Introduction
Barrett's esophagus (BE) is defined as a metaplasia
of the lower esophagus, where the normal squamous epithelium is
replaced by intestinalized columnar epithelium in response to
gastroesophageal reflux disease (GERD). Population based studies
estimate that the prevalence of BE is increasing, with the current
incidence at 1.3–1.6% in general population of Sweden and Italy
(1). Notably, BE is a premalignant
lesion of esophageal adenocarcinoma (EAC) and significantly
enhances the risk of EAC development (2). However, the molecular mechanism of BE
development is still largely unknown.
A number of studies focused on the development of BE
have been carried out in the previous decade. Bile acid is a
pathogenic factor of BE (3) and
has been reported to promote its development through the
upregulation of caudal type homeobox 2 expression (4). A recent study also demonstrated that
the hedgehog pathway may contribute to the development of BE via
regulation of the forkhead box A2 gene (5). Furthermore, expression levels of the
intercellular adhesion molecule 1 and Kruppel like factor 4 genes
were demonstrated to be significantly higher in BE tissues compared
with control samples (6,7), whereas Rho-kinase gene expression was
almost unchanged (8). Although
these studies have made progress, most have only focused on
disparate genes, which is not sufficient for providing an overall
mechanism of BE development. Thus, the aim of the current study was
to explore possible molecular mechanisms and potential biomarkers
of BE using bioinformatic methods. Datasets were downloaded from
the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/) and identified
differentially expressed genes (DEGs) and differentially expressed
microRNAs (DEmiRNAs) using the GEO2R program. Subsequently, DEGs
were analyzed using functional and pathway enrichment analysis, and
then their regulatory networks were constructed. Following
prediction of the target genes of the DEmiRNAs, the target genes
were integrated with the relevant DEGs to further identify the key
genes involved in BE.
Materials and methods
Data resources
mRNA expression profiles (GSE26886, GSE13083 and
GSE34619) and the miRNA expression profile of GSE20099 were
downloaded from the GEO data repository. The dataset of GSE26886
contains 20 BE samples and 19 normal esophageal samples. GSE13083
includes 7 BE samples and 7 normal samples. GSE34619 consists of 10
BE samples and 8 normal samples. The miRNA expression dataset of
GSE20099 contains 14 BE samples and 14 normal samples.
Differential expression analysis
GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r/) is an online
tool that performs comparisons on GEO datasets based on the
GEOquery and Limma R packages (9).
The BE group and normal group were selected, and the GEO2R program
was subsequently applied for differential expression analysis. The
genes and miRNAs that met the cut-off criteria of the adjusted
P-value (adj. P) <0.01 and |log fold change| >2 were
considered as DEGs and DEmiRNAs.
Functional and Kyoto Encyclopedia of
Genes and Genomes (KEGG) pathway enrichment analysis
The Database for Annotation, Visualization and
Integrated Discovery (DAVID; http://david.abcc.ncifcrf.gov/) is a gene functional
enrichment program, providing a large series of functional
annotation tools for researchers to decipher the biological
implications behind huge amounts of genes. To understand the deeper
biological meaning of DEGs, Gene Ontology (GO) and KEGG pathway
enrichment analysis of identified DEGs was performed using DAVID
(10). GO functional analysis
consists of three categories: Biological process (BP), cellular
component (CC) and molecular function (MF). P<0.05 was set as
the threshold value.
Protein-protein interaction (PPI)
network
The Search Tool for the Retrieval of Interacting
Genes/Proteins (STRING; http://string.embl.de/) is an online database
containing known and predicted PPI networks. In this research, a
PPI network of identified DEGs in all three datasets was identified
using the STRING database (combined score >0.4) and subsequently
visualized using Cytoscape (http://www.cytoscape.org/) software (version 3.4.0;
The Cytoscape Consortium, San Diego, CA, USA) (11,12).
The Molecular Complex Detection (MCODE) program within Cytoscape
was used to detect modules of the PPI network (13). The cut-off criteria were set as
follows: Degree, ≥2; node score, ≥0.2; k-score, 0.2; maximum depth,
100. The function and pathway enrichment analysis of the identified
modules was then performed using the DAVID database.
Prediction of miRNA targets
miRecords (http://c1.accurascience.com/miRecords/) is an online
program which integrates miRNA target predictions made by 11
established prediction databases (NBmiRTar, RNA22, MicroInspector,
PicTar, PITA, RNAhybrid, and TargetScan, DIANA-microT, miRanda,
MirTarget2 and miTarget) (14). In
this study, potential targets of DEmiRNAs were identified by at
least four databases.
Results
Differential expression analysis
Gene expression profiles GSE26886, GSE13083 and
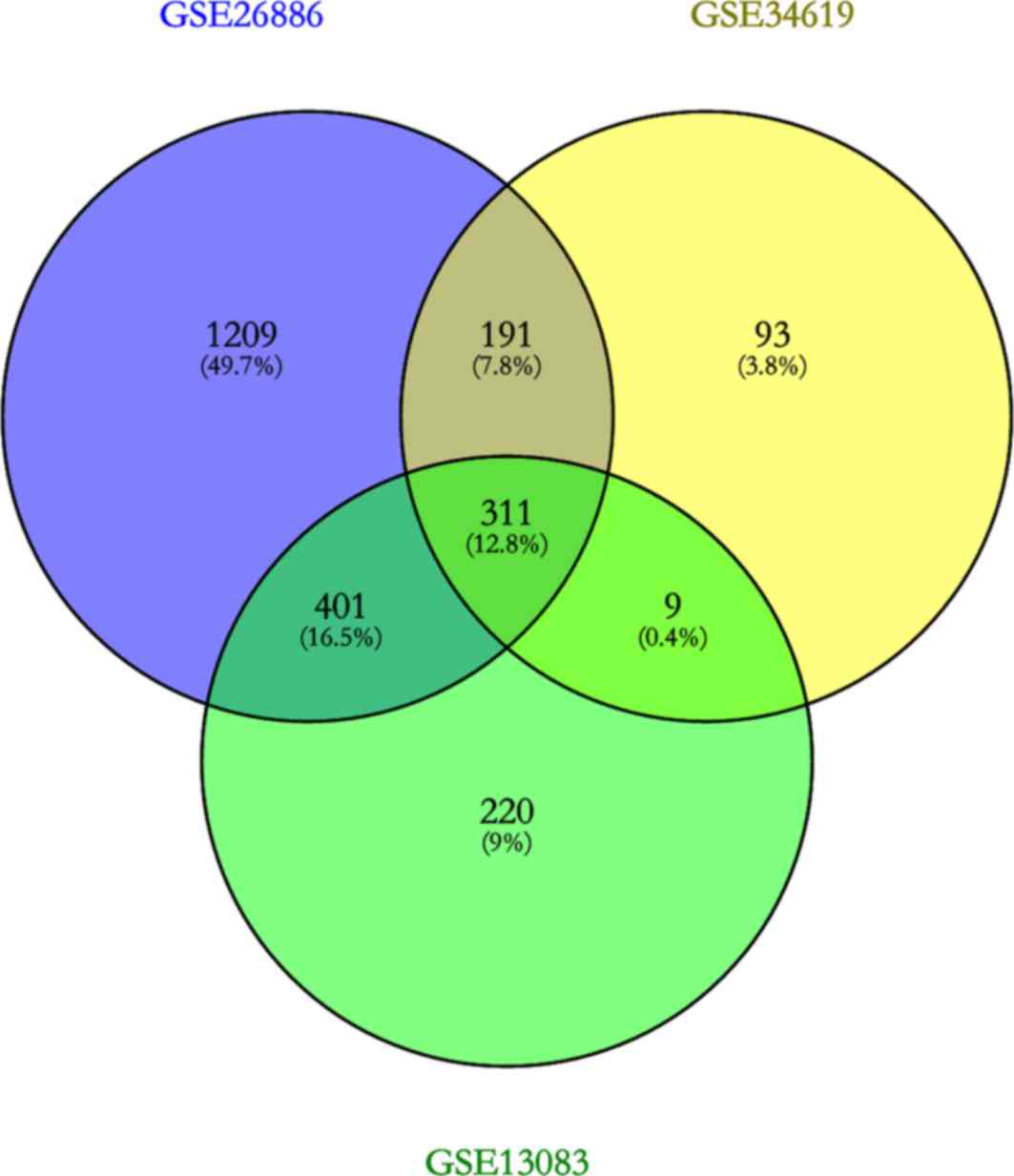
GSE34619 identified 2,112, 941 and 604 DEGs respectively, and 311
DEGs were identified across all three datasets (Fig. 1). Genes present in upregulated and
downregulated groups in different datasets were excluded, and 311
DEGs still presented stable trends in at least two datasets,
including 163 upregulated genes and 148 downregulated genes in BE
samples compared with normal esophageal samples.
Functional and KEGG pathway enrichment
analysis
The upregulated genes enriched in BP terms were
mainly associated with digestion, tissue homeostasis and
microvillus organization, upregulated genes enriched in CC terms
were mainly associated with extracellular vesicles and upregulated
genes enriched in MF terms were mainly associated with actin
binding. Furthermore, KEGG pathway enrichment analysis demonstrated
that the upregulated genes were mainly enriched in pancreatic
secretion, metabolic pathways and salivary secretion pathways
(Table I). The downregulated genes
enriched in BP terms were mainly associated with epidermis
development, keratinocyte differentiation and skin development, in
CC terms the genes were mainly associated with extracellular
regions and in MF terms the genes were mainly associated with
receptor antagonist activity. In addition, KEGG pathway enrichment
analysis revealed that the downregulated genes were mainly
associated with cytochrome P450 drug metabolism and the amoebiasis
pathway (Table II).
 | Table I.Functional and KEGG pathway
enrichment analysis of upregulated genes in BE. |
Table I.
Functional and KEGG pathway
enrichment analysis of upregulated genes in BE.
| A, Biological
process |
|---|
|
|---|
| Term | Description | Count | P-value |
|---|
|
|---|
| GO:0007586 | Digestion | 13 |
3.91×10−8 |
| GO:0001894 | Tissue
homeostasis | 13 |
3.50×10−7 |
| GO:0032528 | Microvillus
organization | 6 |
9.49×10−6 |
| GO:0030277 | Maintenance of
gastrointestinal epithelium | 5 |
1.09×10−5 |
| GO:0016266 | O-glycan
processing | 7 |
1.68×10−5 |
|
| B, Cellular
component |
|
| Term |
Description | Count | P-value |
| GO:1903561 | Extracellular
vesicle | 73 |
1.24×10−15 |
| GO:0043230 | Extracellular
organelle | 73 |
1.26×10−15 |
| GO:0070062 | Extracellular
exosome | 72 |
3.61×10−15 |
| GO:0031988 | Membrane-bounded
vesicle | 78 |
1.55×10−12 |
| GO:0044421 | Extracellular
region part | 80 |
7.07×10−12 |
|
| C, Molecular
function |
|
| Term |
Description | Count | P-value |
|
| GO:0003779 | Actin binding | 14 |
6.49×10−5 |
| GO:0022853 | Active ion
transmembrane transporter activity | 7 |
4.72×10−4 |
| GO:0050839 | Cell adhesion
molecule binding | 13 | 0.001010 |
| GO:0008092 | Cytoskeletal
protein binding | 18 | 0.001705 |
| GO:0022804 | Active
transmembrane transporter activity | 11 | 0.001831 |
|
| D, KEGG |
|
| Term |
Description | Count | P-value |
|
| hsa04972 | Pancreatic
secretion | 8 |
4.20×10−5 |
| hsa01100 | Metabolic
pathways | 27 |
1.16×10−4 |
| hsa04970 | Salivary
secretion | 6 | 0.001815 |
| hsa05110 | Vibrio cholerae
infection | 5 | 0.002050 |
| hsa04976 | Bile secretion | 5 | 0.005352 |
| Table II.Functional and KEGG pathway
enrichment analysis of downregulated genes in BE. |
Table II.
Functional and KEGG pathway
enrichment analysis of downregulated genes in BE.
| A, Biological
process |
|---|
|
|---|
| Term | Description | Count | P-value |
|---|
|
|---|
| GO:0008544 | Epidermis
development | 21 |
3.50×10−13 |
| GO:0030216 | Keratinocyte
differentiation | 14 |
1.24×10−11 |
| GO:0043588 | Skin
development | 17 |
7.84×10−11 |
| GO:0009913 | Epidermal cell
differentiation | 15 |
1.71×10−10 |
| GO:0031424 | Keratinization | 8 |
1.40×10−7 |
|
| B, Cellular
component |
|
| Term |
Description | Count | P-value |
|
| GO:0005576 | Extracellular
region | 73 |
5.14×10−9 |
| GO:0044421 | Extracellular
region part | 63 |
7.59×10−8 |
| GO:0070062 | Extracellular
exosome | 50 |
3.22×10−7 |
| GO:1903561 | Extracellular
vesicle | 50 |
3.76×10−7 |
| GO:0043230 | Extracellular
organelle | 50 |
3.81×10−7 |
|
| C, Molecular
function |
|
| Term |
Description | Count | P-value |
|
| GO:0048019 | Receptor antagonist
activity | 3 | 0.002808 |
| GO:0030547 | Receptor inhibitor
activity | 3 | 0.004791 |
| GO:0005149 | Interleukin-1
receptor binding | 3 | 0.006381 |
| GO:0005198 | Structural molecule
activity | 14 | 0.010380 |
| GO:0005509 | Calcium ion
binding | 13 | 0.013760 |
|
| D, KEGG |
|
| Term |
Description | Count | P-value |
|
| hsa00982 | Drug
metabolism-cytochrome P450 | 4 | 0.013555 |
| hsa05146 | Amoebiasis | 4 | 0.042967 |
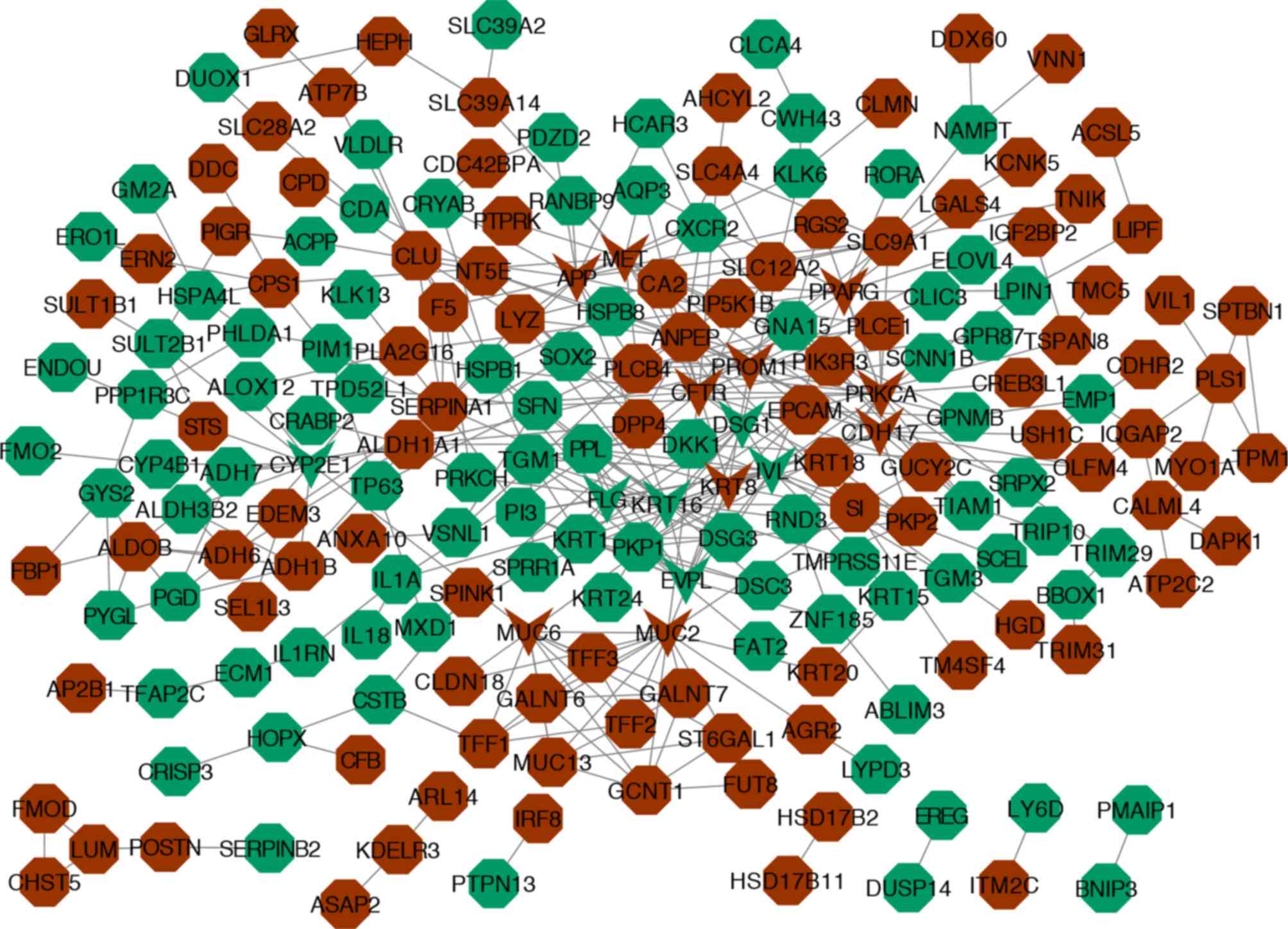
PPI network and modules analysis
A total of 204 nodes and 382 edges were obtained
from the PPI network program. A degree of >10 was set as the
cut-off criterion for hub gene identification. A total of 16 hub
genes were identified: Keratin 16 (KRT16), cystic fibrosis
transmembrane conductance regulator (CFTR), involucrin
(IVL), protein kinase C α (PRKCA), mucin 2,
oligomeric mucus/gel-forming (MUC2), amyloid beta precursor
protein (APP), cadherin 17 (CDH17), mucin 6,
oligomeric mucus/gel-forming (MUC6), MET proto-oncogene,
receptor tyrosine kinase (MET), envoplakin (EVPL),
desmoglein 1 (DSG1), keratin 8 (KRT8), peroxisome
proliferator activated receptor g (PPARG), prominin 1
(PROM1), cytochrome P450 family 2 subfamily E member 1
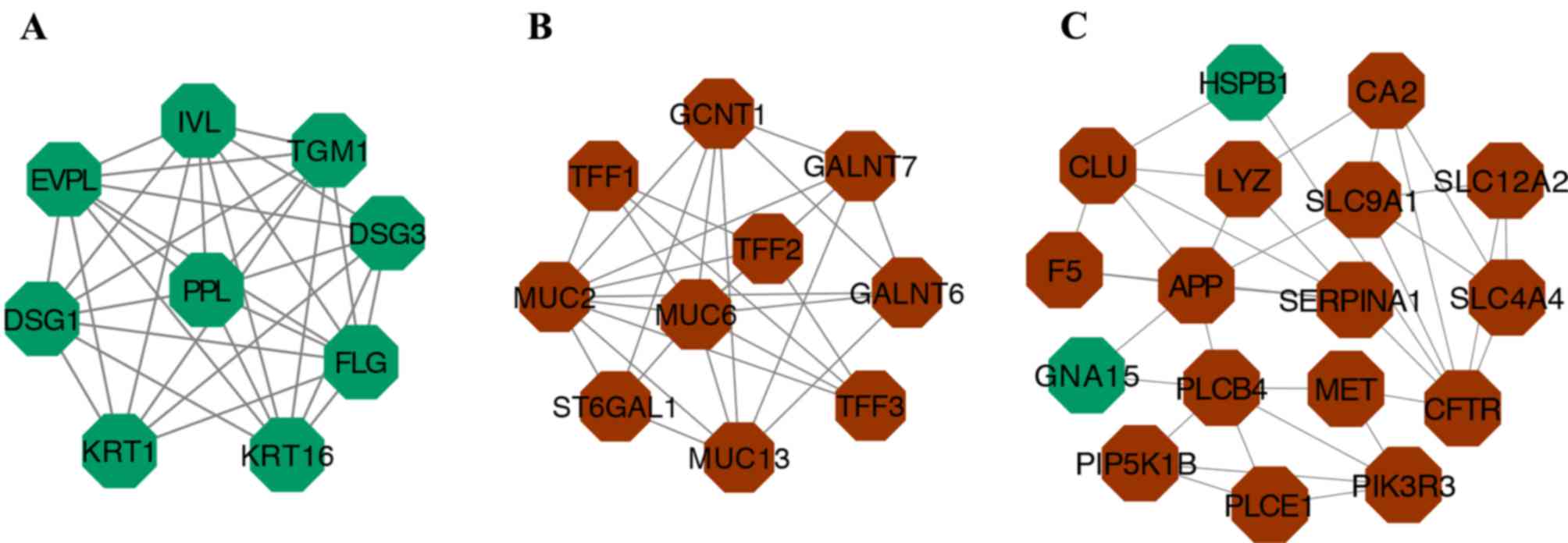
(CYP2E1) and filaggrin (FLG; Table III and Fig. 2). The three most significant
modules were then selected from the PPI network using MCODE
(Fig. 3). Notably, not all of the
hub genes were present in these modules. Functional enrichment
analysis of these modules was then performed. Genes in module A
were mainly enriched in skin development, keratinocyte
differentiation and keratinization. Genes in module B were
predominantly enriched in O-glycan processing, mucin type O-glycan
biosynthesis and metabolic pathways. Finally, genes in module C
were mainly enriched in homeostatic processes, pancreatic secretion
and the bile secretion pathway (Table
IV).
| Table III.Identified hub genes in the
protein-protein interaction network. |
Table III.
Identified hub genes in the
protein-protein interaction network.
|
| GSE26686 | GSE13083 | GSE34619 |
|---|
|
|
|
|
|
|---|
| Gene | logFC | adj. P | logFC | adj. P | logFC | adj. P |
|---|
| KRT16 | −4.62 |
3.33×10−4 | −5.07 |
5.63×10−3 | −3.52 |
2.61×10−5 |
| CFTR | 2.35 |
8.6×10−5 | 5.33 |
1.08×10−6 | 4.31 |
1.61×10−11 |
| IVL | −6.29 |
2.10×10−6 | −4.35 |
2.53×10−4 | −4.27 |
1.79×10−11 |
| PRKCA | 2.79 |
6.77×10−7 | 3.31 |
2.07×10−5 | 2.36 |
2.63×10−11 |
| MUC2 | 4.21 |
4.34×10−5 | 6.72 |
3.16×10−6 | 2.20 |
2.77×10−3 |
| APP | 2.41 |
4.04×10−6 | 3.56 |
1.08×10−5 | 2.09 |
2.31×10−8 |
| CDH17 | 5.58 |
8.00×10−5 | 7.36 |
5.66×10−8 | 4.31 |
2.77×10−4 |
| MUC6 | 6.11 |
1.02×10−6 | 3.10 |
7.45×10−3 | 5.94 |
1.46×10−11 |
| MET | 2.60 |
7.40×10−9 | 4.21 |
1.27×10−6 | 2.32 |
9.04×10−9 |
| EVPL | −3.66 |
2.23×10−7 | −3.55 |
6.81×10−4 | −2.10 |
7.23×10−12 |
| DSG1 | −3.85 |
2.13×10−3 | −4.84 |
4.51×10−5 | −5.48 |
1.06×10−7 |
| KRT8 | 5.73 |
5.13×10−15 | 6.42 |
2.18×10−9 | 4.01 |
3.50×10−11 |
| PPARG | 4.56 |
2.02×10−12 | 3.37 |
1.48×10−6 | 3.02 |
1.66×10−11 |
| PROM1 | 8.47 |
1.02×10−17 | 7.37 |
4.24×10−11 | 5.44 |
2.03×10−14 |
| CYP2E1 | −4.34 |
1.66×10−7 | −4.88 |
1.43×10−6 | −2.06 |
1.59×10−8 |
| FLG | −4.40 |
5.87×10−4 | −6.16 |
3.09×10−5 | −4.96 |
1.14×10−10 |
| Table IV.Functional and KEGG pathway
enrichment analysis of gene modules. |
Table IV.
Functional and KEGG pathway
enrichment analysis of gene modules.
| A, Module A |
|---|
|
|---|
| Term | Description | Count | P-value |
|---|
|
|---|
| GO:0043588 | Skin
development | 7 |
2.13×10−10 |
| GO:0030216 | Keratinocyte
differentiation | 6 |
9.43×10−10 |
| GO:0031424 | Keratinization | 5 |
4.99×10−9 |
| GO:0009913 | Epidermal cell
differentiation | 6 |
7.20×10−9 |
| GO:0008544 | Epidermis
development | 6 |
9.84×10−8 |
|
| B, Module
B |
|
| Term |
Description | Count | P-value |
|
| GO:0016266 | O-glycan
processing | 7 |
1.41×10−13 |
| GO:0006493 | Protein O-linked
glycosylation | 7 |
4.24×10−12 |
| GO:0005975 | Carbohydrate
metabolic process | 9 |
1.91×10−10 |
| GO:0043413 | Macromolecule
glycosylation | 7 |
1.65×10−9 |
| GO:0006486 | Protein
glycosylation | 7 |
1.65×10−9 |
| hsa00512 | Mucin type O-Glycan
biosynthesis | 3 |
1.16×10−4 |
| hsa01100 | Metabolic
pathways | 4 | 0.019425 |
|
| C, Module
C |
|
| Term |
Description | Count | P-value |
|
| GO:0042592 | Homeostatic
process | 10 |
5.77×10−6 |
| GO:0019725 | Cellular
homeostasis | 8 |
6.83×10−6 |
| GO:0030003 | Cellular cation
homeostasis | 7 |
1.09×10−5 |
| GO:0006873 | Cellular ion
homeostasis | 7 |
1.24×10−5 |
| GO:0055080 | Cation
homeostasis | 7 |
2.09×10−5 |
| hsa04972 | Pancreatic
secretion | 6 |
1.07×10−6 |
| hsa04976 | Bile secretion | 4 |
3.98×10−4 |
| hsa04971 | Gastric acid
secretion | 4 |
4.70×10−4 |
| hsa04970 | Salivary
secretion | 4 |
7.60×10−4 |
| hsa04070 |
Phosphatidylinositol signaling system | 4 | 0.001112 |
Integrated analysis of DEmiRNAs and
paired DEGs
The miRNA expression profiles identified five
DEmiRNAs, including two upregulated miRNAs [miRNA-215
(miR-215) and miR-192] and three downregulated miRNAs
(miR-205, miR-203 and miR-486-5p) in BE
samples compared with normal esophageal samples. Among them,
miR-215 and miR-205 were the most significantly
upregulated and downregulated miRNAs, respectively. In addition,
predicted targets of DEmiRNAs were obtained from miRecords
database. Considering the fact that an inverse association exists
between the expression of miRNA and its target mRNA, DEmiRNAs with
target genes that were identified as DEGs were selected.
Remarkably, 33 pairs of DEmiRNAs and DEGs with an inverse
relationship of expression met this criterion. Among these target
genes, PRKCA was the target of two downregulated miRNAs
(miR-203 and miR-205), while epiregulin (EREG)
was the target of two upregulated miRNAs (miR-215 and
miR-192). Furthermore, two hub genes (PRKCA and
CDH17) were predicted as targets of miR-203 (Table V).
| Table V.Differentially expressed miRs and
their paired DEGs in Barret's esophagus. |
Table V.
Differentially expressed miRs and
their paired DEGs in Barret's esophagus.
| miR | Adjusted
P-value | Log fold
change | Paired DEGs |
|---|
|
hsa-miR-215 |
1.05×10−11 | 5.972098 | EREG |
|
hsa-miR-192 |
1.89×10−6 | 2.665345 | EREG |
|
hsa-miR-205 |
1.76×10−3 | −3.301436 | PRKCA,
ABHD2, |
|
|
|
| KDELR3,
DPP4, |
|
|
|
| HSD17B11,
MLPH |
|
hsa-miR-203 |
3.04×10−3 | −2.784608 | PRKCA,
CDH17, |
|
|
|
| CPS1,
TOX3, |
|
|
|
| CPD, STS,
PIGR, |
|
|
|
| FUT8,
KRT20, |
|
|
|
| PBLD |
|
hsa-miR-486-5p |
2.77×10−4 | −2.506280 | TM4SF20 |
Discussion
BE is the precursor lesion of EAC and is still not
completely understood (2).
Therefore, the aim of the current study was to provide an overall
view of the molecular mechanism and biomarkers in BE. In the
present research, a total of 311 DEGs and five DEmiRNAs were
identified from GEO databases using the GEO2R program. The
functional analysis of 163 upregulated DEGs demonstrated these
genes were mainly associated with digestion, tissue homeostasis and
microvillus organization, while the functional analysis of 148
downregulated DEGs demonstrated that they were associated with
epidermis development, keratinocyte differentiation and skin
development. Following construction of the PPI network, 16 hub
genes were identified. The target genes of DEmiRNAs were
subsequently integrated with the DEGs, leading to the
identification of three key genes that may be regulated by
miRNAs.
KEGG pathway enrichment analysis revealed that the
upregulated DEGs were mainly associated with pancreatic secretion,
metabolic pathways, salivary secretion, vibrio cholera infection
and bile secretion. Many clinical studies have focused on these
pathways. Greer et al (15)
reported that the insulin/insulin-like growth factor (IGF) pathway,
which is associated with pancreatic secretion, may have a role in
the development of BE. Regarding the importance of metabolic
pathways, accumulating evidence suggests that obesity is a central
risk factor for BE (16–18). Furthermore, high serum leptin and
adiponectin have been shown to be associated with the development
of BE (19–21).
As mentioned previously, BE is a columnar metaplasia
in response to GERD. GERD can be a result of the impairment of the
anti-reflux barrier, which in turn is associated with salivary
secretion function (22,23). In addition, bile exposure has
confirmed effects in the development of BE (24,25).
Reveiller et al (26)
demonstrated that the expression of squamous differentiation genes
such as IVL and DSG1 was inhibited by bile exposure
in human esophageal epithelial cells. Notably, IVL and
DSG1 are among our identified hub genes.
The downregulated DEGs identified were mainly
associated with the cytochrome P450 drug metabolism pathway.
Supporting this finding, a study previously demonstrated that
cytochrome P450 family 1 subfamily A member 2 (CYP1A2),
CYP3A4 and CYP2E1 were highly expressed in regions of
active cell proliferation in BE (27).
The results of the functional and pathway analysis
performed on the gene modules are largely similar to the DEGs
mentioned above. The module enrichment analysis also indicated that
skin development, mucin type O-glycan biosynthesis and homeostatic
processes were associated with the development of BE. The normal
esophageal epithelium is made up of squamous epithelium, which also
constitutes the skin. The genes associated skin development were
all downregulated. Clinical manifestation of this change in gene
expression is observed in the replacement of squamous epithelium
with columnar epithelium that is typical of BE (1). Notably, genes associated with mucin
type O-glycan in module B were all upregulated in BE samples, which
was correlated with the degree of intestinal metaplasia in BE.
Mucin type O-glycan has been reported to have important roles in
intestinal homeostasis (28). A
previous study indicated that dysfunction of homeostatic processes
may be vital in the development of BE (29). Therefore, a focus on these pathways
may provide us with greater knowledge of BE development.
miRNAs are a group of endogenous non-coding RNA
molecules that act as negative regulators in post-transcriptional
gene regulation. Recent studies reported that aberrant miRNA
expression is associated with the development of BE (30,31).
The current study found five differentially expressed miRNAs,
including two upregulated DEmiRNAs and three downregulated
DEmiRNAs. Among them, miR-215 and miR-205 were the
most significantly upregulated and downregulated DEmiRNAs,
respectively. Other studies have also confirmed the aberrant
expression of these two miRNAs in BE and EAC (32,33).
miR-215 is involved in many diseases, including diabetic
nephropathy, ovarian cancer, non-small cell lung cancer and
multiple carcinomas. miR-215 commonly acts as a tumor
suppressor by promoting apoptosis (34–36).
miR-205 also has an effect in various diseases and may act
as a prognostic marker for many malignant neoplasms (37,38).
This integrated study identified that two hub genes (PRKCA
and CDH17) were target genes of miR-203, and
EREG was a target of two upregulated miRNAs (miR-215
and miR-192).
As a member of the serine/threonine-specific protein
kinase family, PRKCA is involved in various biological processes,
including the innate immune response, angiogenesis, insulin
secretion, cell proliferation, adhesion and migration. In addition,
PRKCA was also identified to be involved in several pathways,
including the mitogen-activated protein kinase (MAPK) pathway,
phosphoinositide 3-kinase (PI3K)-Akt pathway and Wnt pathway.
Previous studies have demonstrated that the MAPK and PI3K pathways
are involved in the proliferation of BE-associated EAC (39), while the insulin/IGF and Wnt
pathways have important roles in BE development (15,40).
CDH17, which is located on chromosome 8,
encodes liver-intestinal cadherin (LI-cadherin), which is involved
in cell adhesion and oligopeptide transport. LI-cadherin is
localized at cell junctions and is selectively expressed in
enterocytes and intestinal goblet cells, but not in the esophagus
(41). Multiple studies have
reported that aberrant LI-cadherin expression is a sensitive marker
for detection of various metaplastic diseases such as gastric
intestinal metaplasia and BE (42,43).
EREG, located on chromosome 4, encodes the
epiregulin protein, which is involved in various biological
processes, including the innate immune response, cytokine
production, wound healing and epithelial cell proliferation. A
previous study also demonstrated that epiregulin regulates the
differentiation of airway epithelial cells through the epidermal
growth factor receptor pathway (44). Furthermore, epiregulin has roles in
a series of other malignant diseases as well as esophageal cancer,
and can be used as biomarker in certain diseases (45–47).
Taken together, this data indicates that the genes PRCKA,
CDH17 and EREG may be involved in the development of
BE through various pathways.
There are certain limitations to the present study.
Due to the fact that there is limited research available on the
pathways involved in BE, the identified KEGG pathways could not be
discussed in detail. Additionally, excluding miRNA data, the
datasets together provided only 37 BE samples and 34 control
samples. Samples obtained from different platforms may also suffer
from some bias. The high level of redundancy in the GO database was
also a limiting factor in the study. Further research should focus
on methods to evaluate possible bias that may arise in this type of
research.
In conclusion, the present study performed a
comprehensive bioinformatics analysis of identified DEGs to
identify potential pathways and biomarkers involved in the
development of BE. In the current research, 311 DEGs and 5 DEmiRNAs
were identified. Following integration of the DEGs with any
corresponding DEmiRNAs, three key genes (PRKCA, CDH17
and EREG) were identified that may be associated with BE.
This research provides a novel insight into molecular mechanisms
that may underlie the development of BE. However, biochemical
analysis and further research are still necessary to validate these
results.
Acknowledgements
The present study is supported by The National
Natural Science Foundation of China (grant no. 81370485).
References
|
1
|
Falk GW, Jacobson BC, Riddell RH,
Rubenstein JH, El-Zimaity H, Drewes AM, Roark KS, Sontag SJ,
Schnell TG, Leya J, et al: Barrett's esophagus:
Prevalence-incidence and etiology-origins. Ann N Y Acad Sci.
1232:1–17. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Solaymani-Dodaran M, Logan RF, West J,
Card T and Coupland C: Risk of oesophageal cancer in Barrett's
oesophagus and gastro-oesophageal reflux. Gut. 53:1070–1074. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tatsugami M, Ito M, Tanaka S, Yoshihara M,
Matsui H, Haruma K and Chayama K: Bile acid promotes intestinal
metaplasia and gastric carcinogenesis. Cancer Epidemiol Biomarkers
Prev. 21:2101–2107. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hu Y, Williams VA, Gellersen O, Jones C,
Watson TJ and Peters JH: The pathogenesis of Barrett's esophagus:
Secondary bile acids upregulate intestinal differentiation factor
CDX2 expression in esophageal cells. J Gastrointest Surg.
11:827–834. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang DH, Tiwari A, Kim ME, Clemons NJ,
Regmi NL, Hodges WA, Berman DM, Montgomery EA, Watkins DN, Zhang X,
et al: Hedgehog signaling regulates FOXA2 in esophageal
embryogenesis and Barrett's metaplasia. J Clin Invest.
124:3767–3780. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Alasehirli B, Oğuz E, Oksuzler E, Koruk I,
Oztuzcu S, Ozkara E, Karakok M, Erbagcı AB and Demiryurek AT:
Investigation of intercellular adhesion molecules (ICAMs) gene
expressions in patients with Barrett's esophagus. Tumour Biol.
35:4907–4912. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kazumori H, Ishihara S, Takahashi Y, Amano
Y and Kinoshita Y: Roles of Kruppel-like factor 4 in oesophageal
epithelial cells in Barrett's epithelium development. Gut.
60:608–617. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Demiryürek S, Koruk I, Bozdag Z, Ozkara E,
Kaplan DS, Oztuzcu S, Cetinkaya A, Alasehirli B and Demiryürek AT:
Investigation of the esophageal Rho-kinase expression in patients
with Barrett's esophagus. Ultrastruct Pathol. 37:284–289. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Barrett T, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, Holko M, et al: NCBI GEO: Archive for functional
genomics data sets-update. Nucleic Acids Res. 41(Database Issue):
D991–D995. 2013.PubMed/NCBI
|
|
10
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Szklarczyk D, Franceschini A, Wyder S,
Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos
A, Tsafou KP, et al: STRING v10: Protein-protein interaction
networks, integrated over the tree of life. Nucleic Acids Res.
43(Database Issue): D447–D452. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bader GD and Hogue CW: An automated method
for finding molecular complexes in large protein interaction
networks. BMC Bioinformatics. 4:22003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xiao F, Zuo Z, Cai G, Kang S, Gao X and Li
T: miRecords: An integrated resource for microRNA-target
interactions. Nucleic Acids Res. 37(Database Issue): D105–D110.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Greer KB, Thompson CL, Brenner L,
Bednarchik B, Dawson D, Willis J, Grady WM, Falk GW, Cooper GS, Li
L and Chak A: Association of insulin and insulin-like growth
factors with Barrett's oesophagus. Gut. 61:665–672. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee SW, Lien HC, Chang CS, Lee TY, Peng YC
and Yeh HZ: Association of metabolic syndrome with erosive
esophagitis and Barrett's esophagus in a Chinese population. J Chin
Med Assoc. 80:15–18. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Crews NR, Johnson ML, Schleck CD, Enders
FT, Wongkeesong LM, Wang KK, Katzka DA and Iyer PG: Prevalence and
predictors of gastroesophageal reflux complications in community
subjects. Dig Dis Sci. 61:3221–3228. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Singh S, Sharma AN, Murad MH, Buttar NS,
El-Serag HB, Katzka DA and Iyer PG: Central adiposity is associated
with increased risk of esophageal inflammation, metaplasia, and
adenocarcinoma: A systematic review and meta-analysis. Clin
Gastroenterol Hepatol. 11:1399–1412.e7. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kendall BJ, Macdonald GA, Hayward NK,
Prins JB, Brown I, Walker N, Pandeya N, Green AC, Webb PM and
Whiteman DC: Study of Digestive Health: Leptin and the risk of
Barrett's oesophagus. Gut. 57:448–454. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Thompson OM, Beresford SA, Kirk EA,
Bronner MP and Vaughan TL: Serum leptin and adiponectin levels and
risk of Barrett's esophagus and intestinal metaplasia of the
gastroesophageal junction. Obesity (Silver Spring). 18:2204–2211.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rubenstein JH, Morgenstern H, McConell D,
Scheiman JM, Schoenfeld P, Appelman H, McMahon LF Jr, Kao JY, Metko
V, Zhang M and Inadomi JM: Associations of diabetes mellitus,
insulin, leptin, and ghrelin with gastroesophageal reflux and
Barrett's esophagus. Gastroenterology. 145:1237–1244.e1-5. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cassiani RA, Mota GA, Aprile LR and Dantas
RO: Saliva transit in patients with gastroesophageal reflux
disease. Dis Esophagus. 28:673–677. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Skoczylas T, Yandrapu H, Poplawski C,
Asadi M, Wallner G and Sarosiek J: Salivary bicarbonate as a major
factor in the prevention of upper esophageal mucosal injury in
gastroesophageal reflux disease. Dig Dis Sci. 59:2411–2416. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sun D, Wang X, Gai Z, Song X, Jia X and
Tian H: Bile acids but not acidic acids induce Barrett's esophagus.
Int J Clin Exp Pathol. 8:1384–1392. 2015.PubMed/NCBI
|
|
25
|
Takahashi Y, Amano Y, Yuki T, Mishima Y,
Tamagawa Y, Uno G, Ishimura N, Sato S, Ishihara S and Kinoshita Y:
Impact of the composition of gastric reflux bile acids on Barrett's
oesophagus. Dig Liver Dis. 43:692–697. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Reveiller M, Ghatak S, Toia L, Kalatskaya
I, Stein L, D'Souza M, Zhou Z, Bandla S, Gooding WE, Godfrey TE and
Peters JH: Bile exposure inhibits expression of squamous
differentiation genes in human esophageal epithelial cells. Ann
Surg. 255:1113–1120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hughes SJ, Morse MA, Weghorst CM, Kim H,
Watkins PB, Guengerich FP, Orringer MB and Beer DG: Cytochromes
P450 are expressed in proliferating cells in Barrett's metaplasia.
Neoplasia. 1:145–153. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bergstrom KS and Xia L: Mucin-type
O-glycans and their roles in intestinal homeostasis. Glycobiology.
23:1026–1037. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rosekrans SL, Baan B, Muncan V and van den
Brink GR: Esophageal development and epithelial homeostasis. Am J
Physiol Gastrointest Liver Physiol. 309:G216–G228. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cabibi D, Caruso S, Bazan V, Castiglia M,
Bronte G, Ingrao S, Fanale D, Cangemi A, Calò V, Listì A, et al:
Analysis of tissue and circulating microRNA expression during
metaplastic transformation of the esophagus. Oncotarget.
7:47821–47830. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bus P, Kestens C, Ten Kate FJ, Peters W,
Drenth JP, Roodhart JM, Siersema PD and van Baal JW: Profiling of
circulating microRNAs in patients with Barrett's esophagus and
esophageal adenocarcinoma. J Gastroenterol. 51:560–570. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Slaby O, Srovnal J, Radova L, Gregar J,
Juracek J, Luzna P, Svoboda M, Hajduch M and Ehrmann J: Dynamic
changes in microRNA expression profiles reflect progression of
Barrett's esophagus to esophageal adenocarcinoma. Carcinogenesis.
36:521–527. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wijnhoven BP, Hussey DJ, Watson DI, Tsykin
A, Smith CM and Michael MZ; South Australian Oesophageal Research
Group, : MicroRNA profiling of Barrett's oesophagus and oesophageal
adenocarcinoma. Br J Surg. 97:853–861. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jia Y, Guan M, Zheng Z, Zhang Q, Tang C,
Xu W, Xiao Z, Wang L and Xue Y: miRNAs in urine extracellular
vesicles as predictors of early-stage diabetic nephropathy. J
Diabetes Res. 2016:79327652016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ge G, Zhang W, Niu L, Yan Y, Ren Y and Zou
Y: miR-215 functions as a tumor suppressor in epithelial ovarian
cancer through regulation of the X-chromosome-linked inhibitor of
apoptosis. Oncol Rep. 35:1816–1822. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hou Y, Zhen J, Xu X, Zhen K, Zhu B, Pan R
and Zhao C: miR-215 functions as a tumor suppressor and directly
targets ZEB2 in human non-small cell lung cancer. Oncol Lett.
10:1985–1992. 2015.PubMed/NCBI
|
|
37
|
Xu H, Yao Y, Meng F, Qian X, Jiang X, Li
X, Gao Z and Gao L: Predictive Value of Serum miR-10b, miR-29c, and
miR-205 as promising biomarkers in esophageal squamous cell
carcinoma screening. Medicine (Baltimore). 94:e15582015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yue X, Lan F, Hu M, Pan Q, Wang Q and Wang
J: Downregulation of serum microRNA-205 as a potential diagnostic
and prognostic biomarker for human glioma. J Neurosurg.
124:122–128. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Vona-Davis L, Frankenberry K, Cunningham
C, Riggs DR, Jackson BJ, Szwerc MF and McFadden DW: MAPK and PI3K
inhibition reduces proliferation of Barrett's adenocarcinoma in
vitro. J Surg Res. 127:53–58. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen X, Jiang K, Fan Z, Liu Z, Zhang P,
Zheng L, Peng N, Tong J and Ji G: Aberrant expression of Wnt and
Notch signal pathways in Barrett's esophagus. Clin Res Hepatol
Gastroenterol. 36:473–483. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gessner R and Tauber R: Intestinal cell
adhesion molecules. Liver-intestine cadherin. Ann N Y Acad Sci.
915:136–143. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mokrowiecka A, Zonnur S, Veits L, Musial
J, Kordek R, Lochowski M, Kozak J, Malecka-Panas E, Vieth M and
Hartmann A: Liver-intestine-cadherin is a sensitive marker of
intestinal differentiation during Barrett's carcinogenesis. Dig Dis
Sci. 58:699–705. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Grötzinger C, Kneifel J, Patschan D,
Schnoy N, Anagnostopoulos I, Faiss S, Tauber R, Wiedenmann B and
Gessner R: LI-cadherin: A marker of gastric metaplasia and
neoplasia. Gut. 49:73–81. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Vermeer PD, Panko L, Karp P, Lee JH and
Zabner J: Differentiation of human airway epithelia is dependent on
erbB2. Am J Physiol Lung Cell Mol Physiol. 291:L175–L180. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sun L, Pan J, Yu L, Liu H, Shu X, Sun L,
Lou J, Yang Z and Ran Y: Tumor endothelial cells promote metastasis
and cancer stem cell-like phenotype through elevated Epiregulin in
esophageal cancer. Am J Cancer Res. 6:2277–2288. 2016.PubMed/NCBI
|
|
46
|
Amsterdam A, Shezen E, Raanan C, Slilat Y,
Ben-Arie A, Prus D and Schreiber L: Epiregulin as a marker for the
initial steps of ovarian cancer development. Int J Oncol.
39:1165–1172. 2011.PubMed/NCBI
|
|
47
|
Watanabe T, Kobunai T, Yamamoto Y,
Kanazawa T, Konishi T, Tanaka T, Matsuda K, Ishihara S, Nozawa K,
Eshima K, et al: Prediction of liver metastasis after colorectal
cancer using reverse transcription-polymerase chain reaction
analysis of 10 genes. Eur J Cancer. 46:2119–2126. 2010. View Article : Google Scholar : PubMed/NCBI
|