Introduction
Stroke is the second most prevalent cause of
mortality worldwide and ~8 million succumb to it annually (1). Ischemic stroke accounts for ~80% of
strokes, and results from the occlusion of a major intracerebral
artery by a thrombus or embolism (2). Re-canalization of occluded cerebral
blood vessels is the effective approach to restore the cerebral
flow and minimize deleterious effects of ischemia on neurons
(3). However, several studies have
indicated that ischemic-reperfusion (I/R) usually induces reactive
oxygen species overproduction, which are highly deleterious and
result in neuronal cell injury or death (4,5).
Therefore, it has become important to explore ways to prevent
cerebral I/R injuries.
Bone morphogenetic proteins (BMPs) are members of
the transforming growth factor-β (TGF-β) superfamily that were
originally identified as factors that induce the formation of bone
and cartilage (6). However, recent
studies indicate BMPs are also expressed in the central nervous
system and involved in the growth, differentiation and apoptosis of
neuronal cells under normal and pathological conditions, including
cerebral I/R (7–9). BMP family members include the BMP-2/4
group (BMP2 and BMP4), osteogenic protein-1 group (BMP5, BMP6, BMP7
and BMP8) and BMP-9/10 group (BMP9 and BMP10) (10). Of these, certain members have been
demonstrated to exert protective roles for cerebral I/R injuries.
For example, Xu et al (11)
demonstrated that the administration of rhBMP7 via a femoral vein
injection 30 min prior to reperfusion significantly increases
neurological function, decreases brain water content and
pathological and morphological damage, and may be associated with
reduced nuclear factor-κB activity. Using the same procedure, Pei
et al (12) also
demonstrated that BMP-7 significantly improves neurological
deficiency and reduces the infarct volume via attenuating oxidative
stress and inhibiting neuronal apoptosis following cerebral
ischemia. By in vitro and vivo experimentation, Wang
et al (13) demonstrated
that BMP6 treatment may reduce cerebral ischemia/reperfusion injury
by alleviating neuronal apoptosis, with decreased immunoreactivity,
enzymatic activity of caspase-3, and density of terminal
deoxynucleotidyl transferase dUTP nick end labeling-positive cells
in the ischemic cortex. However, there have been no studies, to the
best of the authors' knowledge, investigating the role of BMP9, a
newly identified factor for developing basal forebrain cholinergic
neurons (14), in cerebral I/R
injuries.
The aim of the present study was to investigate
whether BMP9 has a protective role in preventing focal cerebral
(IR) injuries and their underlying mechanisms. The results may
provide insight into the underlying functional role of BMP9 in the
nervous system lesion and repair.
Materials and methods
Animals and experimental groups
Adult male Sprague-Dawley rats (n=40; 280–330 g;
aged between 16 and 17 weeks) were obtained from the Experiment
Animal Center of Chongqing Medical University. They were housed in
rooms with controlled temperature (22–25°C), humidity (50–60%) and
a 12-h light/dark cycle, with free access to food and water. All
rats were randomly divided into four groups (n=10): i) Normal
control; ii) sham surgery group; iii) I/R group; and iv) Ad-BMP9 +
I/R group. All animal experiments were approved by the
Institutional Animal Care and Use Committee of Chongqing Medical
University and conducted in accordance with the principles and
procedures outlined in the National Institutes of Health Guide for
the Care and Use of Laboratory Animals (https://grants.nih.gov/grants/olaw/Guide-for-the-Care-and-use-of-laboratory-animals.pdf).
Construction of focal cerebral I/R
model
The transient focal cerebral I/R injury model was
induced via the right middle cerebral artery occlusion (MCAO) as
described by Longa et al (15). Briefly, rats were anesthetized by
intraperitoneal injection of 350 mg/kg chloral hydrate
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) following an
overnight fast and placed in a supine position. A midline neck
incision was made to expose the right common carotid artery,
internal carotid artery (ICA) and external carotid artery (ECA). A
4-0 monofilament nylon suture (Ethicon, Inc., Osaka, Japan) with a
rounded tip was inserted from the lumen of the ECA to that of the
right ICA until a resistance was felt to occlude the origin of the
right middle cerebral artery. Throughout the procedure, body
temperature was maintained within the physiological range by a
thermostatically controlled infrared lamp. Reperfusion was
performed 2 h following MCAO via thread withdrawal. Sham-surgery
rats underwent the same vessel exposure, however without MCAO.
Adenoviral vector (Ad)-BMP9 expression
vector construction
The adenoviral vectors carrying BMP9 and/or GFP were
obtained as a gift from Molecular Oncology Laboratory, the
University of Chicago Medical Center (Chicago, IL, USA). They were
constructed using the AdEasy System (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) according to the manufacturer's
protocol. The recombinant adenoviruses were further packaged and
amplified in 293 cells (ATCC, Manassas, VA, USA) followed by
storage at −80°C (16).
Intracerebroventricular infusion of
Ad-BMP9
Ad-BMP9 was intracerebroventricularly injected into
the rat brain 2 days prior to MCAO. In brief, rats were
anesthetized with 10% chloral hydrate (3 ml/kg) and placed in a
stereotaxic frame. Ad-BMP9 (0.1 ml) was injected into the right
lateral ventricle via a 1 mm burr hole drilled 1.5 mm caudal to the
bregma and 2 mm lateral to the midline. The needle was kept in the
ventricle for 10 min, and then withdrawn. Following the closure of
the wound, the rats were returned to their cages and fed as
normal.
Evaluation of neurological
deficits
Neurological deficits were evaluated 24 h following
reperfusion according to the method of Longa et al (15). Neurological findings were scored on
a 5-point scale: 0 points, no deficit; 1 point, failure to extend
left forepaw; 2 points, circling to the left; 3 points, paresis to
the left; 4 points, no walking spontaneously with depressed level
of consciousness. The cerebral I/R model was considered to be
successfully constructed with a neurological deficits score
>3.
Estimation of cerebral infarct
volume
The cerebral infarct volume was determined by
2,3,5-triphenyltetrazolium chloride (TTC) staining (17). Briefly, the rats were deeply
anesthetized with chloral hydrate (10% chloral hydrate; 3 ml/kg) 24
h following reperfusion and then decapitated, following which the
brains were rapidly removed and frozen at −20°C for 20 min. Brain
tissue was sliced into five 2 mm sections and stained with 2% TTC
solution for 30 min at 37°C in the dark followed by overnight
immersion in 4% paraformaldehyde. The tissue slices were digitally
photographed and the volume of the infarction (indicated as a white
color) was analyzed and expressed as percentage relative to the
total brain volume using the ImageJ software (version 1.37;
National Institutes of Health, Bethesda, MD, USA).
Isolation and culture of
astrocytes
Astrocytes were dissociated from eight neonatal
Sprague-Dawley rats (Experiment Animal Center of Chongqing Medical
University) (18). Briefly, the
head of neonatal rats was cut off following anesthesia (10% chloral
hydrate; 3 ml/kg) and the brains were removed from the skulls.
Following careful removal of the meninges and pia mater, the brain
tissues were cut into pieces by scissors and digested with trypsin
for 10 min at 37°C, then plated on poly-L-lysine-coated culture
dishes with Dulbecco's modified Eagle's medium (DMEM) (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 10% fetal bovine
serum (Gibco; Thermo Fisher Scientific, Inc.) in a humidified
atmosphere of 5% CO2 at 37°C. The culture medium was
changed every 2 days. When the primary cells reached 80–95%
confluence, the cells were detached by trypsinization, centrifuged
at 300 × g for 5 min at room temperature, and replaced. The
subsequent passage cells were used for the following experiment
when 85–100% confluence was achieved.
Simulation of oxygen-glucose
deprivation and reoxygenation model
An oxygen-glucose deprivation and reoxygenation
(OGD/R) model was constructed as previously described (19). Having being washed twice,
astrocytes were immersed in glucose-free DMEM and placed in an
incubator with a premixed gas (1% O2, 95% N2
and 5% CO2) for 4 h. Then, cells were maintained in
normal DMEM (including 5.6 mmol/l glucose) and transferred to a 5%
CO2 incubator at 37 °C for 24 h. For the non-OGD/R
group, cultures were incubated in normal DMEM and placed in 5%
CO2 in air at 37°C for 28 h. Astrocytes were also
treated with Ad-GFP, Ad-BMP9, Ad-BMP9 + extracellular
signal-regulated kinases (ERK) inhibitor PD098059 (20 µM;
Sigma-Aldrich; Merck KGaA), Ad-BMP9 + c-Jun N-terminal kinase (JNK)
inhibitor SP600125 (20 µm; Cell Signaling Technology, Inc.,
Danvers, MA, USA), Ad-BMP9 + p38 inhibitor SB203580 (20 µm; Cell
Signaling Technology, Inc.), PD098059 (20 µm), SP600125 (20 µm),
and SB203580 (20 µm) for 24 h and then subjected to OGD/R
treatment.
Cell viability analysis
Cell viability was assessed by
3-(4,5-dimethylthiazol-2yl)-5-(3-carboxymethoxyphenyl)-(4-sulfophenyl)-2H-tetrazolium
(MTS) method. Astrocytes were cultured in 96-well plates and MTS
solution added. Following 3 h in the dark, cells were measured on a
microplate reader at a wavelength of 490 nm (Bio-Rad Laboratories,
Inc., Hercules, CA, USA).
Cell death analysis
A lactate dehydrogenase release assay was used to
indirectly evaluate cytotoxicity (cell death) of astrocytes
following OGD/R, which was performed using a commercially available
kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Absorbance was detected at a wavelength of 450 nm on microplate
reader (Bio-Rad Laboratories, Inc.).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RT-qPCR was used to investigate the mRNA expression
of BMP9, ERK, P38 and JNK. Total RNA was isolated from the brain
tissues or astrocytes using RNAiso Plus (Takara Biotechnology Co.,
Ltd., Dalian, China) according to the manufacturer's protocol. cDNA
was synthesized with PrimeScript RT reagent kit (Takara
Biotechnology Co., Ltd.). The PCR primers used were: BMP9 forward,
5′-CCTTCTTTCATTCCCTCTGTGA-3′ and reverse,
5′-CCCAACCTTTTGACCCTTTTTA-3′, 143 bp; ERK forward,
5′-TGAAGGATGACGACTTTGAGAA-3′ and reverse,
5′-CTGTAGAACGCACCATAGAAGC-3′, 217 bp; P38 forward,
5′-GAGCGTTACCAGAACCTGTCTC-3′ and reverse,
5′-TGAATGATGGACTGAAATGGTC-3′, 128 bp; JNK forward,
5′-GACACGAAGACGAACTTGAGC-3′ and reverse,
5′-CGTAATAGGCAGGAAAGACACC-3′ 126 bp; and β-actin forward,
5′-CACCCGCGAGTACAACCTTC-3′ and reverse, 5′-CCCATACCCACCATCACACC-3′,
207 bp). Real-time qPCR was run on a Bio-Rad CFX-96 real-time PCR
system (Bio-Rad Laboratories, Inc.) using SYBR Premix Ex Taq kit
(Takara Biotechnology Co., Ltd.) at 95°C for 30 sec, followed by 39
cycles of 95°C for 5 sec, and 60°C for 30 sec. The gene expression
levels relative to internal standard β-actin were calculated by the
2−ΔΔCq method (20).
Western blotting
Western blotting was used to investigate the protein
expression of BMP9, ERK, P38 and JNK. The protein samples were
extracted from brain tissues or astrocytes using RIPA lysis buffer
(BioTeke Corporation, Beijing, China) and then protein levels were
determined using the bichioninic acid method. A 20 µg protein
sample was subjected to 12% SDS-PAGE gel and transferred onto
polyvinylidene fluoride membranes. Blots were blocked with 5%
non-fat milk solution for 1 h at room temperature and then
incubated overnight with anti-BMP9 (cat. no. sc514211; 1:2,000;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA), anti-p-ERK (cat.
no. BS74621; 1:2,000; Bioworld Technology, Inc., St. Louis Park,
MN, USA), anti-p-P38 (cat. no. BS6381; 1:2,000; Bioworld
Technology, Inc.), or anti-p-JNK (cat. no. Bs4763; 1:2,000;
Bioworld Technology, Inc.) at 4°C followed by incubation with
horseradish peroxidase-conjugated goat anti-rabbit IgG secondary
antibodies (cat. no. ZB2301; 1:3,000; Beijing Zhongshan Jinqiao
Biotechnology Co., Ltd., Beijing, China) for 1 h. The blots were
visualized with the ECL system (GE Healthcare Life Sciences, Little
Chalfont, UK) and the intensity of each band quantified by using
the Chemi Doc XRS system.
Statistical analysis
RNA and protein assay data are presented as the mean
± standard error of the mean, which were repeated three times.
Differences between groups were analyzed using Tukey's post hoc
test following one-way analysis of variance through SPSS software,
version 18.0 (SPSS, Inc., Chicago, IL, USA). A t-test was used to
compare the differences between two groups. P<0.05 was
considered to indicate a statistically significant difference. The
area of TTC staining and cerebral infarction was photographed with
a digital camera, and results analyzed using Image J software
(version 1.37).
Results
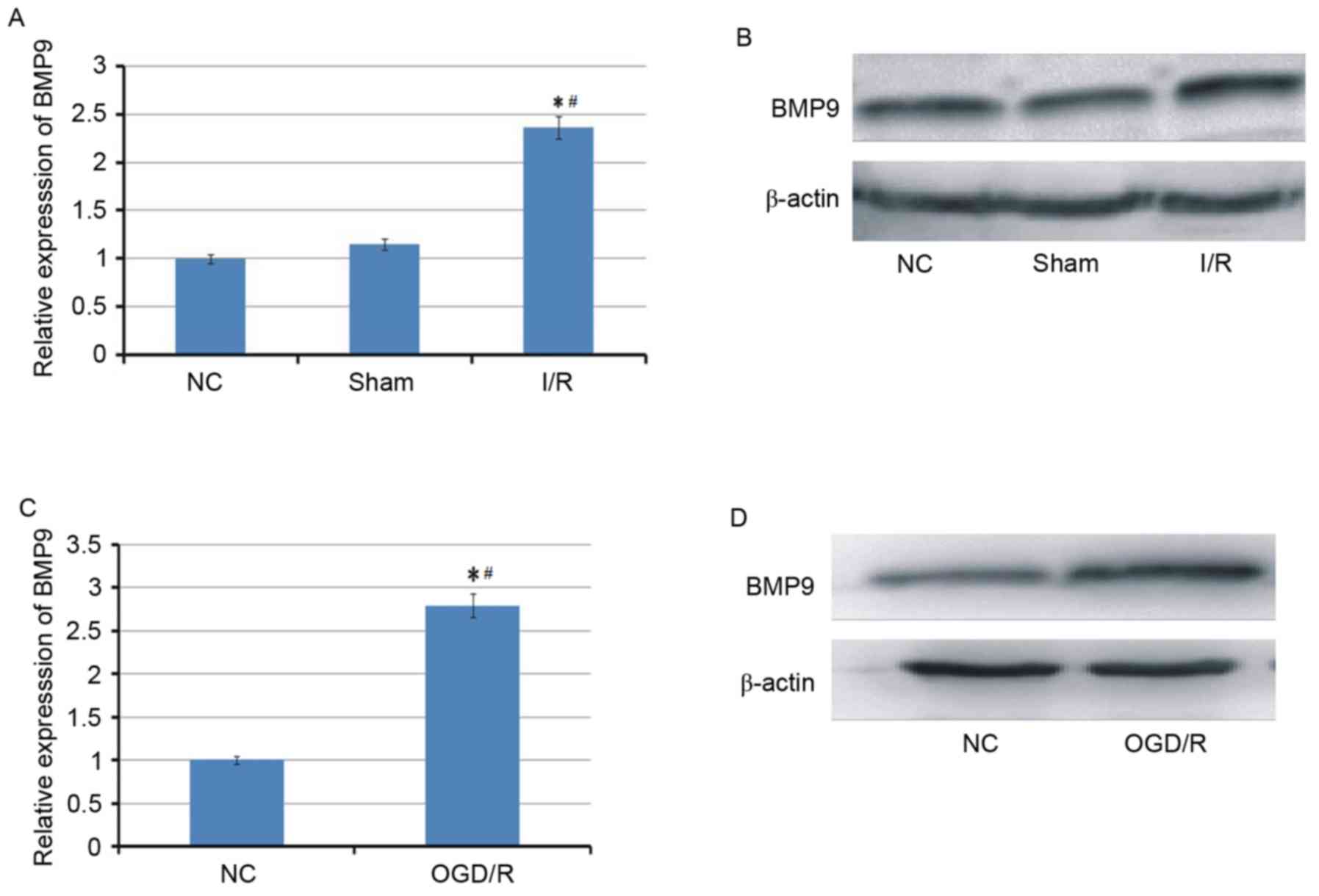
Cerebral I/R induces the upregulated
expression of BMP9
To investigate whether BMP9 is involved in cerebral
I/R, the expression of BMP9 was first examined in cerebral I/R
animal and cell models. As demonstrated in Fig. 1, BMP9 was identified to be
upregulated at mRNA and protein levels in the cerebral I/R animal
model triggered by MACO and the cell model following OGD/R
treatment.
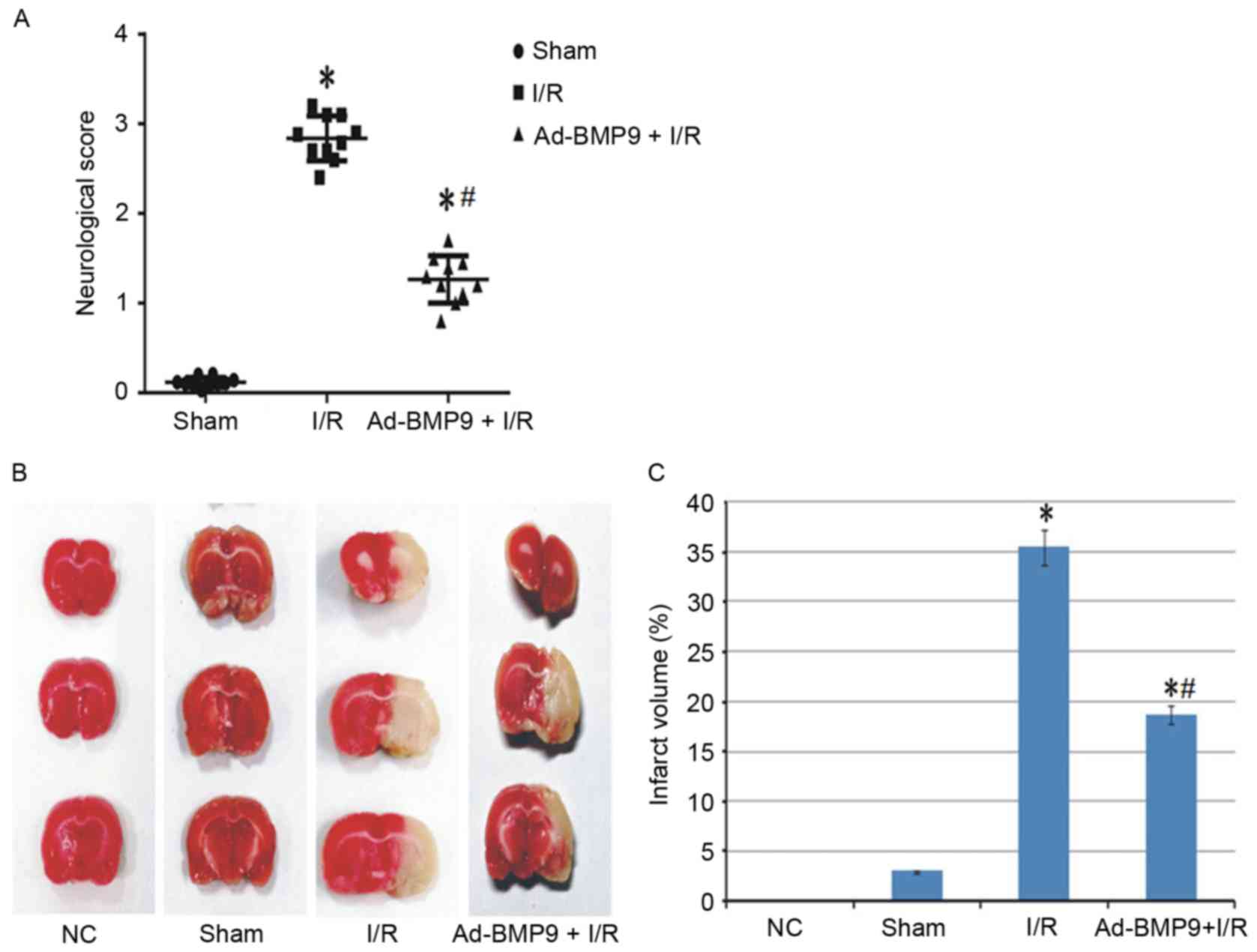
Effect of BMP9 overexpression on
neurological function and infarction
To confirm whether the role of upregulated BMP9 is
protective or deteriorative, BMP9 was overexpressed via adenovirus
vector mediation followed by cerebral I/R treatment. Previous
studies have suggested that cerebral I/R leads to neurological
deficit and cerebral infarction (21,22).
Thus, the role of BMP9 overexpression on neurological function was
assessed according to the score defined by Longa et al
(15) and infarct volume stained
by TTC. As a result, normal and sham surgery rats presented almost
no neurological deficit and infarct volume, however this
significantly increased in the I/R group. Furthermore, BMP9
pretreatment significantly reduced the neurological score and
infarct volume compared with I/R rats (Fig. 2).
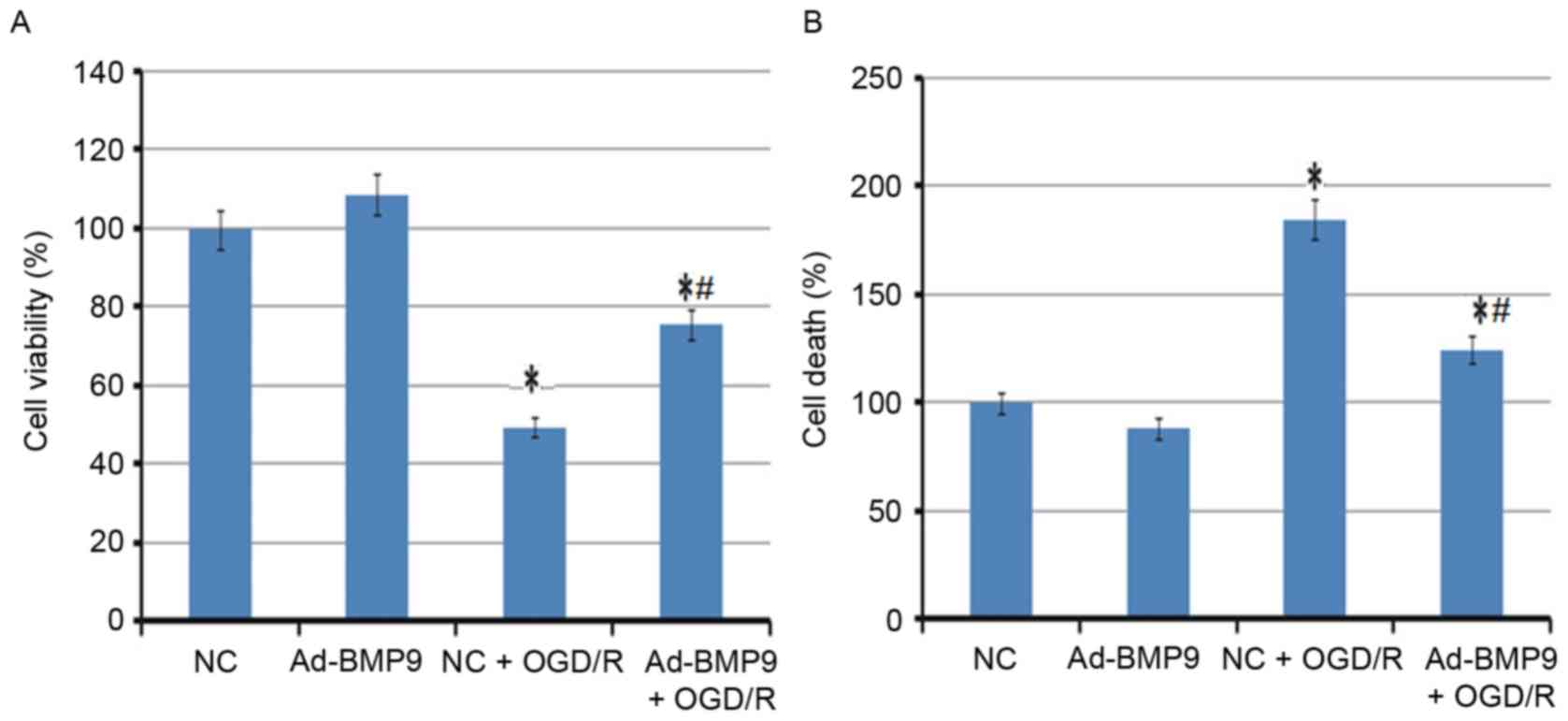
Effect of BMP9 overexpression on cell
viability and death of astrocytes
In addition to the animal model, the overexpression
of BMP9 on astrocytes was also analyzed. In agreement with the
animal experiment, cell viability of astrocytes was significantly
reduced, however the death of astrocytes was enhanced following
OGD/R treatment. However, the BMP9 pretreatment significantly
alleviated this condition, with the increased cell viability and
lower cell death rate (Fig.
3).
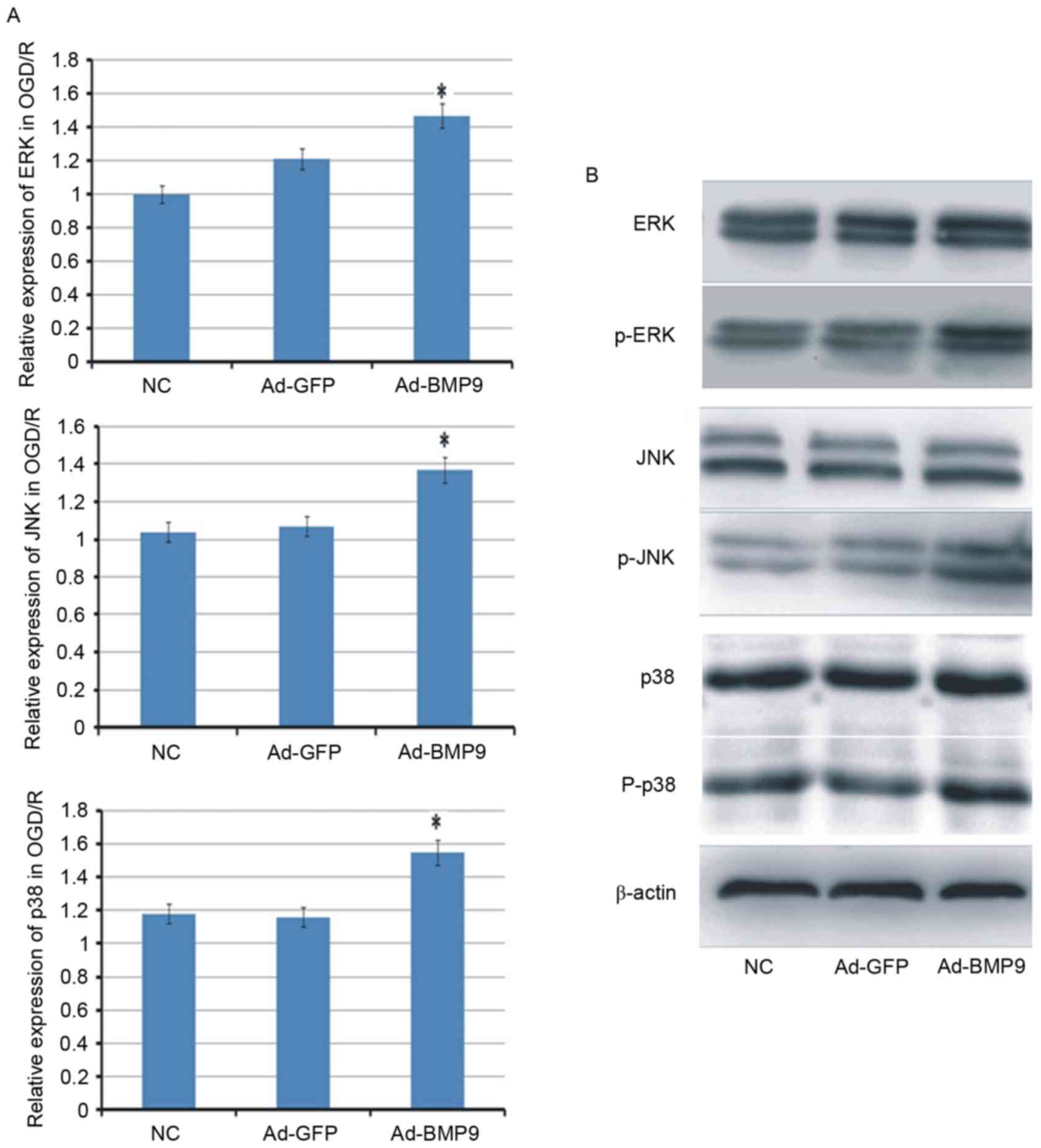
Underlying alleviation mechanism of
BMP9 for cerebral I/R
To further explore the mechanism of BMP9 on cerebral
I/R, three signaling pathways (p38, ERK and JNK) that usually serve
important roles in cell survival, were determined. The results
indicated that the mRNA expression levels of p38, ERK and JNK were
all increased in OGD/R-treated astrocytes. At the protein levels,
the expression of p38, ERK and JNK appeared to be similar in
different groups, however, their phosphorylation levels were
improved by BMP9 with different degrees (Fig. 4).
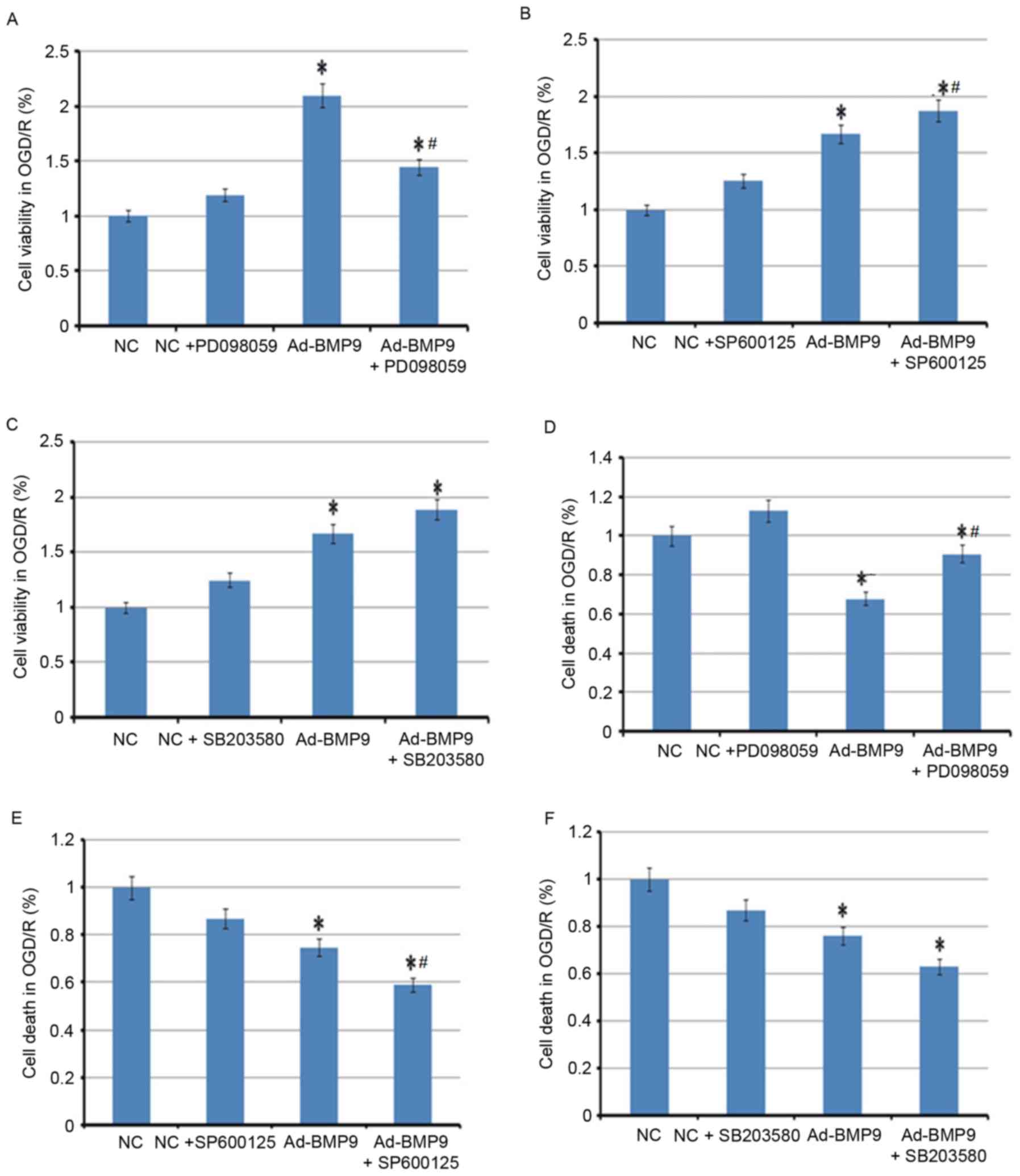
In addition, inhibitors of p38, ERK and JNK were
also used to further demonstrate that the protective role of BMP9
in cerebral I/R is p38-, ERK- and JNK-mediated. Notably, only ERK
inhibitor PD098059 was identified as reversing the protective role
of BMP9, with the reduced cell viability and increased death of
astrocytes compared with BMP9 alone, whereas the JNK inhibitor
SP600125 and p38 inhibitor SB203580 further improved the protective
role of BMP9, indicating the parallel mechanism with BMP9 (Fig. 5).
Discussion
Although accumulating evidence has demonstrated that
BMP9 is expressed in brain neurons (14,23,24),
this is the first study, to the best of the authors' knowledge, to
explore its functional mechanism in cerebral I/R injuries. The
results demonstrated that BMP9 is a protective response molecule
that is significantly upregulated in the cerebral I/R animal and
cell models. Pretreatment with BMP9 effectively ameliorates
neurological defect and brain infarct volume, which may be
associated with its ability to reduce death and improve viability
of astrocytes. The findings of the present study appeared to be in
line with previous studies on the other members of the BMP family
(9,13,25).
BMP9 has been reported to exert its function on
osteogenesis and the maintenance of bone cell survival by
activating the downstream mitogen-activated protein kinase (MAPK)
signaling pathway (26,27). It is also clear that MAPK pathway
members, including ERK, JNK and p38, serve important roles in
neuronal cells as a response to the stress stimuli induced by I/R
(28). Inhibition of ERK by U0126
or PD98059 significantly decreases the number of surviving cells in
hippocampal CA1 subfield and cortex (29), whereas activation of ERK
significantly ameliorates neurological deficit and histopathology
alterations in rats exposed to 90 min MCAO-induced ischemia and 24
h reperfusion (30,31). The alterations in JNK and p38
appeared to be contrary to ERK, with the inhibition of JNK and p38
attenuating infarction volume in I/R brains in vivo
(32,33).
To further confirm whether the protective role of
BMP9 in cerebral I/R injuries is MAPK signaling pathway mediated,
the expression levels of ERK, JNK and p38 were measured in addition
to the use of ERK, JNK and p38 inhibitors in the OGD/R injured
neurons. Although the expression of ERK, JNK and p38 appeared to be
increased following BMP9 treatment, only the ERK inhibitor
partially reversed the effect of BMP9 on the death and viability of
astrocytes, indicating that activation of ERK may be a mechanism
underlying the neuroprotective effects of BMP9.
The present study has certain limitations. First,
the investigation of the neuroprotection effect of BMP9 in
vivo was relatively simple. Apoptosis and proliferation of
neurons and the effect of ERK inhibitor were not analyzed. Second,
the neurological deficit score and infarct volume were evaluated
only at 24 h of reperfusion and the long-term effect of BMP9 on
neurological outcome remains unclear. Third, the downstream
mediators of ERK were not assessed in vitro and in
vivo.
In conclusion, the present study preliminarily
demonstrated that pretreatment with BMP-9 may be a promising option
for the prevention of cerebral I/R injuries, and may reduce death
and maintain the growth of neurons by stimulating the ERK signaling
pathway.
References
|
1
|
Roger VL, Go AS, Lloyd-Jones DM, Adams RJ,
Berry JD, Brown TM, Carnethon MR, Dai S, de Simone G, Ford ES, et
al: Heart disease and stroke statistics-2011 update: A report from
the American Heart Association. Circulation. 123:e18–e209. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stoll G, Kleinschnitz C and Nieswandt B:
Molecular mechanisms of thrombus formation in ischemic stroke:
Novel insights and targets for treatment. Blood. 112:3555–3562.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Deb P, Sharma S and Hassan KM:
Pathophysiologic mechanisms of acute ischemic stroke: An overview
with emphasis on therapeutic significance beyond thrombolysis.
Pathophysiology. 17:197–218. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sanderson TH, Reynolds CA, Kumar R,
Przyklenk K and Hüttemann M: Molecular mechanisms of
ischemia-reperfusion injury in brain: Pivotal role of the
mitochondrial membrane potential in reactive oxygen species
generation. Mol Neurobiol. 47:9–23. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang M, Li YJ, Ding Y, Zhang HN, Sun T,
Zhang K, Yang L, Guo YY, Liu SB, Zhao MG and Wu YM: Silibinin
prevents autophagic cell death upon oxidative stress in cortical
neurons and cerebral ischemia-reperfusion Injury. Mol Neurobiol.
53:932–943. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Reddi AH: Regulation of cartilage and bone
differentiation by bone morphogenetic proteins. Curr Opin Cell
Biol. 4:850–855. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu A and Niswander LA: Bone morphogenetic
protein signalling and vertebrate nervous system development. Nat
Rev Neurosci. 6:945–954. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chalazonitis A and Kessler JA: Pleiotropic
effects of the bone morphogenetic proteins on development of the
enteric nervous system. Dev Neurobiol. 72:843–856. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang R, Pei H, Ru L, Li H and Liu G: Bone
morphogenetic protein 7 upregulates the expression of nestin and
glial fibrillary acidic protein in rats with cerebral
ischemia-reperfusion injury. Biomed Rep. 1:895–900. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Miyazono K, Kamiya Y and Morikawa M: Bone
morphogenetic protein receptors and signal transduction. J Biochem.
147:35–51. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu JH, Zhang TZ, Zhao YY, Wang JK and Yuan
ZG: Protective effects of recombinant human bone morphogenetic
protein-7 on focal cerebral ischemia-reperfusion injury. Int J
Neurosci. 123:375–384. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pei H, Cao D, Guo Z, Liu G, Guo Y and Lu
C: Bone morphogenetic protein-7 ameliorates cerebral ischemia and
reperfusion injury via inhibiting oxidative stress and neuronal
apoptosis. Int J Mol Sci. 14:23441–23453. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Y, Chang CF, Morales M, Chou J, Chen
HL, Chiang YH, Lin SZ, Cadet JL, Deng X, Wang JY, et al: Bone
morphogenetic protein-6 reduces ischemia-induced brain damage in
rats. Stroke. 32:2170–2178. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schnitzler AC, Mellott TJ, Lopez-Coviella
I, Tallini YN, Kotlikoff MI, Follettie MT and Blusztajn JK: BMP9
(bone morphogenetic protein 9) induces NGF as an
autocrine/paracrine cholinergic trophic factor in developing basal
forebrain neurons. J Neurosci. 30:8221–8228. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
craniectomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhao C, Wu N, Deng F, Zhang H, Wang N,
Zhang W, Chen X, Wen S, Zhang J, Yin L, et al: Adenovirus-mediated
gene transfer in mesenchymal stem cells can be significantly
enhanced by the cationic polymer polybrene. PLoS One. 9:e929082014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bederson JB, Pitts LH, Germano SM,
Nishimura MC, Davis RL and Bartkowski HM: Evaluation of
2,3,5-triphenyltetrazolium chloride as a stain for detection and
quantification of experimental cerebral infarction in rats. Stroke.
17:1304–1308. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen H, Tian M, Jin L, Jia H and Jin Y:
PUMA is invovled in ischemia/reperfusion-induced apoptosis of mouse
cerebral astrocytes. Neuroscience. 284:824–832. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dong YF, Chen ZZ, Zhao Z, Yang DD, Yan H,
Ji J and Sun XL: Potential role of microRNA-7 in the
anti-neuroinflammation effects of nicorandil in astrocytes induced
by oxygen-glucose deprivation. J Neuroinflammation. 13:602016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Moskowitz MA, Lo EH and Iadecola C: The
science of stroke: Mechanisms in search of treatments. Neuron.
67:181–198. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lo EH: Experimental models, neurovascular
mechanisms and translational issues in stroke research. Br J
Pharmacol. 153 Suppl 1:S396–S405. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
López-Coviella I, Berse B, Krauss R, Thies
RS and Blusztajn JK: Induction and maintenance of the neuronal
cholinergic phenotype in the central nervous system by BMP-9.
Science. 289:313–316. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lopez-Coviella I, Follettie MT, Mellott
TJ, Kovacheva VP, Slack BE, Diesl V, Berse B, Thies RS and
Blusztajn JK: Bone morphogenetic protein 9 induces the
transcriptome of basal forebrain cholinergic neurons. Proc Natl
Acad Sci USA. 102:pp. 6984–6989. 2005; View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Luan L, Yang X, Zhou C, Wang K and Qin L:
Post-hypoxic and ischemic neuroprotection of BMP-7 in the cerebral
cortex and caudate-putamen tissue of rat. Acta Histochem.
117:148–154. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fong D, Bisson M, Laberge G, Mcmanus S,
Grenier G, Faucheux N and Roux S: Bone morphogenetic protein-9
activates Smad and ERK pathways and supports human osteoclast
function and survival in vitro. Cell Signal. 25:717–728. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ye G, Li C, Xiang X, Chen C, Zhang R, Yang
X, Yu X, Wang J, Wang L, Shi Q and Weng Y: Bone morphogenetic
protein-9 induces PDLSCs osteogenic differentiation through the ERK
and p38 signal pathways. Int J Med Sci. 11:1065–1072. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kovalska M, Kovalska L, Pavlikova M,
Janickova M, Mikuskova K, Adamkov M, Kaplan P, Tatarkova Z and
Lehotsky J: Intracellular signaling MAPK pathway after cerebral
ischemia-reperfusion injury. Neurochem Res. 37:1568–1577. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhu YM, Wang CC, Chen L, Qian LB, Ma LL,
Yu J, Zhu MH, Wen CY, Yu LN and Yan M: Both PI3K/Akt and ERK1/2
pathways participate in the protection by dexmedetomidine against
transient focal cerebral ischemia/reperfusion injury in rats. Brain
Res. 1494:1–8. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang PR, Wang JS, Zhang C, Song XF, Tian N
and Kong LY: Huang-Lian-Jie-Du-Decotion induced protective
autophagy against the injury of cerebral ischemia/reperfusion via
MAPK-mTOR signaling pathway. J Ethnopharmacol. 149:270–280. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang S, Yuan Y, Jiao S, Luo Q and Yu J:
Calcitonin gene-related peptide protects rats from cerebral
ischemia/reperfusion injury via a mechanism of action in the MAPK
pathway. Biomed Rep. 4:699–703. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gong J, Sun F, Li Y, Zhou X, Duan Z, Duan
F, Lei Z, Chen H, Qi S and Shen J: Momordica charantia
polysaccharides could protect against cerebral ischemia/reperfusion
injury through inhibiting oxidative stress mediated c-Jun
N-terminal kinase 3 signaling pathway. Neuropharmacology.
91:123–134. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang W, Tang L, Yong L and Yong W:
Biochanin a protects against focal cerebral ischemia/reperfusion in
rats via inhibition of p38-mediated inflammatory responses. J
Neurol Sci. 348:121–125. 2015. View Article : Google Scholar : PubMed/NCBI
|