;
Fig. 4C). These data suggested
that TASK-1 may be a direct target of miR-138 in HPASMCs.
Discussion
In the present study, evidence for the role for
miR-138 in regulating HPASMCs proliferation, membrane potential and
apoptosis in hypoxic PAH, along with the underlying mechanisms has
been demonstrated. The importance of miRNAs in pathological
processes is being recognized, particularly in cardiovascular
disease (24), and a number of
miRNAs have been implicated in signal transduction pathways
relevant to PAH (25,26). miR-190 regulates hypoxic pulmonary
vascular contractility by targeting a voltage-gated potassium
channel (27). miR-190
demonstrated negative correlation with the expression of a
voltage-dependent K+ channel protein under hypoxia
(12). miR-21 in PAH was
previously demonstrated to regulate cell proliferation and
apoptosis by regulating the expression of proteins that regulate
Bcl-2 and Akt signaling pathways (28). These previous data suggested that
miRNAs are important in vascular cell fate and are involved in the
progression of PAH. The present study focused on miR-138 and
demonstrated its effects on proliferation, membrane potential and
apoptosis in HPASMCs.
The results of the present study indicated that
miR-138 promoted proliferation and attenuated mitochondrial
depolarization in HPASMCs. The deregulation of miR-138 is
frequently associated with the inhibition of proliferation and
migration of smooth muscle cells (29). The signaling pathway underlying the
miR-138-associated anti-apoptosis of HPASMCs was also examined in
the present study, and it was demonstrated that miR-138 induces
ERK1/2 phosphorylation that leads to the cleavage of procaspase-3
and the upregulation of Bcl-2. Caspase activity is known to induce
mitochondrial damage during apoptosis (30).
According to bioinformatics-based analysis and
luciferase assay, TASK-1 was identified as a direct target of
miR-138 in HPASMCs. TASK-1 has been demonstrated previously to
serve a role in neuronal apoptosis (31), which further suggested that
K+ two-pore channels may exhibit a certain role as
modulators of cell proliferation. The mechanism by which
K+ channels may alter the proliferative state of cells
has been an open question for several years. TASK-1 is expressed in
HPASMCs and is hypoxia-sensitive, and controls the resting membrane
potential (15), therefore
suggesting an important role for TASK-1 K+ channels in
the regulation of pulmonary vascular tone.
In conclusion, the results from the present study
indicated that miR-138 may serve an important role in the
proliferation, membrane potential and apoptosis of HPASMCs. miR-138
overexpression was demonstrated to suppress TASK-1 expression,
activate ERK1/2 signaling, and induce of Bcl-2 expression and
decrease caspase-3 expression. Therefore, the stabilization of the
miR-138 level may be a novel strategy for the clinical treatment of
PAH.
Acknowledgements
The present study was supported by research grants
from The National Natural Science Foundation of China (grant no.
81170046), The Anhui Province Education General Projects (grant no.
KJ2015B012by), The Key Project of Top-Notch Talent of Discipline
(specialty) of the Higher Education Institute of Anhui Province
2016, China and from The Anhui Province Education Key Projects
(grant no. KJ2015A159).
References
|
1
|
Rosenblum WD: Pulmonary arterial
hypertension: Pathobiology, diagnosis, treatment, and emerging
therapies. Cardiol Rev. 18:58–63. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pak O, Aldashev A, Welsh D and Peacock A:
The effects of hypoxia on the cells of the pulmonary vasculature.
Eur Respir J. 30:364–372. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sommer N, Dietrich A, Schermuly RT,
Ghofrani HA, Gudermann T, Schulz R, Seeger W, Grimminger F and
Weissmann N: Regulation of hypoxic pulmonary vasoconstriction:
Basic mechanisms. Eur Respir J. 32:1639–1651. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sarkar J, Gou D, Turaka P, Viktorova E,
Ramchandran R and Raj JU: MicroRNA-21 plays a role in
hypoxia-mediated pulmonary artery smooth muscle cell proliferation
and migration. Am J Physiol Lung Cell Mol Physiol. 299:L861–L871.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cha ST, Chen PS, Johansson G, Chu CY, Wang
MY, Jeng YM, Yu SL, Chen JS, Chang KJ, Jee SH, et al: MicroRNA-519c
suppresses hypoxia-inducible factor-1alpha expression and tumor
angiogenesis. Cancer Res. 70:2675–2685. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jiang Y, Yin H and Zheng XL: MicroRNA-1
inhibits myocardin-induced contractility of human vascular smooth
muscle cells. J Cell Physiol. 225:506–511. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Guo L, Qiu Z, Wei L, Yu X, Gao X, Jiang S,
Tian H, Jiang C and Zhu D: The microRNA-328 regulates hypoxic
pulmonary hypertension by targeting at insulin growth factor 1
receptor and L-type calcium channel-α1C. Hypertension.
59:1006–1013. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Courboulin A, Paulin R, Giguère NJ,
Saksouk N, Perreault T, Meloche J, Paquet ER, Biardel S, Provencher
S, Côté J, et al: Role for miR-204 in human pulmonary arterial
hypertension. J Exp Med. 208:535–548. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li S, Ran Y, Zhang D, Chen J, Li S and Zhu
D: MicroRNA-138 plays a role in hypoxic pulmonary vascular
remodelling by targeting Mst1. Biochem J. 452:281–291. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brock M, Samillan VJ, Trenkmann M,
Schwarzwald C, Ulrich S, Gay RE, Gassmann M, Ostergaard L, Gay S,
Speich R and Huber LC: AntagomiR directed against miR-20a restores
functional BMPR2 signalling and prevents vascular remodelling in
hypoxia-induced pulmonary hypertension. Eur Heart J. 35:3203–3211.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Caruso P, Dempsie Y, Stevens HC, McDonald
RA, Long L, Lu R, White K, Mair KM, McClure JD, Southwood M, et al:
A role for miR-145 in pulmonary arterial hypertension: Evidence
from mouse models and patient samples. Circ Res. 111:290–300. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li SS, Ran YJ, Zhang DD, Li SZ and Zhu D:
MicroRNA-190 regulates hypoxic pulmonary vasoconstriction by
targeting a voltage-gated K+ channel in arterial smooth muscle
cells. J Cell Biochem. 115:1196–1205. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
He S, Liu P, Jian Z, Li J, Zhu Y, Feng Z
and Xiao Y: miR-138 protects cardiomyocytes from hypoxia-induced
apoptosis via MLK3/JNK/c-jun pathway. Biochem Biophys Res Commun.
441:763–769. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li GW, Xing WJ, Bai SZ, Hao JH, Guo J, Li
HZ, Li HX, Zhang WH, Yang BF, Wu LY, et al: The calcium-sensing
receptor mediates hypoxia-induced proliferation of rat pulmonary
artery smooth muscle cells through MEK1/ERK1,2 and PI3K pathways.
Basic Clin Pharmacol Toxicol. 108:185–193. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Olschewski A, Li Y, Tang B, Hanze J, Eul
B, Bohle RM, Wilhelm J, Morty RE, Brau ME, Weir EK, et al: Impact
of TASK-1 in human pulmonary artery smooth muscle cells. Circ Res.
98:1072–1080. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bryan RM Jr, You J, Phillips SC, Andresen
JJ, Lloyd EE, Rogers PA, Dryer SE and Marrelli SP: Evidence for
two-pore domain potassium channels in rat cerebral arteries. Am J
Physiol Heart Circ Physiol. 291:H770–H780. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Davie N, Haleen SJ, Upton PD, Polak JM,
Yacoub MH, Morrell NW and Wharton J: ET(A) and ET(B) receptors
modulate the proliferation of human pulmonary artery smooth muscle
cells. Am J Respir Crit Care Med. 165:398–405. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang XF, Zhu J, Geng WY, Zhao SJ, Jiang
CW, Cai SR, Cheng M, Zhou CY and Liu ZB: Electroacupuncture at
Feishu (BL13) and Zusanli (ST36) down-regulates the expression of
orexins and their receptors in rats with chronic obstructive
pulmonary disease. J Integr Med. 12:417–424. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu X, Jiang L, Wang A, Yu J, Shi F and
Zhou X: MicroRNA-138 suppresses invasion and promotes apoptosis in
head and neck squamous cell carcinoma cell lines. Cancer Lett.
286:217–222. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang YH, Wang Y, Yusufali AH, Ashby F,
Zhang D, Yin ZF, Aslanidi GV, Srivastava A, Ling CQ and Ling C:
Cytotoxic genes from traditional Chinese medicine inhibit tumor
growth both in vitro and in vivo. J Integr Med.
12:483–494. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Perelman A, Wachtel C, Cohen M, Haupt S,
Shapiro H and Tzur A: JC-1: Alternative excitation wavelengths
facilitate mitochondrial membrane potential cytometry. Cell Death
Dis. 3:e4302012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Corsten MF, Dennert R, Jochems S,
Kuznetsova T, Devaux Y, Hofstra L, Wagner DR, Staessen JA, Heymans
S and Schroen B: Circulating microRNA-208b and microRNA-499 reflect
myocardial damage in cardiovascular disease. Circ Cardiovasc Genet.
3:499–506. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim J, Kang Y, Kojima Y, Lighthouse JK, Hu
X, Aldred MA, McLean DL, Park H, Comhair SA, Greif DM, et al: An
endothelial apelin-FGF link mediated by miR-424 and miR-503 is
disrupted in pulmonary arterial hypertension. Nat Med. 19:74–82.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gou D, Ramchandran R, Peng X, Yao L, Kang
K, Sarkar J, Wang Z, Zhou G and Raj JU: miR-210 has an
antiapoptotic effect in pulmonary artery smooth muscle cells during
hypoxia. Am J Physiol Lung Cell Mol Physiol. 303:L682–L691. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li SS, Ran YJ, Zhang DD, Li SZ and Zhu D:
MicroRNA-190 regulates hypoxic pulmonary vasoconstriction by
targeting a voltage-gated K+ channel in arterial smooth muscle
cells. J Cell Biochem. 115:1196–1205. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang S, Banerjee S, De Freitas Ad, Cui H,
Xie N, Abraham E and Liu G: miR-21 regulates chronic
hypoxia-induced pulmonary vascular remodeling. Am J Physiol Lung
Cell Mol Physiol. 302:L521–L529. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xu J, Li L, Yun HF and Han YS: MiR-138
promotes smooth muscle cells proliferation and migration in db/db
mice through down-regulation of SIRT1. Biochem Biophys Res Commun.
463:1159–1164. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Takahashi A, Masuda A, Sun M, Centonze VE
and Herman B: Oxidative stress-induced apoptosis is associated with
alterations in mitochondrial caspase activity and Bcl-2-dependent
alterations in mitochondrial pH (pHm). Brain Res Bull. 62:497–504.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Patel AJ and Lazdunski M: The 2P-domain K+
channels: Role in apoptosis and tumorigenesis. Pflugers Arch.
448:261–273. 2004. View Article : Google Scholar : PubMed/NCBI
|