Introduction
Alzheimer's disease (AD) is one of the most severe
types of neurodegenerative disease, characterized by a decline in
memory capacity and other cognitive abilities (1). The incidence of AD has grown rapidly
in the past few decades. It was estimated in 2010 that ~40 million
people worldwide were living with AD, and this figure is expected
to double every 20 years (2). At
present, there are no treatments that are able to completely cure
AD and only symptomatic therapeutics are available (3,4). The
brains of patients with AD have a number of hallmark features. For
instance, twisted strands of hyperphosphorylated tau would cause
neurodegeneration as a consequence of structural incompleteness of
microtubules, which is indeed observed in the widespread
intraneuronal fibrillary tangles and intracellular formation of
neurofibrillary tangles (NFTs) (5). Senile plaques consist of modified β
amyloid (Aβ) peptides which are able to induce neuronal apoptosis.
Aβ is rapidly degraded by a number of proteases which maintain its
concentration in normal neuronal cells (6). Impaired balance between Aβ production
and clearance contribute to the amyloidogenic pathway (6). NFTs primarily consist of
phosphorylated tau protein (7).
Therefore, the degree of plaque deposition and synaptic plasticity
in the hippocampus are critical indices for evaluating the anti-AD
efficacy of potential therapeutics.
Type 2 diabetes is a risk factor for AD development
and may exacerbate the progression of the disease (8). Overlapping mechanisms between type 2
diabetes and brain disorders suggest that antidiabetic drugs may
have beneficial effects on brain-cell metabolism, which may be of
clinical importance for the treatment of brain complications in
diabetes and AD (9). Dipeptidyl
peptidase-IV (DPP4) inhibitors are a class of hypoglycemic drugs
used in clinical practice that have been demonstrated to improve
memory in various animal models of neurodegeneration (10–12).
The results of the GDMD Study in China confirmed a positive
association between plasma DPP4 activity and mild cognitive
impairment in elderly patients with type 2 diabetes. DPP4
inhibitors have been demonstrated to ameliorate cognitive
impairment through the suppression of inflammation and oxidative
stress in mouse models (13).
Vildagliptin is a DPP4 inhibitor that effectively inhibits the
degradation of glucagon-like peptide-1 (GLP-1). Vildagliptin was
demonstrated to increase the expression and activity of GLP-1 in
the peripheral blood and to reduce Aβ, phosphorylated tau and
inflammation in an AD mouse model (14). Thus, the use of vildagliptin to
reduce plaque formation and improve synaptic plasticity warrants
further investigation. In the present study, a rat model of AD was
used to investigate the effects of vildagliptin on cognitive
function and to examine its underlying mechanisms. In combination
with other evidence (14), the
present study indicates that vildagliptin may inhibit plaque
formation and regulate synaptic plasticity in AD, providing novel
evidence to suggest the development of vildagliptin as a potential
therapy for AD.
Materials and methods
Animals
Male Sprague-Dawley rats (n=40; 210–230 g) were
obtained from the Animal Center of Suzhou University (Suzhou,
China) and maintained in plastic cages (5 rats per cage) at 20–24°C
and 50±10% humidity, in a 12-h light-dark cycle with free access to
food and water. All animal experiments were approved by The
Institutional Review Board of Soochow University (Suzhou, China)
and experimental procedures were performed in accordance with
guidelines for The Care and Use of Laboratory Animals, and The
National Institutes of Health (NIH) Guide for the Care and Use of
Laboratory Animals (NIH Publication no. 80–23, revised 1996; NIH,
Bethesda, MD, USA).
Preparation of animal models
Rats were randomly divided into the following four
groups: Sham, AD, AD + low-dose vildagliptin (AD + Vilda-L) and AD
+ high-dose vildagliptin (AD + Vilda-H). Rats were anesthetized
with 1% pentobarbital sodium (40 mg/kg) via intraperitoneal
injection and were subsequently administered 10 µg/µl Aβ1-40 via
intracerebral ventricular injection. The sham group was injected
with the equivalent volume of saline. All rats were subjected to
the Morris water maze test to evaluate whether the AD model was
successfully established from day 11–15 following the induction of
AD and their behavioral indexes were recorded once daily. There was
one mortality incidence in the AD group during the model
establishment. Rats in the vildagliptin groups were treated with 5
(AD + Vilda-L) or 10 (AD + Vilda-H) mg/kg vildagliptin (Gulvus;
Novartis International AG, Basel, Switzerland) once a day by oral
gavage for 4 consecutive weeks. Following vildagliptin treatment,
behavioral tests and biochemical analysis was performed.
Morris water maze (MWM) test
The spatial learning and memory of the rats was
tested by MWM assessment following treatment with vildagliptin.
Rats were placed in a black circular water tank (diameter, 150 cm;
depth, 60 cm; 25°C). Reference objects were placed around the pool
as visual hints and were left unaltered throughout the MWM test.
During the 4-day training period, rats were randomly placed into
the water at any point in the four quadrants, with one test
performed in every quadrant each morning for 1 min at every turn.
The rats that did not locate the platform were placed on the
platform and allowed to stand for 15 sec. Rats that successfully
located the platform were additionally permitted to stand on it for
15 sec. On the 5th day, the platform was removed and the rats were
allowed to swim for 1 min. Maze performance was recorded with a
video camera located above the pool, interfaced with a video
tracking system (HVS Image, Buckingham, UK). The average escape
latency and time spent in each quadrant in a total of five trials
was recorded.
Histology
Following completion of the MWM test, rats were
anesthetized with sodium pentobarbital (60 mg/kg) and euthanized by
transcardial perfusion with cold PBS. The hippocampus of brain was
rapidly isolated, part of the tissues was subsequently fixed with
cold 4% paraformaldehyde containing 0.2% saturated picric acid for
24 h at 4°C. The remaining samples were stored at −80°C.
Paraffin-embedded sections were cut in a coronal plane at a
thickness of 5 µm with a microtome. Paraffin-embedded brain
sections were de-paraffinized with xylene (3 times × 5 min) and
subsequently rehydrated prior to Nissl staining with 0.1% (w/v)
cresyl violet to investigate the degree of neuronal damage in the
hippocampus. The mean number of morphologically-intact neurons per
100 µm length in the CA1 hippocampal area was calculated to
accurately estimate the extent of neuronal damage in comparison to
controls.
Western blot analysis
Frozen samples were obtained and lysed in Tissue
Protein Lysis Solution (Thermo Fisher Scientific, Inc., Waltham,
MA, USA) containing 5% Proteinase Inhibitor Cocktail
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). A total of 25 µg
extracted protein/lane was separated on a 12% SDS-PAGE gel
(Beyotime Institute of Biotechnology, Haimen, China) and
transferred onto a nitrocellulose membrane (Beyotime Institute of
Biotechnology) using wet transfer. The membranes were blocked in 5%
non-fat milk for 2 h at room temperature and washed three times in
PBS with Tween 20 (PBST). The membrane was subsequently incubated
with the following primary antibodies overnight at 4°C: Rabbit
polyclonal caspase-3 (1:1,000; cat. no. AB13847; Abcam, Cambridge,
UK), rabbit polyclonal B cell lymphoma 2 (Bcl-2; 1:1,000; cat. no.
AB59348; Abcam), rabbit monoclonal Bcl-2 associated X protein (Bax;
1:1,000; cat. no. AB32503; Abcam), rabbit polyclonal phosphorylated
(p) protein kinase B (p-Akt; 1:1,000; cat. no. AB38449; Abcam),
rabbit polyclonal Akt (1:1,000; cat. no. AB8805; Abcam), rabbit
monoclonal glycogen synthase kinase 3β (GSK3β; 1:1,000; cat. no.
AB32391; Abcam), rabbit polyclonal p-GSK3β (1:1,000; cat. no.
AB131356; Abcam), rabbit polyclonal postsynaptic density protein 95
(PSD-95; 1:1,000; cat. no. AB18258; Abcam), rabbit monoclonal
synaptophysin (1:1,000; cat. no. AB32127; Abcam), rabbit polyclonal
p-tau (1:1,000; cat. no. AB109390; Abcam), rabbit monoclonal tau
(1:1,000; cat. no. AB32057; Abcam), amyloid precursor protein (APP;
1:1,000; cat. no. AB32136; Abcam) and rabbit polyclonal β-actin
(1:1,000; cat. no. AB8227; Abcam). Following incubation with
primary antibodies, the membranes were washed with PBST three times
for 10 min each and subsequently incubated with polyclonal goat
anti-rabbit secondary antibody (1:1,000; Abcam; cat. no. AB205718)
for 2 h at room temperature. Protein levels were analyzed using
Image J software version 1.41 (NIH) following exposure to an ECL
kit (BOSTER Biological Technology, Pleasanton, CA, USA).
Statistical analysis
The data are presented as the mean ± standard
deviation. Each experiment was repeated a minimum of 3 times. The
statistical analysis was performed using one-way analysis of
variance, followed by the Bonferroni post hoc test, with SPSS 21.0
(IBM Corp., Armonk, NY, USA). P<0.05 was considered to indicate
a statistically significant difference.
Results
Vildagliptin improves spatial learning
and memory impairment in the AD group
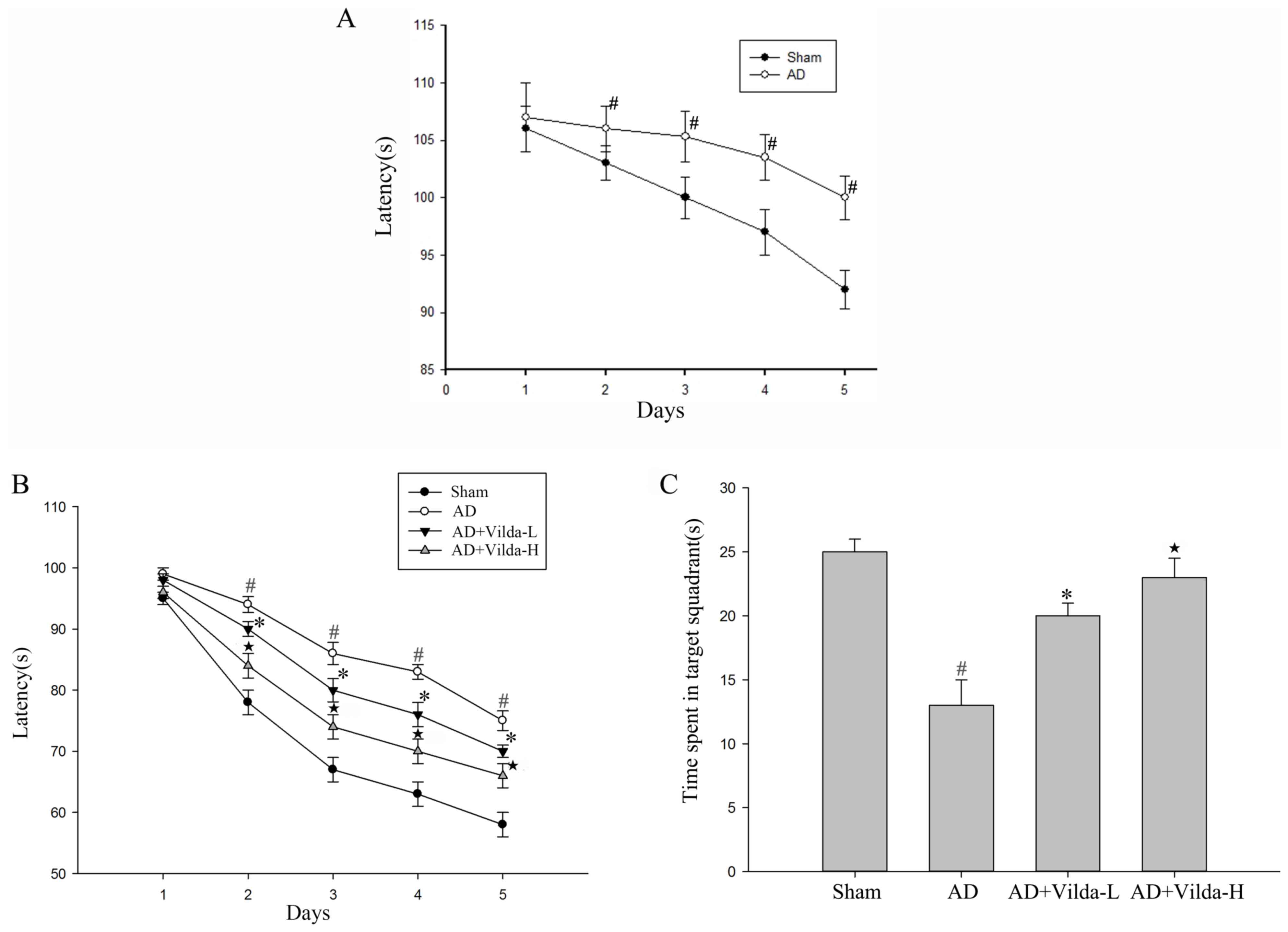
The MWM test was used to assess spatial learning
function. Compared with the Sham group, the escape latency in rats
were administered 10 µg/µl Aβ1-40 via intracerebral ventricular
injection was significantly longer (P<0.01, Fig. 1A) from day 11–15 following the
induction of AD. It indicated that the AD model is modeled
successfully. After four weeks the AD group still exhibited a
significant spatial learning deficit compared with the sham group,
and vildagliptin administration significantly reduced the escape
latency compared with the AD group, particularly in the Vilda-H
group (Fig. 1B). Following the
4-day training period, the platform was removed. The time spent in
the target quadrant was significantly lower in the AD group
compared with the sham group (Fig.
1C). The time spent in the target quadrant by the AD + Vilda-L
group was significantly increased compared with the AD group. The
Vilda-H group demonstrated the most significant improvement in the
spatial probe test compared to the AD group (P<0.05).
Treatment with vildagliptin attenuates
the expression of apoptosis-associated proteins and prevents
neuronal cell loss
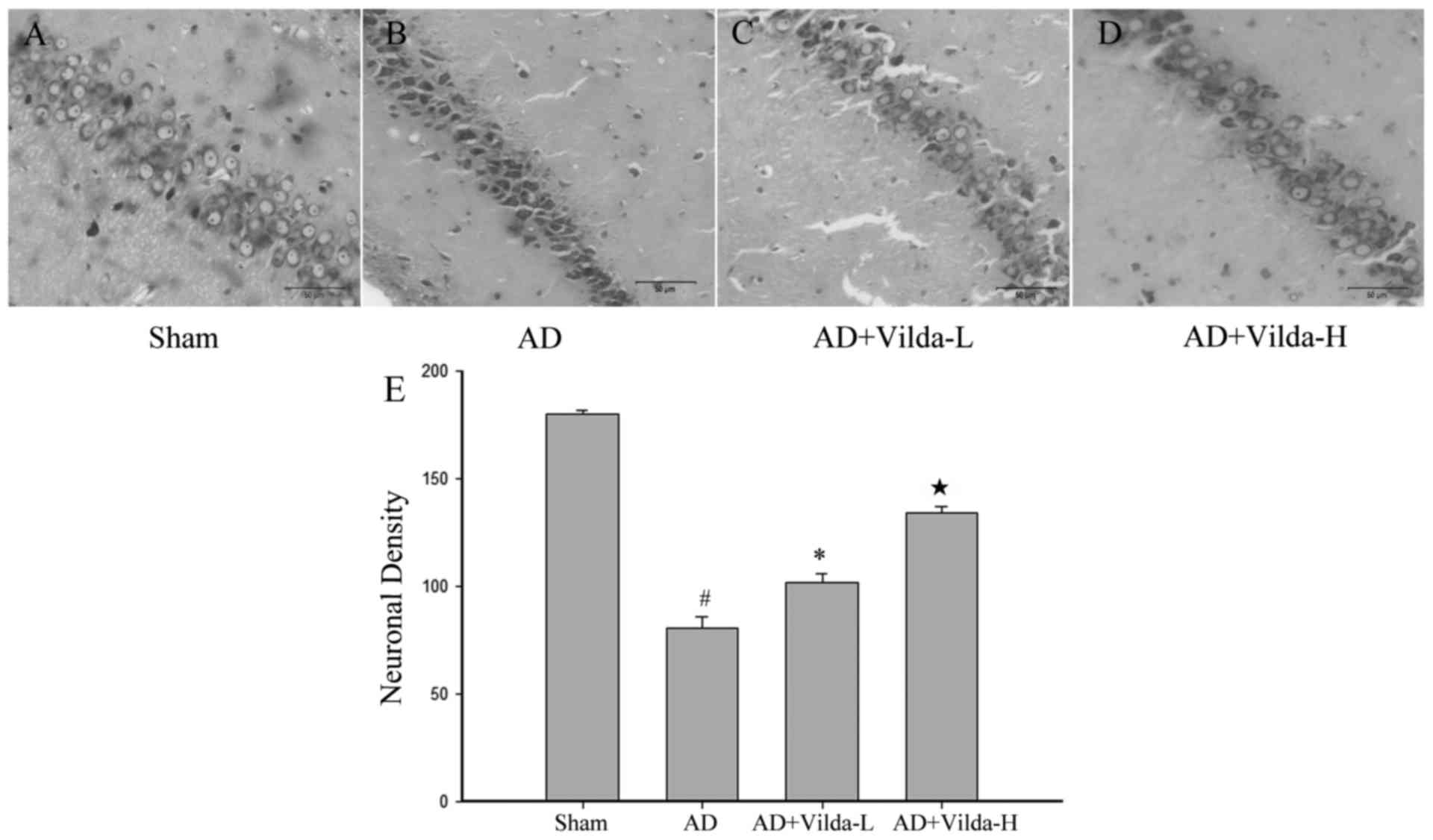
The Nissl staining method was performed to
investigate neuronal alterations in the hippocampal CA1 region of
rats of different groups. Hippocampal neurons in the sham group
were large and conical with well-demarcated amphophilic cytoplasm
and round vesicular nuclei with prominent nucleoli (Fig. 2A). Neurons in the AD group were
characterized by pyknotic pyramidal cells and pronounced neuronal
body shrinkage with nuclear loss (Fig.
2B). Treatment with vildagliptin reduced the AD model-induced
cell loss and pyknosis, although degenerating cells with altered
morphology were still observed (Fig.
2C and D). Treatment with vildagliptin exerted significant
protection against AD-induced neurotoxicity, as determined by
neuronal density (Fig. 2E).
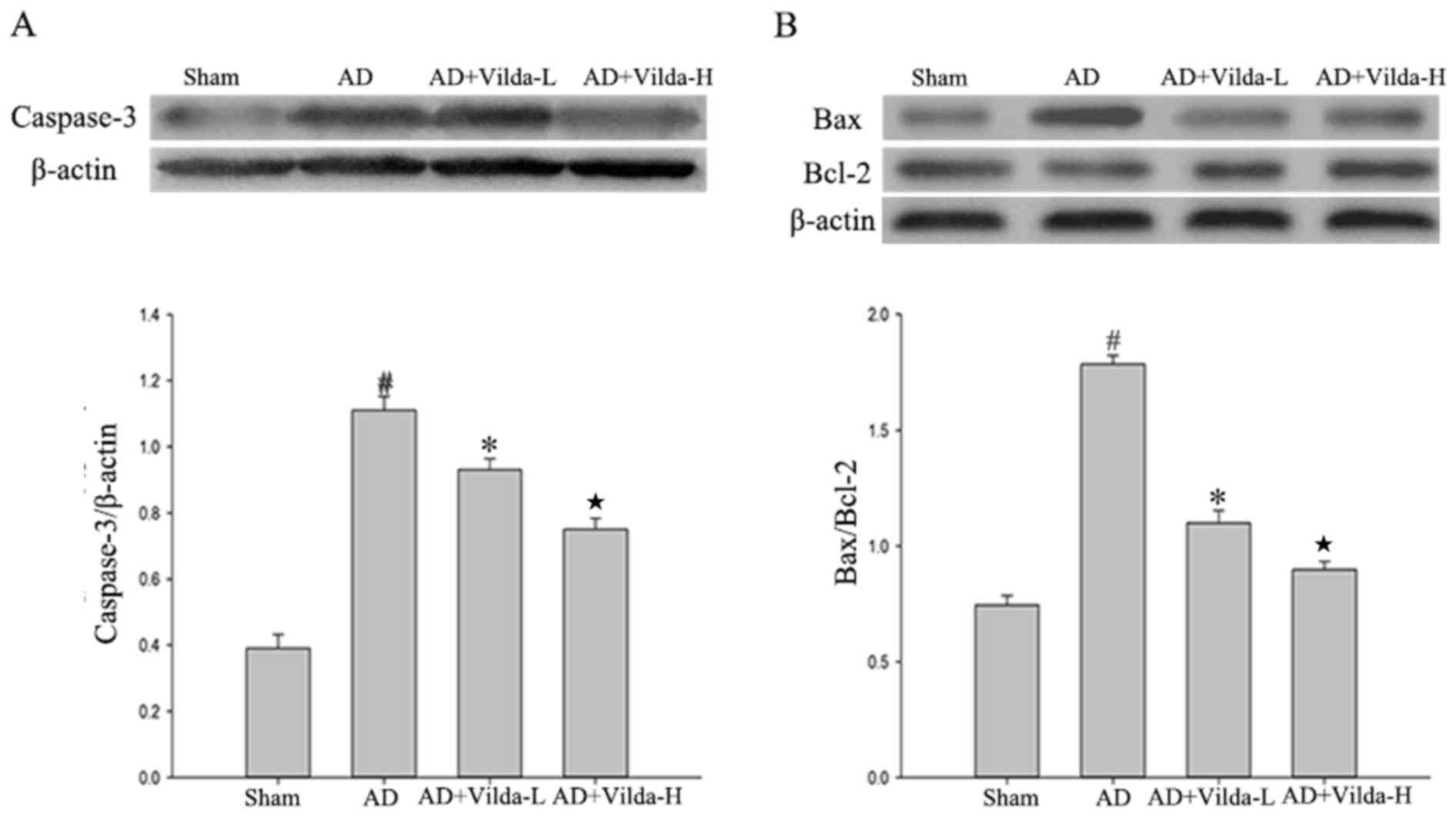
Hippocampal caspase-3, Bax and Bcl-2
expression
The expression of caspase-3 and Bax increased in the
AD model compared with the sham group (Fig. 3A and B; P<0.01) and treatment
with vildagliptin downregulated this expression. The expression
level of Bcl-2 appeared to decrease in the AD model compared with
the sham group (Fig. 3B). However,
vildagliptin-treated rats exhibited a dose-dependent increase in
hippocampal Bcl-2 expression levels by comparison with the AD group
(Vilda-L and Vilda-H, P<0.05).
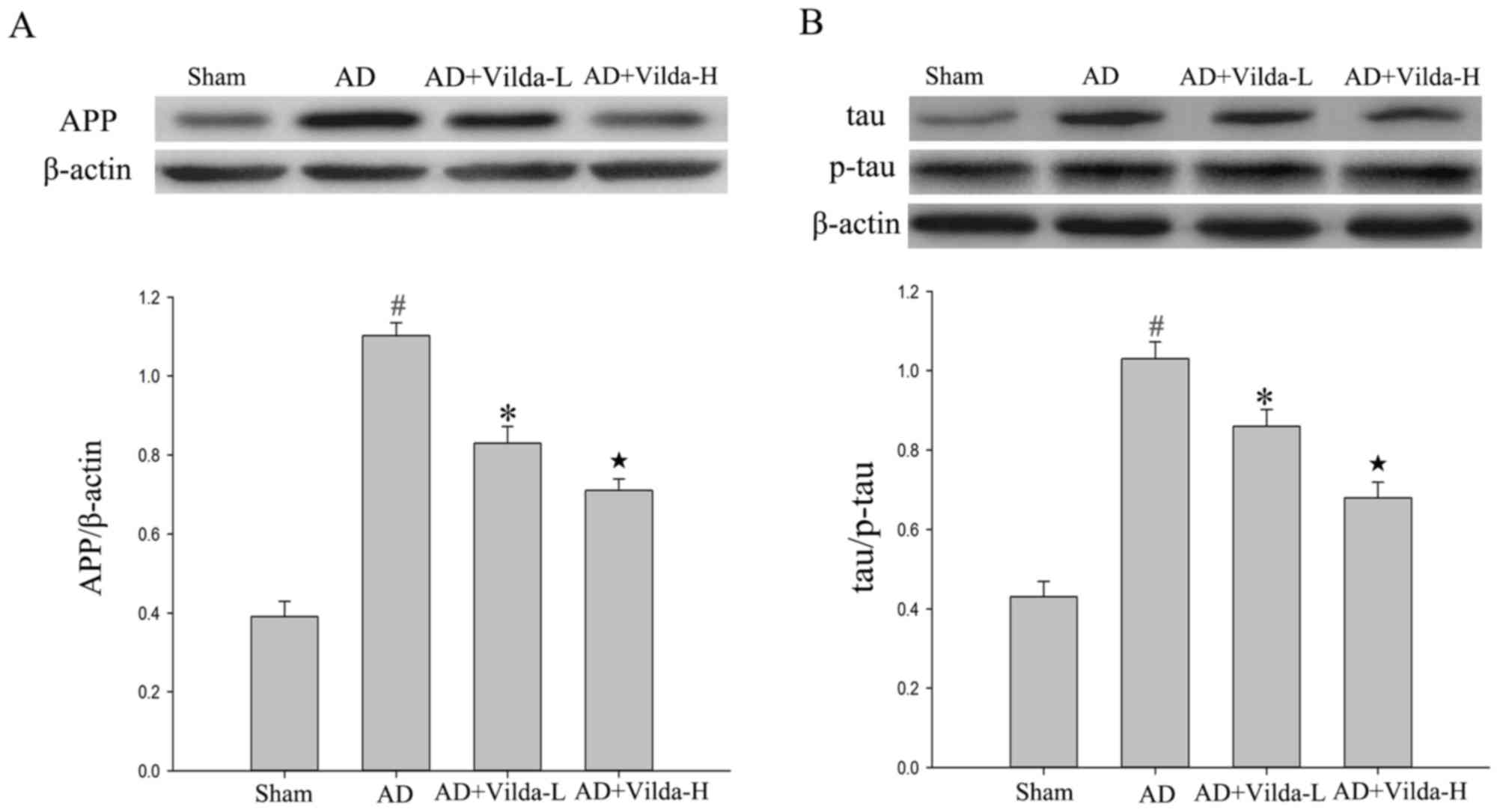
Vildagliptin treatment reduces the
expression of AD-associated proteins
The expression levels of APP in the hippocampus were
determined by western blot analysis. The expression of APP
increased in the AD model group compared with the sham group
(Fig. 4A; P<0.05) and APP
expression was downregulated by treatment with vildagliptin
(Vilda-L and Vilda-H, P<0.05). Additionally, p-tau expression
increased in the AD group compared with the sham group (Fig. 4B; P<0.05). The expression levels
of p-tau were reduced following treatment with vildagliptin
(Vilda-L and Vilda-H, P<0.05).
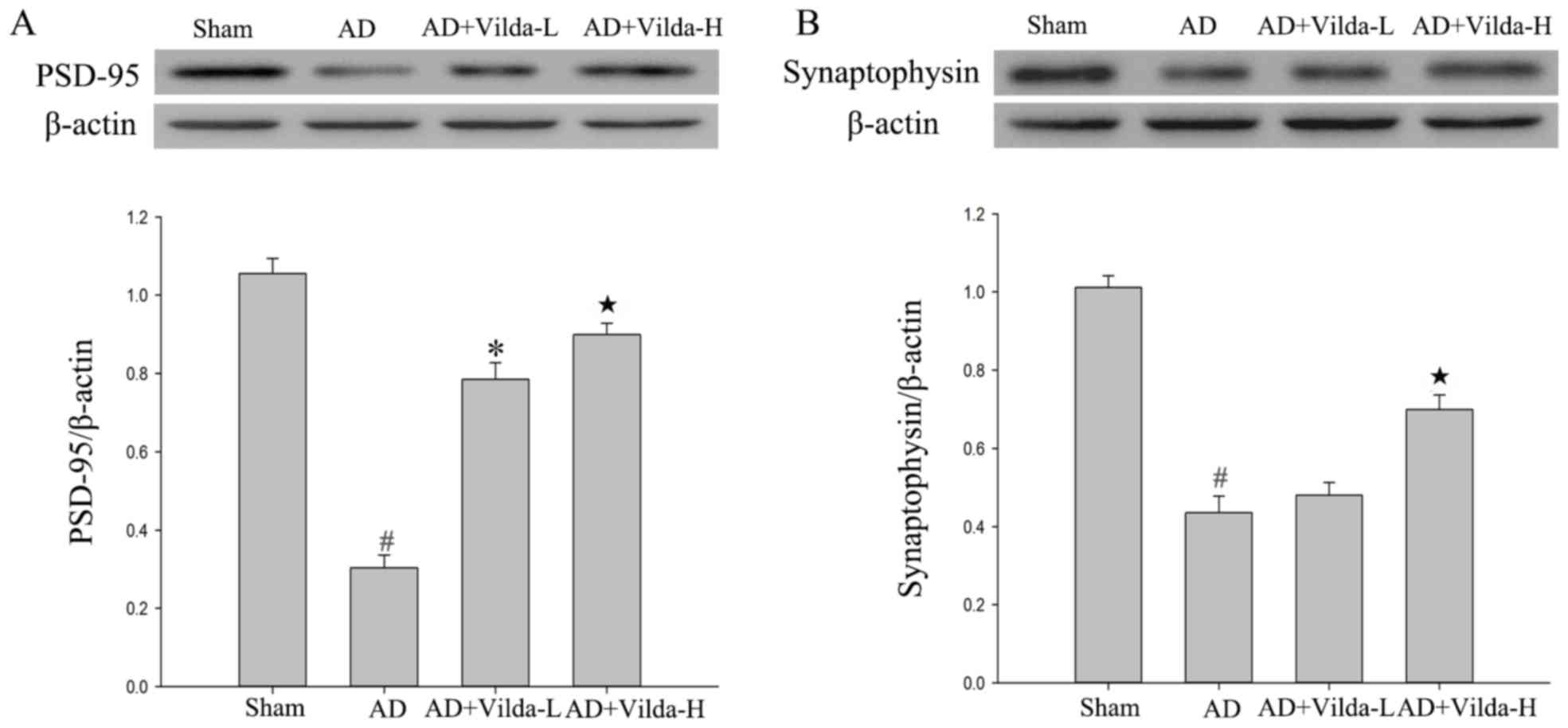
Treatment with vildagliptin enhances
the expression of proteins associated with synaptic plasticity
The hippocampal expression of the synapse-associated
proteins synaptophysin and PSD-95 was analyzed. Levels of PSD-95
and synaptophysin in the hippocampus were significantly decreased
(P<0.01) in the AD group compared with the sham group (Fig. 5). Treatment with vildagliptin
(Vilda-H) significantly enhanced PSD-95 and synaptophysin
expression levels in the hippocampus compared with the AD group
(P<0.01). However, the low dose of vildagliptin did not
significantly increase synaptophysin expression (Fig. 5B).
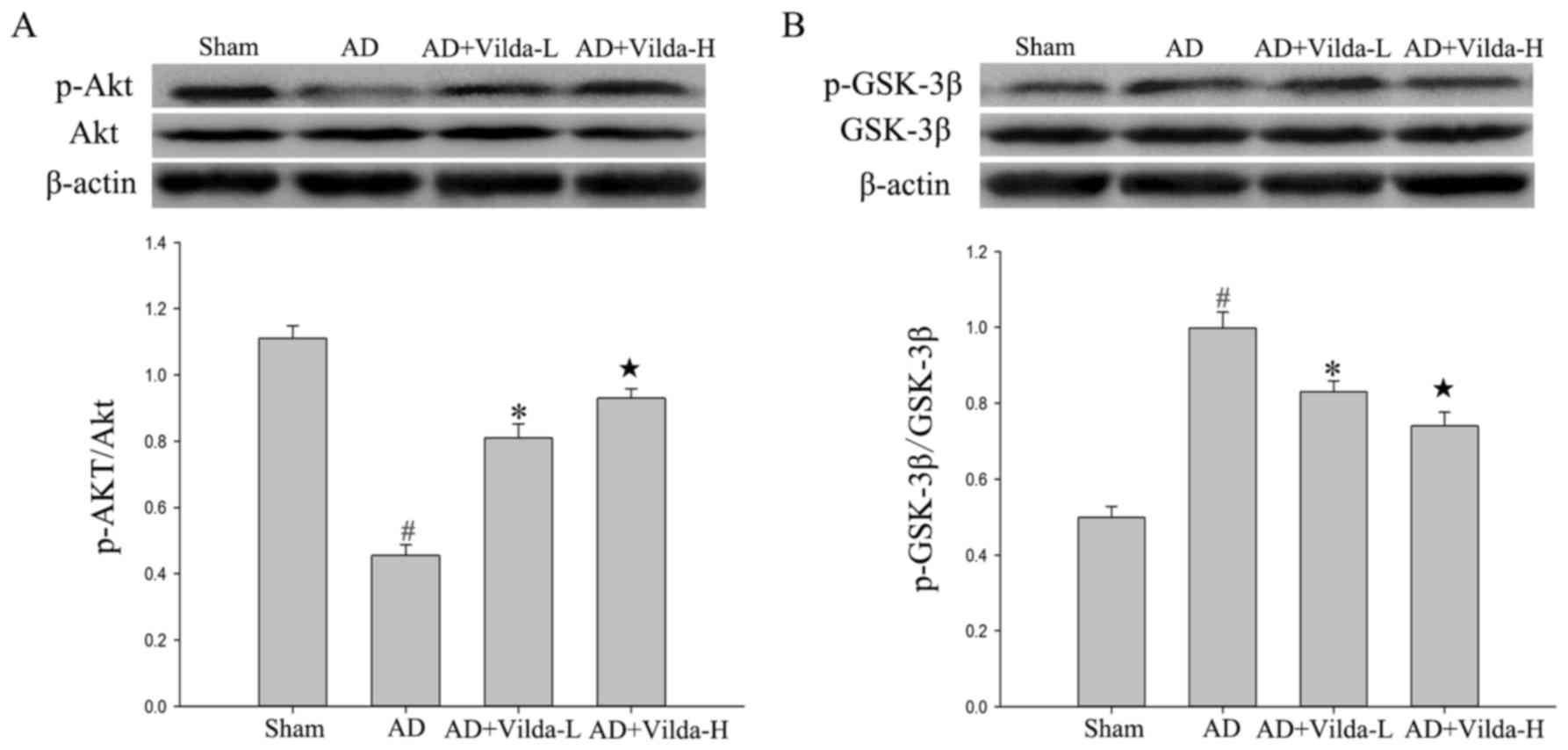
Vildagliptin increases Akt/GSK3β
pathway activity
The total protein levels of Akt and GSK3β did not
vary notably between groups. However, levels of p-Akt were
significantly decreased in the hippocampus of the compared with the
sham group (P<0.05). Rats in the AD group had increased p-GSK3β
levels compared with the rats in sham group (P<0.05). The
administration of vildagliptin significantly increased p-Akt
(Vilda-L, P<0.05; Vilda-H, P<0.01) and reduced p-GSK3β levels
(Vilda-L, P<0.05; Vilda-H, P<0.01), in a dose-dependent
manner (Fig. 6).
Discussion
AD is a progressive neurodegenerative disease that
leads to memory impairment, aphasia, disability, visual impairment,
administrative dysfunction, and personality and behavioral changes.
In 1907, Alois Alzheimer's experimental observations first
identified the pathological modifications of Aβ and tau, which are
the hallmark features of AD (15).
The deposition of prefibrillar and fibrillar oligomeric Aβ, tau
protein phosphorylation, synaptic loss, inflammation and glial cell
activation contribute to AD pathogenesis (16). Various hypotheses have been
proposed to explain the different causes of AD, but the exact
mechanism remains unknown. Currently, FDA approved drugs for AD
only offer symptomatic relief through the control of
neurotransmitter levels and the activity of neurotransmitters,
including donepezil and memantine, which affect the cholinergic and
glutamatergic systems, respectively (17,18).
Thus, the discovery and identification of novel and effective
treatments is required.
DPP4 inhibitors are a class of oral hypoglycemic
agents used in monotherapy or in combination with other
antidiabetic compounds. DPP4 inhibitors bind reversibly and
competitively to DPP4, which indirectly enhances the levels of
incretin hormones, particularly GLP-1 and gastric inhibitory
polypeptide. Numerous studies have investigated the effect of DPP4
inhibitors on cognitive function. Treatment with sitagliptin was
demonstrated to significantly improve the working and reference
memories of diabetic rats (19);
it may additionally improve the memory and hippocampal neurogenesis
of high fat-fed mice (20).
Furthermore, Aβ deposition was delayed in an early stage AD
transgenic mouse model following treatment with sitagliptin
(21).
Vildagliptin has been demonstrated to significantly
reduce oxidative stress in the brain, restore brain insulin
sensitivity and mitochondrial function, and improve hippocampal
synaptic plasticity and cognitive function in an obese rat model
(22). In a streptozotocin
(STZ)-induced diabetic rat model, treatment with vildagliptin
significantly improved memory and learning impairments (23). Furthermore, treatment with
vildagliptin ameliorated cognitive deficits in a STZ-induced rat
model of AD (14). This evidence
demonstrates that vildagliptin may effectively improve cognitive
function.
The results of the present study demonstrated that
the established AD rat model exhibited a decline in memory
performance. Treatment for 1 month with vildagliptin was
demonstrated to ameliorate cognitive deficits in the AD model in a
dose-dependent manner. Bcl-2 expression was increased, and Bax and
caspase-3 expression was decreased by treatment with vildagliptin.
The mechanism underlying the anti-apoptotic effect of vildagliptin
was not determined, but may have been mediated by the activation of
the Akt/GSK3β signaling pathway (24,25),
an effect previously reported for vildagliptin in vitro
(26). Consistent with this
finding, vildagliptin administration following AD model-induced
neuronal damage inhibited the activation of caspase-3, decreased
Bax and increased Bcl-2 expression, and therefore may reduce
apoptosis in the brain.
The AD-afflicted brain is characterized by the
presence of senile plaques and NFTs composed of aggregated Aβ
peptides and p-tau, respectively (27–29).
It has been reported that Aβ plaques are present in the brain of
approximately one-third to one-half of individuals aged ≥65, and
tau inclusions are present almost universally (30). In AD, APP is cleaved by β-secretase
and γ-secretase to produce toxic Aβ protein, which is involved in
plaque formation (31). In the
present study, vildagliptin was demonstrated to significantly
reduce APP and p-tau protein expression, with this effect being
more pronounced at the higher 10 mg/kg vildagliptin dose. This
suggested that vildagliptin was able to reduce Aβ aggregation and
tau hyperphosphorylation in AD. Expression of PSD-95 in the AD
group was significantly decreased compared with the sham group, and
this expression was recovered by treatment with vildagliptin,
suggesting that vildagliptin may improve synaptic plasticity, which
is important for improving cognitive function. Synaptophysin
expression levels were not significantly improved in the AD+Vilda-L
group compared with the AD group. However, a significant
improvement was observed in the AD+Vilda-H group. In terms of
adverse effects, a previous study indicated that the most common
adverse events in vildagliptin-treated subjects were mild or
moderate, and suspected to be unrelated to the study medication.
Nausea occurred in certain vildagliptin-treated subjects, although
this was suggested to not be a dose-limiting side effect of DPP-4
inhibition (32).
In conclusion, the present study demonstrated that
vildagliptin improved learning and memory deficits induced in an AD
rat model, through an increase in the expression of proteins
associated with synaptic plasticity, and a decrease in the
expression of apoptosis and AD-associated proteins. Additionally,
activation of the Akt and GSK-3β inhibition may have contributed to
the improvement in cognitive function mediated by vildagliptin. The
present study provides evidence for the development of vildagliptin
as a potential therapeutic for AD.
Acknowledgements
The present study was supported by a Project of
Opening Project of Provincial Key Laboratory of Soochow University
(grant no. KJS1513) and the Suzhou Science and Technology Project
(grant no. XJ201460).
References
|
1
|
Scheltens P, Blennow K, Breteler MM, de
Strooper B, Frisoni GB, Salloway S and Van der Flier WM:
Alzheimer's disease. Lancet. 388:505–517. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Prince M, Bryce R, Albanese E, Wimo A,
Ribeiro W and Ferri CP: The global prevalence of dementia: A
systematic review and metaanalysis. Alzheimers Dement. 9:63–75.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li X, Bao X and Wang R: Neurogenesis-based
epigenetic therapeutics for Alzheimer's disease. Mol Med Rep.
14:1043–1053. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen PY, Tsai CT, Ou CY, Hsu WT, Jhuo MD,
Wu CH, Shih TC, Cheng TH and Chung JG: Computational analysis of
novel drugs designed for use as acetylcholinesterase inhibitors and
histamine H3 receptor antagonists for Alzheimer's disease by
docking, scoring and de novo evolution. Mol Med Rep. 5:1043–1048.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Henry W, Querfurth HW and LaFerla FM:
Mechanisms of disease Alzheimer's disease. New Engl J Med.
362:329–344. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zuroff L, Daley D, Black KL and
Koronyo-Hamaoui M: Clearance of cerebral Aβ in Alzheimer's disease:
Reassessing the role of microglia and monocytes. Cell Mol Life Sci.
74:2167–2201. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kepp KP: Bioinorganic chemistry of
Alzheimer's disease. Chem Rev. 112:5193–5239. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Patrone C, Eriksson O and Lindholm D:
Diabetes drugs and neurological disorders: New views and
therapeutic possibilities. Lancet Diabetes Endocrinol. 2:256–262.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Palleria C, Leporini C, Maida F, Succurro
E, De Sarro G, Arturi F and Russo E: Potential effects of current
drug therapies on cognitive impairment in patients with type 2
diabetes. Front Neuroendocrinol. 42:76–92. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Darsalia V, Olverling A, Larsson M,
Mansouri S, Nathanson D, Nyström T, Klein T, Sjöholm Å and Patrone
C: Linagliptin enhances neural stem cell proliferation after stroke
in type 2 diabetic mice. Regul Pept 190–191. 1–31. 2014.
|
|
11
|
Nassar NN, Al-Shorbagy MY, Arab HH and
Abdallah DM: Saxagliptin: A novel antiparkinsonian approach.
Neuropharmacology. 89:308–317. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Matteucci E and Giampietro O: Mechanisms
of Neurodegeration in Type 2 diabetes and the neuroprotective
potential of dipeptidyl peptidase 4 inhibitors. Curr Med Chem.
22:1573–1581. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zheng T, Qin L, Chen B, Hu X, Zhang X, Liu
Y, Liu H, Qin S, Li G and Li Q: Association of plasma DPP4 activity
with mild cognitive impairment in elderly patients with type 2
diabetes: results from the GDMD study in China. Diabetes Care.
39:1594–1601. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kosaraju J, Murthy V, Khatwal RB, Dubala
A, Chinni S, Muthureddy Nataraj SK and Basavan D: Vildagliptin: An
anti-diabetes agent ameliorates cognitive deficits and pathology
observed in streptozotocin-induced Alzheimer's disease. J Pharm
Pharmacol. 65:1773–1784. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Brion JP, Fraser H, Flament-Durand J and
Dickinson AG: Amyloid scrapie plaques in mice and Alzheimer senile
plaques, share common antigens with tau, a microtubule-associated
protein. Neurosci Lett. 78:113–118. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gonzalez B, Abud EM, Abud AM, Poon WW and
Gylys KH: Tau spread, apolipoprotein E, inflammation and more:
Rapidly evolving basic science in alzheimer disease. Neurol Clin.
35:175–190. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Storr T: Ligand design in medicinal
inorganic chemistry. John Wiley & Sons; New York, NY: 2014,
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jakobroetne R and Jacobsen H: Alzheimer's
disease: From pathology to therapeutic approaches. Angew Chem Int
Ed Enge. 48:3030–3059. 2009. View Article : Google Scholar
|
|
19
|
Pintana H, Apaijai N, Chattipakorn N and
Chattipakorn SC: DPP-4 inhibitors improve cognition and brain
mitochondrial function of insulin-resistant rats. J Endocrinol.
218:1–11. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gault VA, Lennox R and Flatt PR:
Sitagliptin, a dipeptidyl peptidase-4 inhibitor, improves
recognition memory, oxidative stress and hippocampal neurogenesis
and upregulates key genes involved in cognitive decline. Diabetes
Obes Metab. 17:403–413. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
D'Amico M, Di Filippo C, Marfella R,
Abbatecola AM, Ferraraccio F, Rossi F and Paolisso G: Long-term
inhibition of dipeptidyl peptidase-4 in Alzheimer's prone mice. Exp
Gerontol. 45:202–207. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pintana H, Tanajak P, Pratchayasakul W,
Sa-Nguanmoo P, Chunchai T, Satjaritanun P, Leelarphat L,
Chattipakorn N and Chattipakorn SC: Energy restriction combined
with dipeptidyl peptidase-4 inhibitor exerts neuroprotection in
obese male rats. Br J Nutr. 1–9. 2016.PubMed/NCBI
|
|
23
|
El Batsh MM, El Batch MM, Shafik NM and
Younos IH: Favorable effects of vildagliptin on metabolic and
cognitive dysfunctions in streptozotocin-induced diabetic rats. Eur
J Pharmacol. 769:297–305. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gao C, Hölscher C, Liu Y and Li L: GSK3: A
key target for the development of novel treatments for type 2
diabetes mellitus and Alzheimer disease. Rev Neurosci. 23:1–11.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yu T and Lin W: Small-molecule GSK-3
inhibitor rescued apoptosis and neurodegeneration in
anesthetics-injured dorsal root ganglion neurons. Biomed
Pharmacother. 84:395–402. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fan R, Li X, Gu X, Chan JC and Xu G:
Exendin-4 protects pancreatic beta cells from human islet amyloid
polypeptide-induced cell damage: Potential involvement of AKT and
mitochondria biogenesis. Diabetes Obes Metab. 12:815–824. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Savelieff MG, Lee S, Liu Y and Lim MH:
Untangling amyloid-β, tau, and metals in Alzheimer's disease. Acs
Chem Biol. 8:856–865. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nadezhdin KD, Bocharova OV, Bocharov EV
and Arseniev AS: Structural and dynamic study of the transmembrane
domain of the amyloid precursor protein. Acta Naturae. 3:69–76.
2011.PubMed/NCBI
|
|
29
|
Köpke E, Tung YC, Shaikh S, Alonso AC,
Iqbal K and Grundke-Iqbal I: Microtubule-associated protein tau.
Abnormal phosphorylation of a non-paired helical filament pool in
Alzheimer disease. J Biol Chem. 268:24374–24384. 1993.PubMed/NCBI
|
|
30
|
Braak H, Thal DR, Ghebremedhin E and Del
Tredici K: Stages of the pathologic process in Alzheimer disease:
Age categories from 1 to 100 years. J Neuropathol Exp Neurol.
70:960–969. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Do Carmo S, Crynen G, Paradis T, Reed J,
Iulita MF, Ducatenzeiler A, Crawford F and Cuello AC: Hippocampal
proteomic analysis reveals distinct pathway deregulation profiles
at early and late stages in a rat model of alzheimer's-like amyloid
pathology. Mol Neurobiol. May 13–2017.(Epub ahead of print).
View Article : Google Scholar
|
|
32
|
Pratley RE, Jauffret-Kamel S, Galbreath E
and Holmes D: Twelve-week monotherapy with the DPP-4 inhibitor
vildagliptin improves glycemic control in subjects with type 2
diabetes. Horm Metab Res. 38:423–428. 2006. View Article : Google Scholar : PubMed/NCBI
|