Introduction
Systemic lupus erythematosus (SLE), a complex
autoimmune disease, is characterized by multiple immunologic
abnormalities. It has previously been reported that
~7.4–159.4/100,000 people suffer from SLE worldwide, and SLE
predominantly affects women of childbearing age (1–3). The
presence of high titres of autoantibodies against nuclear
components, elevated circulating immune complexes and complement
deficiency are the predominant characteristics of the disease. The
etiology of SLE is incompletely understood; however, genetic
factors are important in the susceptibility to the disease.
Long noncoding RNAs (lncRNAs) are transcript RNA
molecules, longer than 200 nucleotides, which do not encode a
protein and reside in the nucleus or cytoplasm (4). lncRNAs are classified by their
position relative to protein-coding messenger RNAs (mRNAs), and
comprise the long intergenic ncRNA (lincRNA), intronic lncRNA,
antisense lncRNA, transcribed pseudogene lncRNAs and enhancer RNA
(eRNA). Although dysregulation of lncRNA expression has been
characterized predominantly in cancer, it has recently been
evaluated in autoimmune diseases, such as autoimmune thyroid
disease (AITD) and rheumatoid arthritis (RA) (5,6).
Specifically, lncRNAs have been proposed as a regulator of immune
response (7–9) and contribute to the inflammatory
response (10–13). Thus, it is hypothesized that
lncRNAs, in combination with mRNAs, are also involved in the
germination and development of SLE. However, until now, knowledge
regarding the possible association between abnormal lncRNAs and SLE
remains limited.
In the present study, integrative lncRNA-mRNA
microarray analysis was performed to determine the expression
profiles of lncRNAs in SLE samples and normal samples. In addition,
three lncRNAs and three mRNAs were confirmed using reverse
transcription-quantitative polymerase chain reaction (RT-qPCR).
Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes
(KEGG) analysis were used to predict the function and signaling
pathways affected by the differentially expressed mRNAs, which were
target genes of aberrantly expressed lncRNAs. The results of the
present study suggest that lncRNA expression patterns may provide
further insight into the pathogenesis of SLE.
Materials and methods
SLE patients and healthy control
subjects
Whole blood was collected from 29 SLE patients and
34 age- and sex-matched healthy control subjects (HCs), who were
enrolled between July 2015 and May 2016, in the Department of
Rheumatology and Immunology, The First Affiliated Hospital of
Nanchang University (Nanchang, China). HCs were selected based on
no history of autoimmune disease and all SLE patients were
diagnosed according to the American College of Rheumatology
classification criteria for SLE (14). A total of 10 of the already
enrolled 29 patients with SLE and 10 of the already enrolled 34
age- and sex-matched HCs were recruited to isolate the peripheral
blood mononuclear cells (PBMCs) and perform microarray analysis.
Other samples (19 patients with SLE and 24 HCs) were used to verify
the results of microarray by RT-qPCR assay. The study was approved
by the Ethics Committee of the First Affiliated Hospital of
Nanchang University (approval no. 2014003) and was conducted in
accordance with the Declaration of Helsinki. Informed consent was
obtained from all the participants before commencing the study.
Blood sample collection and RNA
isolation
Venous blood samples were obtained from 29 SLE
patients and 34 HCs and stored in EDTA tubes. PBMCs were isolated
from the venous blood by Ficoll-Histopaque (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) density gradient centrifugation (1,000 ×
g, 20 min, 22°C). Total RNA was extracted from PBMCs from each
specimen using TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) according to the manufacturer's
instructions. The concentration and quality of the RNA were
assessed by absorbance spectrometry (260 nm/280 nm) using a
NanoDrop ND-1000 (Thermo Fisher Scientific, Inc.).
Microarray and data analysis
RNA samples were analyzed using Arraystar Human
LncRNA Array v3.0 (Arraystar, Inc., Rockville, MD, USA), according
to the manufacturer's instructions, which includes 33,045 lncRNAs
and 30,215 coding transcripts. Briefly, rRNA was removed from total
RNA and the mRNAs were obtained using an mRNA-ONLY™ Eukaryotic mRNA
Isolation kit (Epicentre; Illumina, Inc., San Diego, CA, USA). The
random priming method was utilized to amplify each sample and mRNA
was transcribed into fluorescent complementary RNAs (cRNAs) without
3′bias. Labeled cRNAs were hybridized to the Human LncRNA
Microarray. After washing the slides using Gene Expression Wash
Buffer 1 (Agilent Technologies, Inc., Santa Clara, CA, USA), and
Gene Expression Wash Buffer 2 (Agilent Technologies, Inc.), the
arrays were scanned using an Agilent G2505C Scanner. Raw data were
extracted using GeneSpring GX v12.0 software package (Agilent
Technologies, Inc.). The microarray work was performed by Kangcheng
Biology Engineering Co., Ltd. (Shanghai, China).
Functional group analysis
GO (www.geneontology.org) and KEGG (www.genome.ad.jp/kegg) databases were used to analyze
the biological functions and signaling pathways affected by
disexpression of mRNAs (the cut-off P-value was 0.05).
RT-qPCR assay
The expression levels of differentially expressed
lncRNAs and mRNAs were confirmed by RT-qPCR. Briefly, total RNA was
extracted from PBMCs using TRIzol reagent (Sigma-Aldrich; Merck
KGaA), and 5 µg samples were used for the synthesis of first strand
cDNA using a PrimeScript™ RT Reagent kit (Takara Biotechnology Co.,
Ltd., Dalian, China) according to the manufacturer's protocol. The
RT reaction was performed in a 10 µl reaction containing 5X
PrimeScript™ Buffer, 1 µl RT specific primer, 0.5 µl PrimeScript™
RT Enzyme Mix and 5 µg of total RNA. The RT assay was set at an
initial denaturation step at 37°C for 15 min, followed by 85°C for
5 sec. Following first strand cDNA synthesis, the PCR reaction was
performed in a 10 µl reaction containing 1X SYBR-Green PCR Master
mix (Takara Biotechnology Co., Ltd.), 0.4 µM of each specific
forward and reverse primer and 0.5 µl of cDNA template. The PCR
assay was set at an initial denaturation step at 95°C for 5 min,
followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min with an
ABI 7500 Real-time PCR System (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The primers are detailed in Table I. Gene expression levels were
normalized to GAPDH in cDNA samples, and all experiments were
performed in triplicate. The relative expression levels of the
genes were determined using the ∆Cq method, where ∆Cq=Cq median
gene-Cq median GAPDH (15,16).
 | Table I.Primer sequence. |
Table I.
Primer sequence.
| Gene name | Primer sequence | Product (bp)
length |
|---|
| AK130076 | F:
CCAACATGCTGACTCACCCTTCC | 185 |
|
| R:
ATGGAGTCTCGCTCTGTCACCCA |
|
| CTC-471J1.2 | F:
ACAAATCTGAAAATACCACCTTG | 106 |
|
| R:
TTTCCTAGAAATCATTTAACCCA |
|
| RP11-875O11.1 | F:
CCCGATGGAATCTTACTCTGTTG | 129 |
|
| R:
CATGCCTGTAATCCCAGCTACTC |
|
| PGLYRP1 | F:
CACATGAAGACACTGGGCTGGT | 147 |
|
| R:
CATGAAGCTGATGCCAATGGAC |
|
| PDCD4 | F:
GGATGAAAGGGCATTTGAGAAGAC | 152 |
|
| R:
CCAATGCTAAGGATACTGCCAACA |
|
| PTEN | F:
ATCATTTCTTCATAGTGCTCCCC | 125 |
|
| R:
CAATAGTAGTTGTACTCCGCTTA |
|
| GAPDH | F:
GCACCGTCAAGGCTGAGAAC | 138 |
|
| R:
TGGTGAAGACGCCAGTGGA |
|
Statistical analysis
Data are presented as mean ± standard deviation. The
Mann-Whitney test or Student's t-test was used to perform
statistical analysis. P<0.05 was considered to indicate a
statistically significant difference.
Results
Aberrant lncRNA expression in PBMCs of
SLE
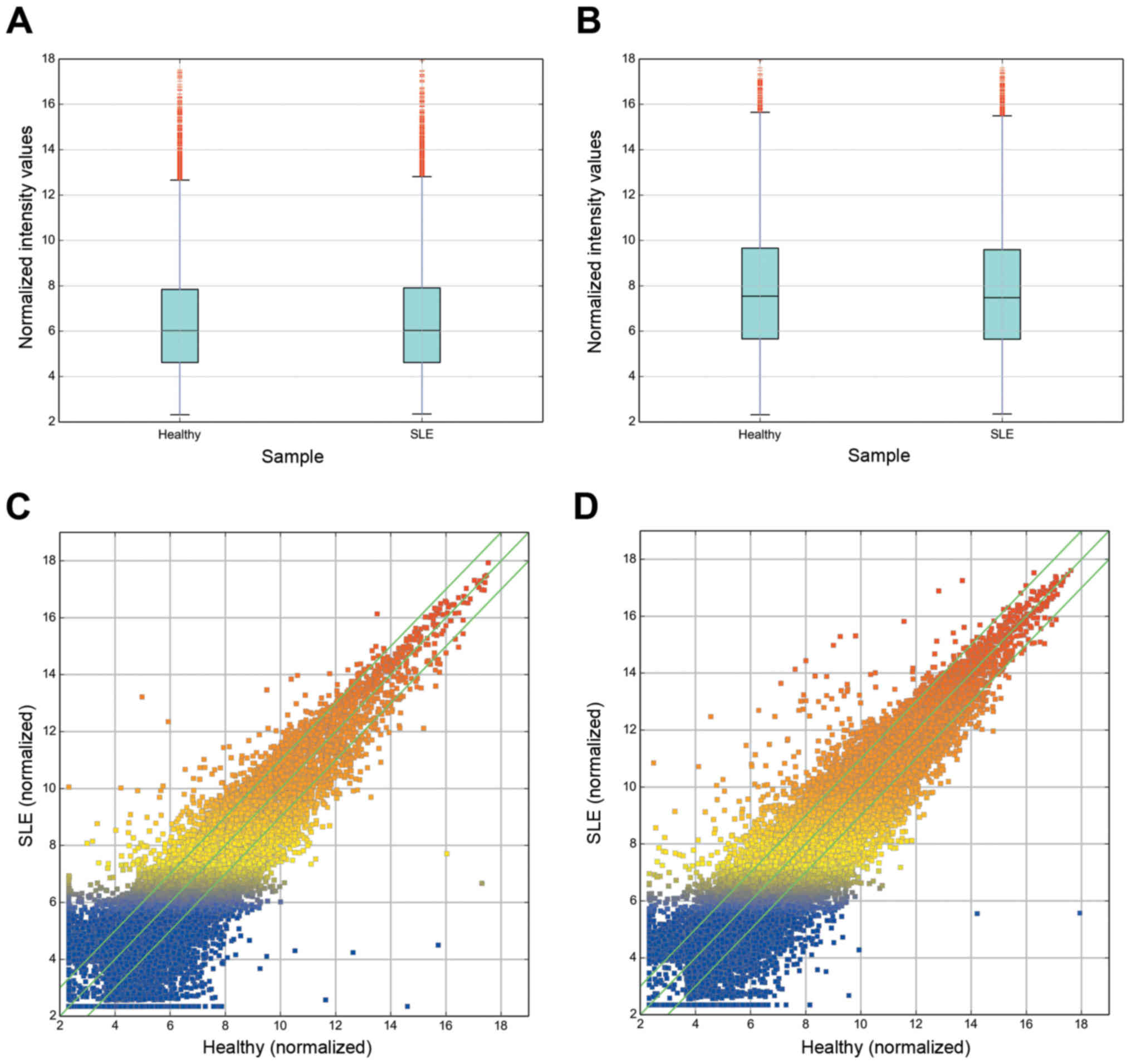
To investigate the possible biological functions of
lncRNAs in SLE, the lncRNA and mRNA expression profiles were
estimated in PBMCs of SLE using microarray analysis (Figs. 1 and 2). The expression profiles of 8,868
(28.9%) lncRNAs indicated that they were differentially expressed
(more than two-fold) between two groups (data no shown). Among
these, 3,657 were upregulated while 5,211 were downregulated in the
SLE group, compared with those in the healthy group. The most
significantly deregulated lncRNAs were ENST00000523884 (fold change
up, 303.1127021) and ENST00000559539 (fold change down,
4868.2903529).
Aberrant mRNA expression in PBMCs of
SLE
To evaluate the involvement of lncRNAs in
transcriptional, epigenetic or posttranscriptional regulation of
gene expression, the expression levels of potential target mRNAs of
the differentially expressed lncRNA data were predicted. In total,
compared with the healthy group, 6,876 mRNAs (2,862 upregulated and
4,014 downregulated) were identified to be differentially expressed
(P<0.05). The most markedly deregulated mRNAs were thyroglobulin
(fold change up, 329.7330982) and potassium calcium-activated
channel subfamily M regulatory beta subunit 3 (fold change down,
5246.5342205).
lncRNA classification and subgroup
analysis
According to previous reports, lncRNAs are
classified into different subgroups, such as lncRNAs with
enhancer-like function (lncRNA-a), antisense lncRNA and lincRNAs
(17). Previous studies
demonstrated that subgroups, such as lincRNA and enhancer-like
lncRNAs are involved in numerous types of disease (18,19).
lncRNA-a were identified using GENCODE annotation (16). Further analysis was performed in
the current study by classifying and stratifying the lncRNAs into
the aforementioned subgroups (17). A total of 847 enhancer-like lncRNAs
were identified to be significantly differentially expressed (fold
change ≥2.0; P<0.05, data not shown), among which 430 lncRNAs
were upregulated and 417 were downregulated in the SLE group,
compared with the healthy group. The expression profiles of 1,911
lincRNAs (1,225 of which were upregulated) indicated that they were
significantly differentially expressed (fold change ≥2.0;
P<0.05, data not shown) between the SLE and healthy groups.
Among these, 1,225 were upregulated and 686 were downregulated. It
was also identified in antisense lncRNA profiling that 449
antisense lncRNAs (147 upregulated and 302 downregulated) were
differentially expressed (fold change ≥2.0; P<0.05, data not
shown) between the groups. In addition, certain nearby coding genes
may be regulated by these subgroup lncRNAs. Furthermore, 474
matched lncRNA-mRNA pairs were identified for 293 differentially
expressed lncRNAs (fold change ≥3.0) and 381 differentially
expressed mRNAs (fold change ≥3.0). Among them, 310 pairs were
differentially expressed unidirectionally (up or down), while 164
pairs were differentially expressed bidirectionally.
GO and KEGG signaling pathway
analysis
Through GO analysis, the downregulated and
upregulated transcripts of lncRNAs were identified to be associated
with biological processes, cellular components and molecular
function. Additionally, the differentially expressed mRNAs between
the SLE and healthy groups were significantly enriched in the
cellular macromolecule metabolic process, intracellular part,
nucleic acid binding, single-multicellular organism process, cell
periphery and sequence-specific DNA binding, amongst others (data
no shown). Using the latest version of the KEGG database, KEGG
pathway analysis was performed to evaluate differentially expressed
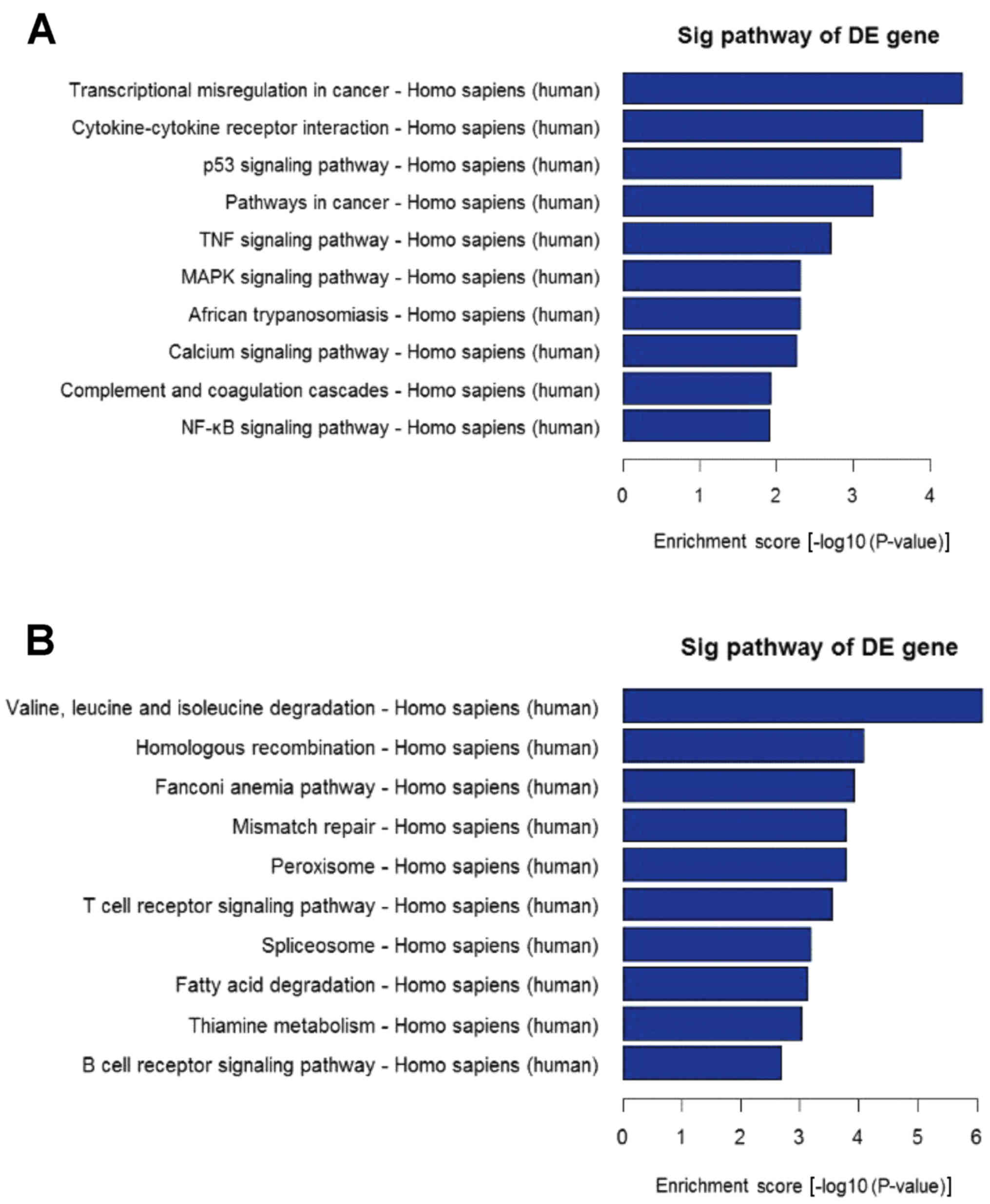
mRNAs. Pathway analysis determined that these lncRNAs may target 65
gene pathways, including 26 upregulated pathways, for example,
‘Transcriptional misregulation in cancer’ and 39 downregulated
pathways, for example, ‘Valine, leucine and isoleucine degradation’
(Fig. 3).
RT-qPCR validation
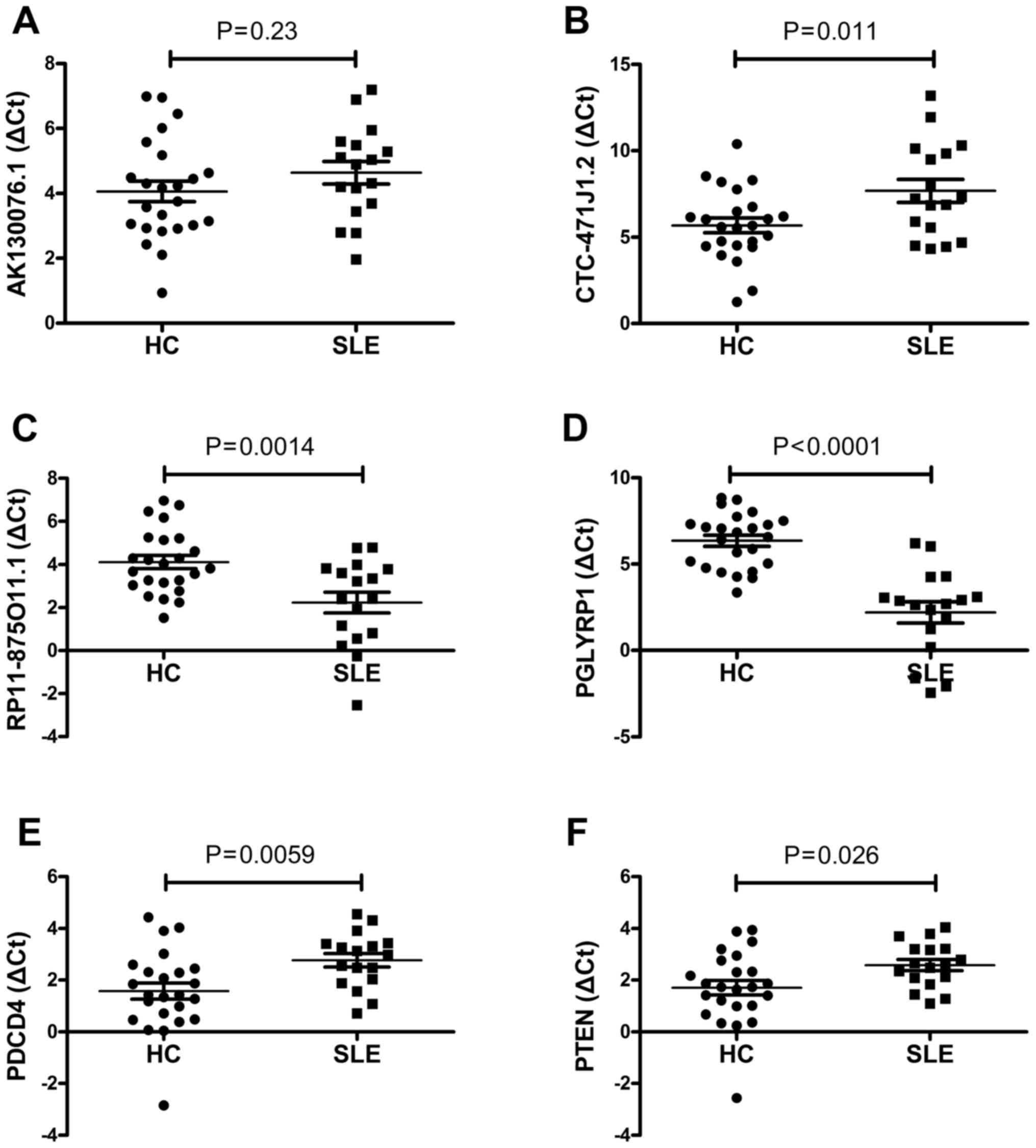
In order to verify the reliability of the microarray
data, a number of interesting candidate lncRNAs (AK130076.1,
CTC-471J1.2 and RP11-875O11.1) and mRNAs [peptidoglycan recognition
protein 1 (PGLYRP1), programmed cell death 4 (PDCD4) and
phosphatase and tensin homolog (PTEN)] were initially identified
for further analysis. The expression levels of these lncRNAs and
mRNAs were confirmed by RT-qPCR. The primers are presented in
Table I. For the lncRNAs, the
results demonstrated that RP11-875O11.1 were upregulated and that
CTC-471J1.2 were downregulated in 17 SLE patients relative to the
healthy group (all P<0.05; Fig. 4B
and C). The expression level of AK130076.1 was not
significantly different between SLE patients and healthy control
subjects (P>0.05; Fig. 4A). For
the mRNAs, the RT-qPCR analysis indicated that the expression
levels of PGLYRP1, PDCD4 and PTEN changed significantly between the
two groups (P<0.05 for each mRNA; Fig. 4). The results demonstrated that the
expression level patterns of RP11-875O11.1, CTC-471J1.2, PGLYRP1,
PDCD4 and PTEN were consistent with those obtained by microarray
analysis.
Discussion
SLE, an autoimmune disease, is characterized by
aberrant lymphocyte activation. It has a prevalence of 40 cases per
100,000 individuals, with onset typically occurring in women of
childbearing age (female:male ratio, 9:1) (20). Current treatment strategies using
immunosuppressive drugs and other medications for SLE are effective
at reducing morbidity and mortality, but fail to effectively cure
the disease (21). However, the
pathogenic mechanisms underlying SLE remain largely unknown;
therefore, further investigations of SLE are considered to be of
great importance.
Evidence from previous research indicated the
aberrant expression of lncRNAs in the pathogenesis of SLE (22,23);
however, fewer studies have examined the expression profile of
lncRNAs in SLE. PBMCs, which include numerous immune cells, are key
in host defense, and are used to identify novel disease mediators,
disease variants and treatment responses (24–26).
PBMCs have been used to discriminate the differences in non-coding
RNA (microRNAs) profiles in SLE using microarray technology
(27,28). Therefore, in an attempt to obtain
the expression pattern of lncRNAs in SLE, lncRNA microarray
technology was used to investigate the lncRNA signatures of 10 SLE
patients.
In the present study, a number of differentially
expressed mRNAs and lncRNAs were detected in PBMCs of SLE patients
to investigate the potential functions of lncRNAs in its
pathogenesis. A total of 8,868 lncRNAs (3,657 upregulated and 5,211
downregulated) and 6,876 mRNAs (2,862 upregulated and 4,014
downregulated) were significantly expressed in the SLE group
compared with the healthy group. lncRNAs include lincRNAs, intronic
lncRNAs, antisense lncRNAs, transcribed pseudogene lncRNAs and
eRNAs. lncRNAs function via a variety of mechanisms; however, a
common and important function of lncRNAs is to alter nearby
encoding gene expression by affecting the process of transcription
(29) or directly performing an
enhancer-like role (17,30). Wu et al (22) demonstrated that lincRNAs were
associated with the pathogenesis of SLE. Furthermore, the results
demonstrated that enhancer-like lncRNAs (430 upregulated and 417
downregulated), lincRNAs (1,225 upregulated and 686 downregulated)
and antisense lncRNAs (147 upregulated and 302 downregulated) were
aberrantly expressed. In addition, results from microarray analysis
revealed that these subgroup lncRNAs regulate certain nearby coding
genes.
To investigate the potential regulatory roles of
lncRNAs, GO category and KEGG pathway annotation were used to
analyze the target gene pool. GO and KEGG pathway analyses
demonstrated that downregulated and upregulated transcripts of
lncRNAs were associated with biological process, cellular
components and molecular function, which were associated with 65
gene pathways that corresponded to transcripts, for example,
‘Cytokine-cytokine receptor interaction’, ‘TNF signaling pathway’,
‘MAPK signaling pathway’ and ‘NF-κB signaling pathways’, which
indicated that the pathology of SLE is predominantly associated
with the regulation of multiple genes. Furthermore, these pathways
are associated with the initiation and development of SLE (31,32).
Subsequently, six altered lncRNAs and mRNAs were
selected, and their expression levels and the microarray results
were assessed via RT-qPCR. The change in mRNA expression level was
confirmed to be concordant with the microarray data, while the
expression tendency of lncRNA demonstrated a similar trend,
although it was not exactly the same. This may be due to the
heterogeneity of SLE, differing sample sizes and variation in the
sensitivity of the methods performed. Generally, the data from
microarray analysis requires confirmation by RT-qPCR, which is
considered to be more accurate.
In conclusion, lncRNA and mRNA expression levels
were analyzed in PBMCs from SLE samples using microarrays, which
revealed a novel and interesting foundation for improving the
understanding of the association between lncRNA homeostasis in
PBMCs and SLE. However, the findings described in the present study
represent a starting point for the investigation of lncRNAs in SLE.
Further investigations are required to evaluate the signaling
pathways identified in the GO and KEGG analyzes with regard to
their role in the development and progression of SLE.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81360459) and the
Jiangxi Provincial Natural Science Foundation of China (grant no.
20151BAB215031). The authors would like to thank Dr Rui Wu at the
Department of Rheumatology, The First Affiliated Hospital of
Nanchang University (Nanchang, China).
References
|
1
|
Bertsias GK, Salmon JE and Boumpas DT:
Therapeutic opportunities in systemic lupus erythematosus: State of
the art and prospects for the new decade. Ann Rheum Dis.
69:1603–1611. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kow NY and Mak A: Costimulatory pathways:
Physiology and potential therapeutic manipulation in systemic lupus
erythematosus. Clin Dev Immunol. 2013:2459282013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kirou KA and Gkrouzman E: Anti-interferon
alpha treatment in SLE. Clin Immunol. 148:303–312. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kowalczyk MS, Higgs DR and Gingeras TR:
Molecular biology: RNA discrimination. Nature. 482:310–311. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shirasawa S, Harada H, Furugaki K, Akamizu
T, Ishikawa N, Ito K, Ito K, Tamai H, Kuma K, Kubota S, et al: SNPs
in the promoter of a B cell-specific antisense transcript,
SAS-ZFAT, determine susceptibility to autoimmune thyroid disease.
Hum Mol Genet. 13:2221–2231. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Song J, Kim D, Han J, Kim Y, Lee M and Jin
EJ: PBMC and exosome-derived Hotair is a critical regulator and
potent marker for rheumatoid arthritis. Clin Exp Med. 15:121–126.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Heward JA and Lindsay MA: Long non-coding
RNAs in the regulation of the immune response. Trends Immunol.
35:408–419. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Willingham AT, Orth AP, Batalov S, Peters
EC, Wen BG, Aza-Blanc P, Hogenesch JB and Schultz PG: A strategy
for probing the function of noncoding RNAs finds a repressor of
NFAT. Science. 309:1570–1573. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Collier SP, Collins PL, Williams CL,
Boothby MR and Aune TM: Cutting edge: Influence of Tmevpg1, a long
intergenic noncoding RNA, on the expression of Ifng by Th1 cells. J
Immunol. 189:2084–2088. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li Z, Chao TC, Chang KY, Lin N, Patil VS,
Shimizu C, Head SR, Burns JC and Rana TM: The long noncoding RNA
THRIL regulates TNFα expression through its interaction with
hnRNPL. Proc Natl Acad Sci USA. 111:pp. 1002–1007. 2014; View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li B, Tsoi LC, Swindell WR, Gudjonsson JE,
Tejasvi T, Johnston A, Ding J, Stuart PE, Xing X, Kochkodan JJ, et
al: Transcriptome analysis of psoriasis in a large case-control
sample: RNA-seq provides insights into disease mechanisms. J Invest
Dermatol. 134:1828–1838. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu Q, Zhang X, Dai L, Hu X, Zhu J, Li L,
Zhou C and Ao Y: Long noncoding RNA related to cartilage injury
promotes chondrocyte extracellular matrix degradation in
osteoarthritis. Arthritis Rheumatol. 66:969–978. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Müller N, Döring F, Klapper M, Neumann K,
Schulte DM, Türk K, Schröder JO, Zeuner RA, Freitag-Wolf S,
Schreiber S and Laudes M: Interleukin-6 and tumour necrosis
factor-α differentially regulate lincRNA transcripts in cells of
the innate immune system in vivo in human subjects with rheumatoid
arthritis. Cytokine. 68:65–68. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tan EM, Cohen AS, Fries JF, Masi AT,
McShane DJ, Rothfield NF, Schaller JG, Talal N and Winchester RJ:
The 1982 revised criteria for the classification of systemic lupus
erythematosus. Arthritis Rheum. 25:1271–1277. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tian M, Chen R, Li T and Xiao B: Reduced
expression of circRNA hsa_circ_0003159 in gastric cancer and its
clinical significance. J Clin Lab Anal. Jun 15–2017.(Epub ahead of
print). View Article : Google Scholar
|
|
16
|
Xu G, Chen J, Pan Q, Huang K, Pan J, Zhang
W, Chen J, Yu F, Zhou T and Wang Y: Long noncoding RNA expression
profiles of lung adenocarcinoma ascertained by microarray analysis.
PLoS One. 9:e1040442014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ørom UA, Derrien T, Beringer M, Gumireddy
K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q,
et al: Long noncoding RNAs with enhancer-like function in human
cells. Cell. 143:46–58. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shi X, Sun M, Liu H, Yao Y and Song Y:
Long non-coding RNAs: A new frontier in the study of human
diseases. Cancer Lett. 339:159–166. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wan ZY, Song F, Sun Z, Chen YF, Zhang WL,
Samartzis D, Ma CJ, Che L, Liu X, Ali MA, et al: Aberrantly
expressed long noncoding RNAs in human intervertebral disc
degeneration: A microarray related study. Arthritis Res Ther.
16:4652014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gurevitz SL, Snyder JA, Wessel EK, Frey J
and Williamson BA: Systemic lupus erythematosus: A review of the
disease and treatment options. Consult Pharm. 28:110–121. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xiong W and Lahita RG: Pragmatic
approaches to therapy for systemic lupus erythematosus. Nat Rev
Rheumatol. 10:97–107. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu Y, Zhang F, Ma J, Zhang X, Wu L, Qu B,
Xia S, Chen S, Tang Y and Shen N: Association of large intergenic
noncoding RNA expression with disease activity and organ damage in
systemic lupus erythematosus. Arthritis Res Ther. 17:1312015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang F, Wu L, Qian J, Qu B, Xia S, La T,
Wu Y, Ma J, Zeng J, Guo Q, et al: Identification of the long
noncoding RNA NEAT1 as a novel inflammatory regulator acting
through MAPK pathway in human lupus. J Autoimmun. 75:96–104. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Toonen EJ, Barrera P, Radstake TR, van
Riel PL, Scheffer H, Franke B and Coenen MJ: Gene expression
profiling in rheumatoid arthritis: Current concepts and future
directions. Ann Rheum Dis. 67:1663–1669. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shaffer AL, Wright G, Yang L, Powell J,
Ngo V, Lamy L, Lam LT, Davis RE and Staudt LM: A library of gene
expression signatures to illuminate normal and pathological
lymphoid biology. Immunol Rev. 210:67–85. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yamagata T, Benoist C and Mathis DA:
Shared gene-expression signature in innate-like lymphocytes.
Immunol Rev. 210:52–66. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Te JL, Dozmorov IM, Guthridge JM, Nguyen
KL, Cavett JW, Kelly JA, Bruner GR, Harley JB and Ojwang JO:
Identification of unique microRNA signature associated with lupus
nephritis. PLoS One. 5:e103442010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu D, Zhao H, Zhao S and Wang X: MicroRNA
expression profiles of peripheral blood mononuclear cells in
patients with systemic lupus erythematosus. Acta Histochem.
116:891–897. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mattick JS and Gagen MJ: The evolution of
controlled multitasked gene networks: The role of introns and other
noncoding RNAs in the development of complex organisms. Mol Biol
Evol. 18:1611–1630. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mattick JS: Lincing long noncoding RNAs
and enhancer function. Dev Cell. 19:485–486. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yougbaré I, Boire G, Roy M, Lugnier C and
Rouseau E: NCS 613 exhibits anti-inflammatory effects on PBMCs from
lupus patients by inhibiting p38 MAPK and NF-κB signalling pathways
while reducing proinflammatory cytokine production. Can J Physiol
Pharmacol. 91:353–361. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhu LJ, Landolt-Marticorena C, Li T, Yang
X, Yu XQ, Gladman DD, Urowitz MB, Fortin PR and Wither JE: Altered
expression of TNF-alpha signaling pathway proteins in systemic
lupus erythematosus. J Rheumatol. 37:1658–1666. 2010. View Article : Google Scholar : PubMed/NCBI
|