Introduction
Cholangiocarcinoma (CCA) is a malignancy of bile
duct epithelium related with a high invasion and metastasis. CCA
can be classified into perihilar/extrahepatic and intrahepatic
type, based on anatomical location. The incidence and mortality
rates of CCA, particularly the intrahepatic type, have been
increasing worldwide (1–3). The prevalence of CCA in Northeast
Thailand, especially in Khon Kaen, has been found about 89% of all
primary liver cancers which is related with liver flukes
[Opisthorchis viverrini (OV)] infection, making the region
as the area of the highest incidence in the world (84 per 100,000
in men and 36 per 100,000 in women) (4–6).
Prolonged parasitic infection possibly induces chronic inflammation
of biliary tract to carcinogenic substances leading to genetic and
epigenetic aberrations in the epithelial cells (7). The prognosis of patients with
unrespectable tumors is poor, of which the overall survival rates
are low and the majority of patients die within a year of diagnosis
because no effective treatment is available (8,9).
Luvira et al (8) found that
the overall median survival of CCA patients in Northeast Thailand
was 4 months (95% CI, 3.3–4.7) and 2-year survival rate was only
8.1% (95% CI, 4.5–12.9).
Our previous study on the genome-wide methylation
using the Infinium HumanMethylation27 BeadChip microarray
(Illumina, Inc., San Diego, CA, USA) (10) demonstrated hypo- and
hyper-methylation status of 28 CCA cases in comparison with 6 match
normal adjacent tissues. The methylation level of individual genes
obtained from methylation array data was presented as β-value
ranging from 0 (unmethylation) to 1 (complete or 100% methylation).
To address whether aberrant methylation of genes involved in
chemotherapeutic response was different between short and long
survival, in this study, 8 of 28 CCA cases treated with
5-fluorouracil (5-FU) consisting of 4 short and 4 long survival
were selected for data analysis based on their differential
β-values of genes which play roles in DNA repair, apoptosis, cell
proliferation, drug metabolism and angiogenesis. Based on previous
study for gene selection (11),
differential β-value of >0.1 (10% fold-change) indicating 1.5
differential fold-change of gene expression was applied.
Accordingly, 10 genes which had differential β-values ranging from
0.1–0.4 in short and long survival were selected. There were DNA
repair: protein phosphatase 4 catalytic subunit (PPP4C)
(12); apoptosis: runt related
transcription factor 3 (RUNX3) (13,14),
interferon regulatory factor 4 (IRF4) (15), ubiquitin C-terminal hydrolase L1
(UCHL1) (16), tumor
protein p53 inducible protein 3 (TP53I3) (17); cell proliferation: cyclin D2
(CCND2) (18), Ras
association domain family member 1 (RASSF1) (19); drug metabolism: aldehyde
dehydrogenase 1 family member A3 (ALDH1A3) (20), solute carrier family 29 member 1
(SLC29A1) (21); and
angiogenesis: human immunodeficiency virus-1 tat interactive
protein 2 (HTATIP2) (22).
RASSF1 plays roles in the apoptotic DNA damage response (DDR), cell
proliferation and regulates XPA-mediated DNA repair (23), however, RASSF1 was
categorized into cell proliferation in this study. RUNX3 and
IRF4 are transcription factor genes which play a role in
apoptosis, they were categorized into apoptosis. These 10 genes
were studied in more detail for their methylation status in CCA
samples using methylation-sensitive high-resolution melting
(MS-HRM). Moreover, their association with clinicopathological data
was also analyzed.
Materials and methods
Samples and DNA preparation
Fifty-four frozen liver tissues of intrahepatic CCA
patients with clinicopathological data and 19 matched adjacent
normal samples were kindly supplied by the Cholangiocarcinoma
Research Institute, Khon Kaen University, Khon Kaen, Thailand.
Written informed consent was obtained from all patients. The
project was approved by the Khon Kaen University Ethics Committee
for Human Research (HE571022).
DNA methylation levels of 10 candidate genes were
quantified in 54 CCA patients that were then divided into two
groups as low and high methylation based on a cut-off value (median
of methylation level of individual genes) for survival analysis.
The survival time is the time after operation date until the date
of death. Genes which were significantly correlated with survival
time were further performed for survival analysis in 42 of 54 CCA
cases that underwent 5-FU therapy after surgery (12 were excluded
as untreated patients). Of 42 5-FU treated cases, 19 normal
adjacent tissues were obtained and quantified for methylation
levels which were used to compare to those of 42 tumor tissues.
DNA from frozen liver tissues was extracted using
Qiagen® Blood & Tissue kit (Qiagen, Hilden, Germany)
following the manufacturer's instructions. Normal male leukocyte
genomic DNA was used as an unmethylated control. For in
vitro methylated DNA, 10 µg of male leukocyte genomic DNA was
treated with 10 units of SssI methyltransferase (New England
Biolabs, Ipswich, MA) containing 160 µM S-adenosylmethionine and 1×
methylase buffer at 37°C for 1 h. The mixture was incubated at 65°C
for 20 min to stop reaction, followed by ethanol precipitation and
resuspended in sterile water. Methylated and unmethylated DNA
controls were spectroscopically quantified using the NanoVue (GE
Healthcare, Buckinghamshire, UK) and stored at −20°C until
bisulfite modification.
Bisulfite modification
Sodium bisulfite modification was performed using
the EZ DNA Methylation-Gold kit (Zymo Research Corp., Irvine, CA,
USA) following the manufacturer's instructions. The eluted DNA was
ethanol precipitated and dissolved into 20 µl of distilled water,
after which was spectroscopically quantified at 260 nm using the
NanoVue (GE Healthcare). Complete bisulfite modified DNA was
validated using modified and wild type Calponin specific
primers as described previously (24). Sample which was not found a
specific band from modified primer (333 bp) and/or given a specific
band with wild type primers (333 bp) was considered unmodified or
incompletely modified. Only complete bisulfite modified DNA was
used as a DNA template for PCR and MS-HRM assay in this study.
Primers
Primers were designed and examined via electronic
PCR (ePCR) using Methyl Primer Express program v1.0 (Applied
Biosystems; http://bisearch.enzim.hu/?m=genompsearch). PCR
conditions were optimized which could amplify both unmethylated and
methylated DNA at the specific CpG islands. The primer sequences of
10 candidate genes including optimal conditions for MS-HRM are
shown in Table I. The two
different HTATIP2 isoforms are encoded by the same promotor. The
alternative splicing is taken place at the C-terminus, as a result,
two isoforms are generated. In this study, we performed DNA
methylation at the gene promotor of HTATIP2. All primers
were supplied by Pacific Science Co. Ltd., Bangkok, Thailand.
 | Table I.Primer sequences for
methylation-sensitive high-resolution melting, optimized conditions
and product details. |
Table I.
Primer sequences for
methylation-sensitive high-resolution melting, optimized conditions
and product details.
|
|
|
|
| Product |
|---|
|
|
|
|
|
|
|---|
| Primer name | Sequences
(5′-3′) | Ta (°C) | MgCl2
(mM) | Size (bp) | CG site |
|---|
| UCHL1-F |
CGAGTGAGATTGTAAGGTTTGG | 55 | 2.5 | 143 | 14 |
| UCHL1-R |
AACGCACTATAAAACCTATACAA |
|
|
|
|
| IRF4-F |
GCGTTGGTTTGGGTTTTAA | 58 | 2.5 | 143 | 13 |
| IRF4-R |
CCGCCTCAACCACTCCT |
|
|
|
|
| CCND2-F |
GAAGCGAGGTTGTTTTGG | 58 | 3.5 | 159 | 15 |
| CCND2-R |
CCCCGACTCTCTTCCTAAC |
|
|
|
|
| HTATIP2-F |
TTCGATTAGGGAAGGTGGGA | 56 | 2.5 | 176 | 15 |
| HTATIP2-R |
CCGACCAAAAAACCTAACC |
|
|
|
|
| TP53I3-F |
GTTTCGTTGTTTTGGTTTGT | 53 | 3.5 | 145 | 14 |
| TP53I3-R |
TACCGCATCCAACCCTAT |
|
|
|
|
| RUNX3-F |
AACGTTTGGAGAGTAGTGTT | 55 | 2.5 | 139 | 14 |
| RUNX3-R |
CGATAAAAAAACCTACCCTC |
|
|
|
|
| RASSF1-F |
GGTTTCGGTATTTAGTATTTAGG | 57 | 3.5 | 120 | 12 |
| RASSF1-R |
ATCGATAAACAACCCCACCC |
|
|
|
|
| ALDH1A3-F |
GGCGAAGTTTTAGGGTTT | 56 | 3.5 | 147 | 17 |
| ALDH1A3-R |
CACGTACCCTACTCTTAAATC |
|
|
|
|
| PPP4C-F |
GGTCGATGTGAGGGGAGG | 60 | 2.5 | 130 | 13 |
| PPP4C-R |
CACCGCACAAAAATCTCCTAA |
|
|
|
|
| SLC29A1-F |
TTCGGGTTTAAAATAGGTTG | 57 | 2.5 | 155 | 13 |
| SLC29A1-R |
CGAACCTCCATCCCCATC |
|
|
|
|
MS-HRM
MS-HRM can detect both methylated and unmethylated
nucleotide difference due to different melting temperature by using
a single primer set. The HRM can determine change of decreasing
fluorescence intensity of PCR products with DNA duplex melting
temperature (melting temperature in CG-rich product is higher than
that of AT-rich product). Therefore, MS-HRM consists of real-time
PCR using bisulfite-converted DNA and melting analysis of PCR
products (HRM) which reflects the thermodynamic behavior of the
MS-HRM amplicon. As a result, the methylation status can be
determined through comparison with melting standard curves created
by different dilution ratios of methylated and unmethylated DNA
controls (25).
PCR amplification and HRM were performed on
LighCycler480® Real time PCR machine (Roche, Mannheim,
Germany). The 20 µl of PCR reaction consisted of 1× PCR buffer (67
mM Tris, Ph 8.4, 16.6 mM ammonium sulfate and 0.1% Tween-20), 300
nM of each primer, 200 µM of each dNTP, 20 ng of bisulfite modified
DNA, 1.5 µM SYTO®9 (Invitrogen, Carlsbad, CA), 0.5 unit
of Platinum Taq DNA polymerase (Invitrogen), and
MgCl2 concentration as described in Table I. The cycling stage was performed
as follows: Holding at 95°C for 10 min to activate enzyme, 40
cycles of denaturation at 95°C for 15 sec, annealing at temperature
as described in Table I for 30 sec
and extension at 72°C for 1 min. After amplification, HRM stage was
initiated by denaturing PCR product at 95°C for 1 min, followed by
reannealing at 40°C for 1 min and slowly warmed by continuous
acquisition to 95°C with 1% ramp rate (°C/s). All DNA samples were
analyzed on a Light Cycler 480® 8-Tube strips with Light
Cycler 480® sealing foil (Roche). Each reaction was
performed in triplicate and no DNA template control was included in
each experiment. To quantify DNA methylation of each assay, a
standard curve was generated by running MS-HRM of standard dilution
series including 100, 50, 25, 10, 5, 1 and 0% methylated controls.
The precision of the assay was performed by including internal
controls (25 and 75% methylated DNA) in each PCR run. The
amplification plot and raw data of MS-HRM were reviewed using The
Light Cycler 480®. MS-HRM data was analyzed using The
Light Cycler 480® Gene Scanning Software. The
calibration curve and linear equation of each MS-HRM were performed
in Microsoft Excel 2016 and used for determination of methylation
level of individual genes in clinical samples.
Statistical analysis
The correlations of clinicopathological data of CCA
patients including age at initial diagnosis, sex, histological
types, 5-FU treatment and the postoperative survival time with
methylation status of individual genes were analyzed using Fisher's
exact test. The difference of methylation levels between two
independent groups was analyzed using Mann-Whitney U test, and
Wilcoxon Matched-Pairs Signed-Ranks test was used for matched pair
samples. Overall survival was analyzed using Kaplan-Meier and
log-rank test, the univariate and multivariate Cox regression
models. The statistical analysis was performed using SPSS version
17.0 for windows (SPSS Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Quantification of DNA methylation by
MS-HRM
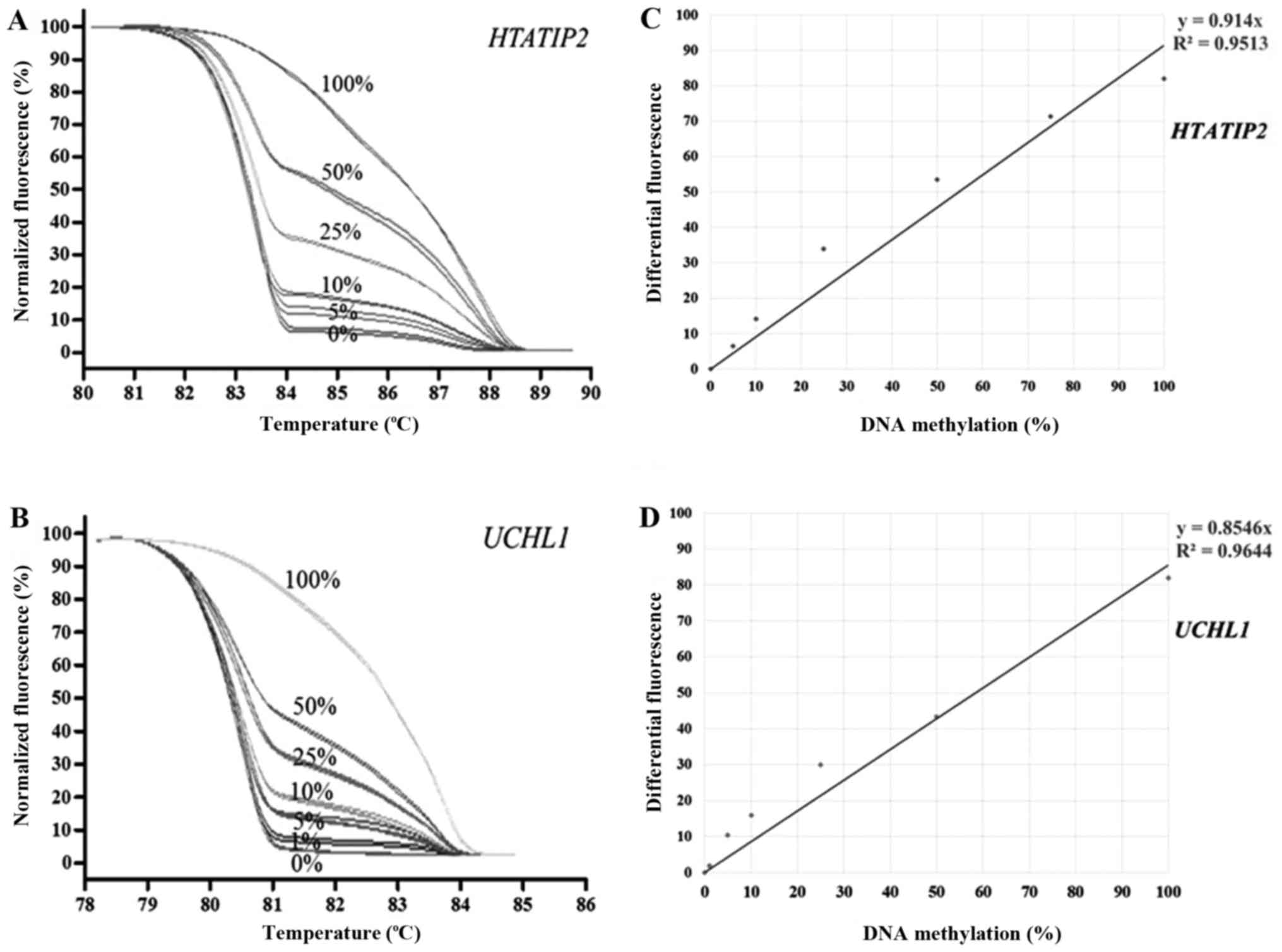
MS-HRM of individual genes was performed using
standard dilution series including 100, 50, 25, 10, 5, 1 and 0%
methylated controls. The lower detection limit of most genes was 5%
except 1% for UCHL1. The MS-HRM representatives of
HTATIP2 and UCHL1 are shown in Fig. 1. The reproducibility of MS-HRM was
performed using internal controls (25 and 75% of methylated DNA in
a background of unmethylated DNA) which were run in triplicate in
every experiment. The coefficient of variation (%CV) of both intra-
and inter-assay of this study was less than 10%.
Aberrant DNA methylation is commonly
found in CCA
DNA methylation levels of 10 candidate genes were
quantified in 54 CCA samples using MS-HRM. As shown in Fig. 2, the distribution of DNA
methylation levels showed high percentage in UCHL1 and
IRF4 with median of 57.29 and 56.32%, respectively. The
moderate DNA methylation levels were found in CCND2,
HTATIP2 and TP53I3 (median; 22.02, 13.59 and 12.41%,
respectively), whereas RUNX3, RASSF1, ALDH1A3,
PPP4C and SLC29A1 had very low DNA methylation levels
(median; 1.69, 4.11, 3.05, 2.63 and 1.87%, respectively).
 | Figure 2.Scatter plot with median line
graphics of the DNA methylation levels of the 10 candidate genes in
the 54 cholangiocarcinoma samples. UCHL1, ubiquitin C-terminal
hydrolase L1; IRF4, interferon regulatory factor 4; CCND2, cyclin
D2; HTATIP2, human immunodeficiency virus-1 tat interactive protein
2; TP53I3, tumor protein p53 inducible protein 3; RUNX3, runt
related transcription factor 3; RASSF1, Ras association domain
family member 1; ALDH1A3, aldehyde dehydrogenase 1 family member
A3; PPP4C, protein phosphatase 4 catalytic subunit; SLC29A1, solute
carrier family 29 member 1. |
DNA methylation of HTATIP2 and UCHL1
was associated with patients' overall survival
According to the median of methylation level of each
candidate gene, 54 CCA patients were divided into two groups,
depending on their methylation level, as low and high methylation
level groups. The median methylation level of high and low
HTATIP2 methylation group was 15.10 and 12.31%,
respectively. The median methylation level of high and low
UCHL1 methylation group was 69.10 and 38.69%, respectively.
Our results showed that CCA patients with high HTATIP2
methylation had significantly longer overall survival than those
with low methylation (median; 59.4 vs. 26.2 weeks, P=0.003)
(Fig. 3A). In contrast, CCA
patients who had low UCHL1 methylation showed significantly
longer overall survival than those who had high methylation
(median; 61.3 vs. 33.3 weeks, P=0.044) (Fig. 3B). Moreover, the multivariate Cox
regression analysis demonstrated that UCHL1 was an
independent factor for CCA with hazard ratio of 1.81 (95%CI,
1.01–3.25) in high methylation group (Table II). However, no significant
differences in correlation between DNA methylation and overall
survival time of CCND2, PPP4C, IRF4,
ALDH1A3, SLC29A1, TP53I3, RASSF1, and
RUNX3 were found. Also, no significant correlations of DNA
methylation status of individual gens with age, sex, histological
type, tumor stage and chemotherapy were observed.
| Table II.Cox regression analysis of
cholangiocarcinoma clinicopathological parameters and the DNA
methylation of 10 genes. |
Table II.
Cox regression analysis of
cholangiocarcinoma clinicopathological parameters and the DNA
methylation of 10 genes.
|
| Univariate | Multivariate |
|---|
|
|
|
|
|---|
| Parameters (n) | HR (95% CI) | P-value | HR (95% CI) | P-value |
|---|
| Age, years |
|
|
|
|
| ≤59
(26) | Reference |
|
|
|
| >59
(28) | 0.951
(0.55–1.65) | 0.857 | – | – |
| Sex |
|
|
|
|
| Male
(33) | Reference |
|
|
|
| Female
(21) | 1.14
(0.65–1.99) | 0.651 | – | – |
| Histopathology |
|
|
|
|
| Well
differentiated (20) | Reference |
|
|
|
| Less
differentiated (11) | 0.75
(0.33–1.69) | 0.483 | – | – |
| Staging |
|
|
|
|
| I–II
(4) | Reference |
|
|
|
| III–IV
(36) | 0.87
(0.30–2.48) | 0.794 | – | – |
| Chemotherapy |
|
|
|
|
|
Treatment (42) | Reference |
|
|
|
| No
treatment (12) | 0.70
(0.36–1.36) | 0.289 | – | – |
| UCHL1 |
|
|
|
|
| Low
methylation (27) | Reference |
| Reference |
|
| High
methylation (27) | 1.84
(1.03–3.29) | 0.040 | 1.81
(1.01–3.25) | 0.048 |
| IRF4 |
|
|
|
|
| Low
methylation (27) | Reference |
|
|
|
| High
methylation (27) | 1.24
(0.72–2.13) | 0.448 | – | – |
| CCND2 |
|
|
|
|
| Low
methylation (27) | Reference |
|
|
|
| High
methylation (27) | 0.77
(0.44–1.33) | 0.342 | – | – |
| HTATIP2 |
|
|
|
|
| Low
methylation (27) | Reference |
|
|
|
| High
methylation (27) | 0.55
(0.32–0.97) | 0.039 | NS | NS |
| TP53I3 |
|
|
|
|
| Low
methylation (27) | Reference |
|
|
|
| High
methylation (27) | 0.92
(0.53–1.58) | 0.755 | – | – |
| RUNX3 |
|
|
|
|
| Low
methylation (27) | Reference |
|
|
|
| High
methylation (27) | 0.55
(0.31–0.99) | 0.044 | NS | NS |
| RASSF1 |
|
|
|
|
| Low
methylation (27) | Reference |
|
|
|
| High
methylation (27) | 1.03
(0.59–1.80) | 0.911 | – | – |
| ALDH1A3 |
|
|
|
|
| Low
methylation (27) | Reference |
|
|
|
| High
methylation (27) | 0.57
(0.32–0.99) | 0.046 | NS | NS |
| PPP4C |
|
|
|
|
| Low
methylation (27) | Reference |
|
|
|
| High
methylation (27) | 1.36
(0.79–2.37) | 0.268 | – | – |
| SLC29A1 |
|
|
|
|
| Low
methylation (27) | Reference |
|
|
|
| High
methylation (27) | 1.02
(0.59–1.76) | 0.939 | – | – |
Of 54 CCA, only 42 cases underwent 5-FU chemotherapy
after surgery and were divided into two groups, depending on their
methylation level, as low and high methylation level groups;
according to the median of methylation level of HTATIP2 and
UCHL1 (median; 13.41 and 58.03%, respectively). The
Kaplan-Meier analysis of 5-FU treated CCA patients showed that
patients with high methylation of HTATIP2 and low
methylation of UCHL1 had longer overall survival (83.8 vs.
22.7 weeks, P<0.001; 65.6 vs. 25.7 weeks, P=0.041, respectively)
as shown in Fig. 3C and D,
respectively. Nevertheless, no significant differences in
correlation between DNA methylation and overall survival time of
CCND2, PPP4C, IRF4, ALDH1A3, SLC29A1, TP53I3, RASSF1, and
RUNX3 were found.
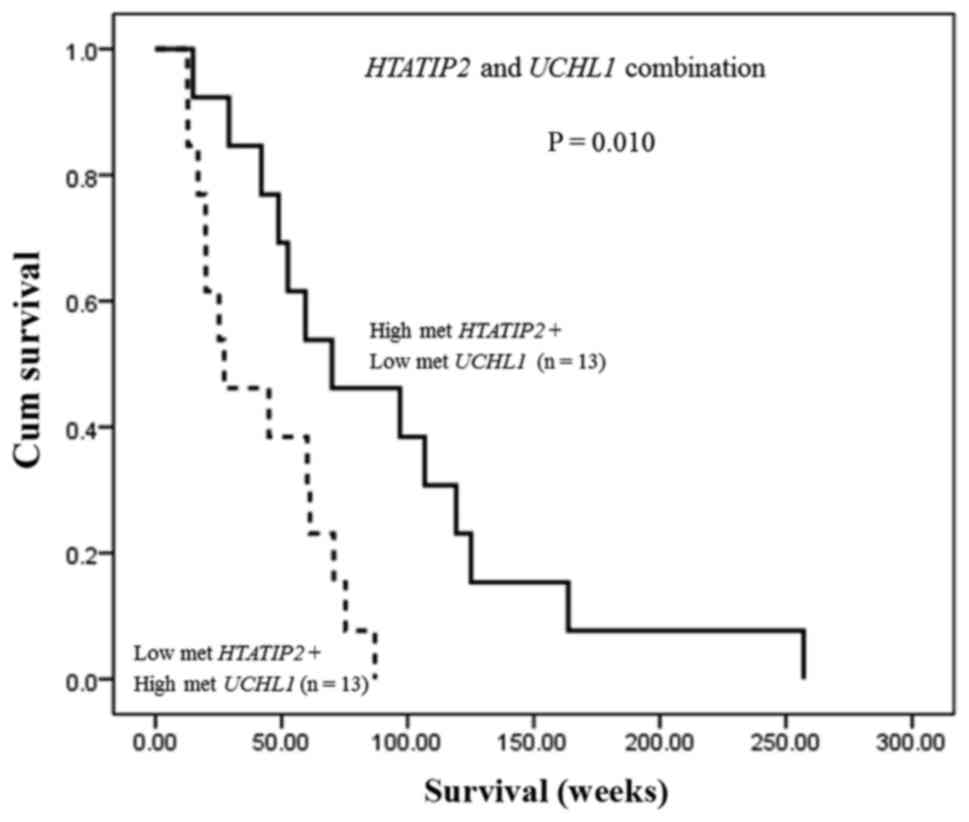
In addition, the combination of HTATIP2 and
UCHL1 methylation status exhibited that CCA patients who had
both high methylation of HTATIP2 and low methylation of
UCHL1 showed significantly longer overall survival time than
those with both low methylation of HTATIP2 and high
methylation of UCHL1 (median; 70.0 vs. 27.3 weeks, P=0.010)
(Fig. 4) indicating their
potential as a predictive biomarker for CCA.
High methylation of HTATIP2 and UCHL1
was found in CCA but not in adjacent normal tissue
To address whether high methylation levels of
UCHL1 and HTATIP2 found in 42 CCA patients treated
with 5-FU were not found in normal tissue, the methylation levels
of these genes were determined in 19 adjacent normal tissues of 42
cases. As shown in Fig. 5A and B,
the methylation levels of HTATIP2 and UCHL1 were
significantly higher in CCA than in adjacent normal tissues.
Matched-pair analysis of 19 cases showed that methylation levels of
HTATIP2 and UCHL1 were significantly higher in CCA
than in adjacent normal tissues (Fig.
5C and D).
Discussion
Our results showed high methylation levels of
multiple genes including UCHL1, IRF4, CCND2,
HTATIP2 and TP53I3 while very low methylation was found
in RUNX3, RASSF1, ALDH1A3, PPP4C and SLC29A1.
This finding suggested that DNA methylation is an epigenetic event
commonly found in CCA. However, we did not perform immunostaining
of the proteins of these genes in CCA samples which could indicate
the effect of DNA methylation on gene silencing. It is conceivable
that DNA methylation of these genes, at least in part, may
potentially lead to gene silencing which may have the great impact
on cancer cell survival and progression. Patients treated with 5-FU
in our pilot study (10) were
divided into short and long survival to select genes differentially
methylated, by which HTATIP2 was one of 10 genes selected.
In this present study, patients with high and low methylated
HTATIP2 were statistically analyzed with overall
survival.
The present study showed that hypermethylation of
two genes, namely HTATIP2 and UCHL1, was not only
tumor specific, but also correlated with patients' overall survival
time. An important corollary of this study is the potential of
methylation status of HTATIP2 and UCHL1 which can
serve as predictive biomarkers for CCA in which HTATIP2
hypermethylation is a favorable predictive marker, whereas
UCHL1 hypermethylation is an unfavorable predictive marker,
and when used in combination may strengthen their potential.
HTATIP2 is implicated in the regulation of tumor
cell growth, metastasis, apoptosis and autophagy related pathways
(26,27). There are 2 isoforms of HTATIP2, in
which isoform 1 is a metastasis suppressor with proapoptotic as
well as anti-angiogenic properties which is oxidoreductase required
for tumor suppression (26,28).
However, small isoform 2 (TC3), 15 kDa, actually has a
death-protective function (29).
Hypermethylation of HTATIP2 was found in 47% of
hepatocellular carcinoma samples but was not detected in normal
liver tissues (30). Another study
showed that 36% of colorectal carcinoma samples had hypermethylated
HTATIP2 (31), whereas
hypermethylation of HTATIP2 was found in 98% (53/54) of CCA
in this study. The high frequency of HTATIP2 and
UCHL1 hypermethylation found in liver fluke related CCA may
be due to chronic inflammation caused by liver fluke infection. It
has been shown that DNA methylation is observed during inflammation
and inflammation-associated carcinogenesis (32). Methylation of both genes may
potentially lead to gene silencing resulting in no cell growth
inhibition and apoptosis induction.
The UCHL1 is a de-ubiquitinating enzyme which
cleaves, recycles ubiquitin molecules, and stabilizes ubiquitin
pool suggesting its functions as tumor suppressor. UCHL1 has been
reported to directly interact with p53 and stabilize p53 by
inhibiting its degradation through the ubiquitination pathway
(33). Restoration expression of
silenced UCHL1 of hepatocellular carcinoma cell lines significantly
inhibited cell growth and colony formation (33). UCHL1 gene silencing by
promoter methylation may deregulate the p53 pathway by reducing its
ability in apoptosis induction caused by a chemotherapeutic agent
in gastric cancer, by which UCHL1 re-expression could induce
apoptosis (34). In the present
study, UCHL1 hypermethylation was observed in CCA, but not
in the adjacent normal epithelium. We speculate that loss of UCHL1
expression due to promoter hypermethylation may result in increased
p53 degradation via ubiquitination pathway, and consequently loss
of p53 function in apoptosis induction. This may in turn lead to
acquired chemotherapeutic drug resistance in CCA. However, further
study of UCHL1 in CCA is required in order to know its function
which may be relevant to chemotherapy response.
We demonstrated that CCA patients with high
methylation level of HTATIP2 and low methylation level of
UCHL1 were associated with longer overall survival. This
finding showed the significant association not only in 54 CCA but
also in 42 5-FU treated CCA suggesting the great value of
UCHL1 and HTATIP2 methylation as a predictive
biomarker for CCA. Based on median overall survival of CCA patients
in Northeast Thailand reported by Luvira et al (8) which was 4 months, it is hard to get
the data of disease-free survival. The meta-analysis of 14 studies
with 1705 patients involved revealed significant association
between high expression of HTATIP2 in cancer patients and a good
overall survival (35). In
contradictory to our results, the methylation of the HTATIP2
promoter did not significantly influence the overall survival.
Moreover, downregulation of HTATIP2 is involved in
progression and aggressiveness of hepatocellular carcinoma
(36) and decreased HTATIP2
expression in laryngeal carcinoma is related to poor prognosis and
shorten survival (37), suggesting
a tumor suppressive function of HTATIP2. Previous study showed that
HTATIP2 might be an upstream regulator of p53 which induces
apoptosis through both p53-dependent and -independent pathways
(38), while mutant HTATIP2
down-regulates the endogenous expression of p53 mRNA and protein
(39). Furthermore, HTATIP2 could
reduce the expression of vascular endothelial growth factor (VEGF)
mRNA which resulted in inhibition of angiogenesis (26). Dong et al (40) studied HTATIP2 methylation
which the CpG sites are the same as our study and protein
expression in glioma patients and observed no significant
association indicating that DNA methylation is not a major
epigenetic event for gene silencing. Accordingly, high methylation
level of HTATIP2 may not reflect low protein expression in
CCA patients. However, further study is required to precisely
identify its roles in CCA.
Bonazzi et al (41) showed that hypermethylation of
UCHL1 is crucial for gene silencing in melanoma, of which
the CpG sites are the same as our study. Poor prognosis was
observed in CCA patients with high methylation of UCHL1
implicating low expression of UCHL1. The expression of UCHL1 could
activate the p14ARF-p53 signaling pathway by deubiquitinating p53
and p14ARF as well as ubiquitinating MDM2 through its two opposing
enzyme activities, hydrolase and ligase (42,43).
In opposite to our finding, high UCHL1 expression was significantly
associated with shorter overall survival in breast cancer (44).
Although IRF4, CCND2, TP53I3
exhibited high methylation levels in CCA patients, no correlation
of their methylation status with patients' survival and clinical
parameters was found. In addition, hypermethylation of RUNX3
(45), RASSF1 (46) and ALDH1A3 (47) was found in multiple tumors such as
meningiomas, gliomas and glioblastoma, but in our study, they
exhibited very low methylation levels which were the same levels of
normal epithelial cells.
In summary, our findings provide an insight into
aberrant DNA methylation of UCHL1, IRF4, CCND2,
HTATIP2 and TP53I3 as an epigenetic event of CCA. The
methylation of HTATIP2 and UCHL1 can served as a
potential predictive biomarker for CCA.
Acknowledgements
This project was supported by the Higher Education
Research Promotion and National Research University Project of
Thailand, the Office of the Higher Education Commission, through
the Center of Excellence in Specific Health Problems in Greater
Mekong Sub-region cluster (SHeP-GMS), Khon Kaen University,
Thailand. C.N. was financially supported by the Centre for Research
and Development of Medical Diagnostic Laboratories, Faculty of
Associated Medical Sciences, Khon Kaen University, Khon Kaen,
Thailand. We thank the Cholangiocarcinoma Research Institute, Khon
Kaen University, Khon Kaen, Thailand for providing samples and
clinical data.
References
|
1
|
Mosconi S, Beretta GD, Labianca R, Zampino
MG, Gatta G and Heinemann V: Cholangiocarcinoma. Crit Rev Oncol
Hemat. 69:259–270. 2009. View Article : Google Scholar
|
|
2
|
Shaib Y and El-Serag HB: The epidemiology
of cholangiocarcinoma. Semin Liver Dis. 24:115–125. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang H, Yang T, Wu M and Shen F:
Intrahepatic cholangiocarcinoma: Epidemiology, risk factors,
diagnosis and surgical management. Cancer Lett. 379:198–205. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bridgewater J, Galle PR, Khan SA, Llovet
JM, Park JW, Patel T, Pawlik TM and Gores GJ: Guidelines for the
diagnosis and management of intrahepatic cholangiocarcinoma. J
Hepatol. 60:1268–1289. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vatanasapt V, Uttaravichien T, Mairiang
EO, Pairojkul C, Chartbanchachai W and Haswell-Elkins M:
Cholangiocarcinoma in north-east Thailand. Lancet. 335:116–117.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shin HR, Oh JK, Masuyer E, Curado MP,
Bouvard V, Fang YY, Wiangnon S, Sripa B and Hong ST: Epidemiology
of cholangiocarcinoma: An update focusing on risk factors. Cancer
Sci. 101:579–585. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sripa B, Bethony JM, Sithithaworn P,
Kaewkes S, Mairiang E, Loukas A, Mulvenna J, Laha T, Hotez PJ and
Brindley PJ: Opisthorchiasis and Opisthorchis-associated
cholangiocarcinoma in Thailand and Laos. Acta Trop. 120 Suppl
1:S158–S168. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Luvira V, Nilprapha K, Bhudhisawasdi V,
Pugkhem A, Chamadol N and Kamsa-ard S: Cholangiocarcinoma patient
outcome in Northeastern Thailand: Single-center prospective study.
Asian Pac J Cancer Prev. 17:401–406. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Malhi H and Gores GJ: Cholangiocarcinoma:
Modern advances in understanding a deadly old disease. J Hepatol.
45:856–867. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sriraksa R, Zeller C, Dai W, Siddiq A,
Walley AJ, Limpaiboon T and Brown R: Aberrant DNA methylation at
genes associated with a stem cell-like phenotype in
cholangiocarcinoma tumors. Cancer Prev Res (Phila). 6:1348–1355.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
McCarthy DJ and Smyth GK: Testing
significance relative to a fold-change threshold is a TREAT.
Bioinformatics. 25:765–771. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang X, Ozawa Y, Lee H, Wen YD, Tan TH,
Wadzinski BE and Seto E: Histone deacetylase 3 (HDAC3) activity is
regulated by interaction with protein serine/threonine phosphatase
4. Genes Dev. 19:827–839. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu Z, Zhang X, Xu X, Chen L, Li W, Yu H,
Sun Y, Zeng J and Jia J: RUNX3 inhibits survivin expression and
induces cell apoptosis in gastric cancer. Eur J cell Biol.
93:118–126. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ju XL, Ishikawa TO, Naka K, Ito K, Ito Y
and Oshima M: Context-dependent activation of Wnt signaling by
tumor suppressor RUNX3 in gastric cancer cells. Cancer Sci.
105:418–424. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Imaizumi Y, Kohno T, Yamada Y, Ikeda S,
Tanaka Y, Tomonaga M and Matsuyama T: Possible involvement of
interferon regulatory factor 4 (IRF4) in a clinical subtype of
adult T-cell leukemia. Jpn J Cancer Res. 92:1284–1292. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xiang T, Li L, Yin X, Yuan C, Tan C, Su X,
Xiong L, Putti TC, Oberst M, Kelly K, et al: The ubiquitin
peptidase UCHL1 induces G0/G1 cell cycle arrest and apoptosis
through stabilizing p53 and is frequently silenced in breast
cancer. PLoS One. 7:e297832012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Moll UM and Slade N: p63 and p73: Roles in
development and tumor formation. Mol Cancer Res. 2:371–386.
2004.PubMed/NCBI
|
|
18
|
Sicinski P, Donaher JL, Geng Y, Parker SB,
Gardner H, Park MY, Robker RL, Richards JS, McGinnis LK, Biggers
JD, et al: Cyclin D2 is an FSH-responsive gene involved in gonadal
cell proliferation and oncogenesis. Nature. 384:470–474. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shivakumar L, Minna J, Sakamaki T, Pestell
R and White MA: The RASSF1A tumor suppressor blocks cell cycle
progression and inhibits cyclin D1 accumulation. Mol Cell Biol.
22:4309–4318. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Koppaka V, Thompson DC, Chen Y, Ellermann
M, Nicolaou KC, Juvonen RO, Petersen D, Deitrich RA, Hurley TD and
Vasiliou V: Aldehyde dehydrogenase inhibitors: A comprehensive
review of the pharmacology, mechanism of action, substrate
specificity and clinical application. Pharmacol Rev. 64:520–539.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Garcia-Manteiga J, Molina-Arcas M, Casado
FJ, Mazo A and Pastor-Anglada M: Nucleoside transporter profiles in
human pancreatic cancer cells: Role of hCNT1 in
2′,2′-difluorodeoxycytidine-induced cytotoxicity. Clin Cancer Res.
9:5000–5008. 2003.PubMed/NCBI
|
|
22
|
Ito M, Jiang C, Krumm K, Zhang X, Pecha J,
Zhao J, Guo Y, Roeder RG and Xiao H: TIP30 deficiency increases
susceptibility to tumorigenesis. Cancer Res. 63:8763–8767.
2003.PubMed/NCBI
|
|
23
|
Donninger H, Clark J, Rinaldo F, Nelson N,
Barnoud T, Schmidt ML, Hobbing KR, Vos MD, Sils B and Clark GJ: The
RASSF1A tumor suppressor regulates XPA-mediated DNA repair. Mol
Cell Biol. 35:277–287. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sriraksa R, Chaopatchayakul P,
Jearanaikoon P, Leelayuwat C and Limpaiboon T: Verification of
complete bisulfite modification using Calponin-specific primer
sets. Clin Biochem. 43:528–530. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wojdacz TK, Dobrovic A and Hansen LL:
Methylation-sensitive high-resolution melting. Nat Protoc.
3:1903–1908. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
NicAmhlaoibh R and Shtivelman E:
Metastasis suppressor CC3 inhibits angiogenic properties of tumor
cells in vitro. Oncogene. 20:270–275. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
King FW and Shtivelman E: Inhibition of
nuclear import by the proapoptotic protein CC3. Mol Cell Biol.
24:7091–7101. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shtivelman E: A link between metastasis
and resistance to apoptosis of variant small cell lung carcinoma.
Oncogene. 14:2167–2173. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Whitman S, Wang X, Shalaby R and
Shtivelman E: Alternatively spliced products CC3 and TC3 have
opposing effects on apoptosis. Mol Cell Biol. 20:583–593. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lu B, Ma Y, Wu G, Tong X, Guo H, Liang A,
Cong W, Liu C, Wang H, Wu M, et al: Methylation of Tip30 promoter
is associated with poor prognosis in human hepatocellular
carcinoma. Clin Cancer Res. 14:7405–7412. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen X, Cao X, Dong W, Luo S, Suo ZH and
Jin Y: Expression of TIP30 tumor suppressor gene is down-regulated
in human colorectal carcinoma. Dig Dis Sci. 55:2219–2226. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hartnett L and Egan LJ: Inflammation, DNA
methylation and colitis-associated cancer. Carcinogenesis.
33:723–731. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yu J, Tao Q, Cheung KF, Jin H, Poon FF,
Wang X, Li H, Cheng YY, Rocken C, Ebert MP, et al: Epigenetic
identification of ubiquitin carboxyl-terminal hydrolase L1 as a
functional tumor suppressor and biomarker for hepatocellular
carcinoma and other digestive tumors. Hepatology. 48:508–518. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Waraya M, Yamashita K, Ema A, Katada N,
Kikuchi S and Watanabe M: Exclusive association of p53 mutation
with super-high methylation of tumor suppressor genes in the p53
pathway in a unique gastric cancer phenotype. PLoS One.
10:e01399022015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xu T, Jin Z, Yuan Y, Zheng H, Li C, Hou W,
Guo Q and Hua B: Tat-Interacting protein 30 (TIP30) expression
serves as a new biomarker for tumor prognosis: A systematic review
and meta-analysis. PLoS One. 11:e01684082016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhu M, Yin F, Fan X, Jing W, Chen R, Liu
L, Zhang L, Liu Y, Liang Y, Bu F, et al: Decreased TIP30 promotes
Snail-mediated epithelial-mesenchymal transition and
tumor-initiating properties in hepatocellular carcinoma. Oncogene.
34:1420–1431. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhu M, Yin F, Yang L, Chen S, Chen R, Zhou
X, Jing W, Fan X, Jia R, Wang H, et al: Contribution of TIP30 to
chemoresistance in laryngeal carcinoma. Cell Death Dis.
5:e14682014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhao J, Zhang X, Shi M, Xu H, Jin J, Ni
HD, Yang SL, Dai JX, Wu MC and Guo YJ: TIP30 inhibits growth of HCC
cell lines and inhibits HCC xenografts in mice in combination with
5-FU. Hepatology. 44:205–215. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jiang C, Pecha J, Hoshino I, Ankrapp D and
Xiao H: TIP30 mutant derived from hepatocellular carcinoma
specimens promotes growth of HepG2 cells through up-regulation of
n-cadherin. Cancer Res. 67:3574–3582. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dong X, Deng Q, Nie X, Zhang M, Jia W,
Chen C, Xu C and Xu R: Downregulation of HTATIP2 expression is
associated with promoter methylation and poor prognosis in glioma.
Exp Mol Pathol. 98:192–199. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bonazzi VF, Nancarrow DJ, Stark MS, Moser
RJ, Boyle GM, Aoude LG, Schmidt C and Hayward NK: Cross-platform
array screening identifies COL1A2, THBS1, TNFRSF10D and UCHL1 as
genes frequently silenced by methylation in melanoma. PLoS One.
6:e261212011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Xiang T, Li L, Yin X, Yuan C, Tan C, Su X,
Xiong L, Putti T, Oberst M, Kelly K, et al: The ubiquitin peptidase
UCHL1 induces G0/G1 cell cycle arrest and apoptosis through
stabilizing p53 and is frequently silenced in breast cancer. PLoS
One. 7:e297832012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li L, Tao Q, Jin H, van Hasselt A, Poon
FF, Wang X, Zeng MS, Jia WH, Zeng YX, Chan AT and Cao Y: The tumor
suppressor UCHL1 forms a complex with p53/MDM2/ARF to promote p53
signaling and is frequently silenced in nasopharyngeal carcinoma.
Clin Cancer Res. 16:2949–2958. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Schroder C, Milde-Langosch K, Gebauer F,
Schmid K, Mueller V, Wirtz RM, Meyer-Schwesinger C, Schluter H,
Sauter G and Schumacher U: Prognostic relevance of ubiquitin
C-terminal hydrolase L1 (UCH-L1) mRNA and protein expression in
breast cancer patients. J Cancer Res Clin Oncol. 139:1745–1755.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Majchrzak-Celinska A, Paluszczak J,
Szalata M, Barciszewska AM, Nowak S and Baer-Dubowska W: DNA
methylation analysis of benign and atypical meningiomas:
Correlation between RUNX3 methylation and WHO grade. J Cancer Res
Clin Oncol. 141:1593–1601. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Majchrzak-Celinska A, Paluszczak J,
Szalata M, Barciszewska AM, Nowak S, Kleszcz R, Sherba A and
Baer-Dubowska W: The methylation of a panel of genes differentiates
low-grade from high-grade gliomas. Tumor Biol. 36:3831–3841. 2015.
View Article : Google Scholar
|
|
47
|
Ni W, Luo L, Ping Z, Yuan HP, Zhao XD and
Xu W: Prognostic value of ALDH1A3 promoter methylation in
glioblastoma: A single center experience in Western China. Asian
Pac J Cancer Prev. 16:591–594. 2015. View Article : Google Scholar : PubMed/NCBI
|