Introduction
Acute myocardial infarction (AMI) may cause
irreversible damage to functional myocardial cells, negative
myocardial remodeling and progressive deterioration of cardiac
function (1). In addition, it
remains the primary cause of disease incidence and mortality in
China (1). According to the Heart
Disease and Stroke Statistics report by the American Heart
Association (2), ~2,200 patients
succumbed to cardiovascular diseases every day in 2008. In
addition, an average of 785,000 cases of coronary atherosclerosis
(3) and an average of 195,000
cases of MI are reported annually in China. AMI is the most serious
type of coronary heart disease, which involves cardiomyocyte
necrosis due to long-term ischemia and oxygen deficit. In addition,
oxidative stress has significant effects on the pathological
alterations associated with MI (4).
AMI may cause aerobic metabolic disorders due to
long-term ischemia and hypoxia caused by coronary occlusion, which
may lead to myocardial cell interstitial hyperemia, edema and
degenerative necrosis, accompanied by increased inflammatory cell
infiltration (5). Oxidative
damage, caused by the excessive generation of free radicals and
oxygen free radicals in cardiac tissue, results in damage to the
structure and function of myocardial cell membranes, mitochondrial
damage and autolysis (6).
Myocardium under accelerated ischemia may progress from reversible
injury to irreversible degenerative necrosis (7). Malignant arrhythmia may also occur,
which may lead to further ventricular remodeling and cardiac
dysfunction. Revascularization and reperfusion therapy are
currently considered the most effective therapeutic methods;
however, ischemia reperfusion can cause further damage to the
remaining myocardium. The important mechanism underlying this
damage may be oxidative stress (7). The longer the duration of ischemic
and hypoxic conditions, the more evident the oxidative stress
response will be in myocardial cells. In accordance, the higher the
degree of myocardial injury, the heavier the state of illness will
be (8).
Great Burdock (Arctium lappa) Achene extract
is used in traditional Chinese medicine to treat anemopyretic cold,
measles, carbuncles, ingested poison and other diseases (9). Modern pharmacological research has
demonstrated that Great Burdock Achene extract possesses
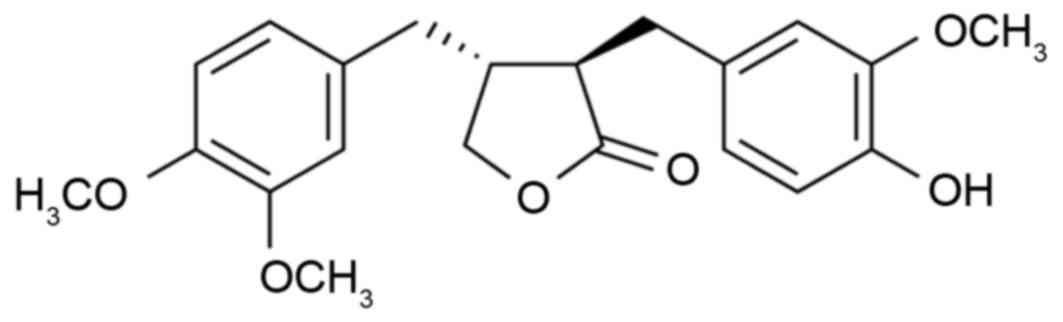
antibiotic, antitumor and hypoglycemic effects (10). Arctigenin (Fig. 1) is one of the active ingredients
extracted from Great Burdock Achene, which has numerous
pharmacological effects, including antitumor and neuroprotective
activities; arctigenin also has strong anti-inflammatory,
immunoregulatory and antiviral activity, and inhibits the
heat-shock response (11,12). The present study aimed to evaluate
the protective effects of arctigenin against MI and the potential
underlying mechanisms.
Materials and methods
AMI model rats
The present study was approved by the Ethics
Committee of Liaocheng People's Hospital of Shandong Province
(Liaocheng, China). A total of 40 male Sprague-Dawley rats (weight,
between 250 and 300 g; age, 10–12 weeks), were purchased from Jinan
Jinfeng Experimental Animal Co., Ltd. (Shandong, China) and housed
at 22–24°C and 55% humidity in an animal room under a 12-h
light/dark cycle with free access to water and food. The present
study was performed in accordance with the recommendations from the
Guide for Animal Management Rules from the Ministry of Health of
Liaocheng People's Hospital of Shandong Province. To generate the
AMI model, rats were anesthetized by peritoneal injection with 1%
pentobarbital (40 mg/kg). Subsequently, coronary arteries were
exposed and the descending branch of the left anterior coronary
artery was marked with silk and ligated for 1 h with hemostatic
forceps; the wound was sutured (1–2 cm) following ligation in order
to generate the AMI model.
Treatment groups
A total of 40 male Sprague-Dawley rats were randomly
assigned into the following subgroups (n=8 rats/group): i) Sham
group, which was used as a control (treated with normal saline);
ii) AMI model group (vehicle control; AMI rats were treated with
normal saline following the generation of the AMI model; iii) AMI
rats treated with 50 µmol/kg arctigenin (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) following the generation of the AMI model; iv)
AMI rats treated with 100 µmol/kg arctigenin following the
generation of the AMI model; and v) AMI rats treated with 200
µmol/kg arctigenin following the generation of the AMI model. Rats
were sacrificed following treatment with arctigenin for 1 week.
Measurement of alanine transaminase
(ALT), creatine kinase (CK)-MB, lactate dehydrogenase (LDH),
malondialdehyde (MDA), glutathione peroxidase (GSH-PX), catalase
(CAT), superoxide dismutase (SOD), interleukin (IL)-1β and IL-6
activity
Following arctigenin treatment, blood was
immediately collected from the abdominal aorta and used to analyze
the serum levels of ALT (cat. no. C009-2), CK-MB (cat. no. H197),
MDA (cat. no. A003-1), GSH-PX (cat. no. A005), CAT (cat. no.
A007-1), SOD (cat. no. A001-1), IL-1β (cat. no. H002) and IL-6
(cat. no. H007) using Commercial ELISA kits (Nanjing Jiancheng
Bioengineering Institute, Nanjing, China) according to the
manufacturer's instructions.
Measurement of caspase-3 activity
Following arctigenin treatment, mice were sacrificed
and their hearts were removed and homogenized in ice-cold
radioimmunoprecipitation lysis buffer (Beyotime Institute of
Biotechnology). Protein concentrations were measured using a
Bicinchoninic Acid Protein Assay kit (Beyotime Institute of
Biotechnology). Proteins (10 µg) were then incubated with the
caspase-3 substrate, Ac-DEVD-pNA (cat. no. C1115; Beyotime
Institute of Biotechnology) for 2 h at 37°C in order to measure
caspase-3 activity, according to the manufacturer's protocol.
Evaluation of infarct size
Following arctigenin treatment, rats were sacrificed
and the hearts were removed. The hearts were sliced into five
sections (1.0 mm) perpendicular to the long axis, and the sections
were incubated with 1% 2,3,5-triphenyl tetrazolium chloride
(Sigma-Aldrich; Merck KGaA) in phosphate solution at 37°C for 10
min. Infarct sizes were determined by computer morphometry using
Image-Pro Plus 6.0 software (Media Cybernetics, Inc., Rockville,
MD, USA).
Western blot analysis
Following arctigenin treatment, mice were sacrificed
and their hearts were removed and homogenized in ice-cold
radioimmunoprecipitation lysis buffer (Beyotime Institute of
Biotechnology). Protein concentrations were measured using the
Bicinchoninic Acid Protein Assay kit (Beyotime Institute of
Biotechnology). Proteins (80 µg/lane) were separated by 10%
SDS-PAGE and were transferred to polyvinylidene fluoride membranes
(EMD Millipore, Billerica, MA, USA). Membranes were blocked with 5%
skim milk powder for 1 h at 37°C and probed with antibodies against
inducible nitric oxide synthase (iNOS; cat. no. sc-649; 1:500;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA), cyclooxygenase 2
(COX-2; cat. no. sc-7951; 1:500; Santa Cruz Biotechnology, Inc.),
phosphorylated-extracellular signal-regulated kinase 1/2 (p-ERK1/2;
cat. no. sc-101761; 1:500; Santa Cruz Biotechnology, Inc.), ERK1/2
(cat. no. 4695; 1:2,000; Cell Signaling Technology, Inc., Danvers,
MA, USA), heme oxygenase-1 (HO-1; cat. no. sc-10789; 1:500; Santa
Cruz Biotechnology, Inc.) and β-actin (cat. no. sc-7210; 1:500;
Santa Cruz Biotechnology, Inc.) at 4°C overnight. Membranes were
then incubated with horseradish peroxidase-conjugated goat
anti-mouse secondary antibodies (cat. no. 7074; 1:5,000; Cell
Signaling Technology, Inc.) for 1 h at room temperature, and the
immune complexes were detected by enhanced chemiluminescence (Cell
Signaling Technology, Inc.). The optical densities of
immunopositive bands were determined by Gene Tools image analysis
(Syngene, Frederick, MD, USA) and Bio-Rad Quantity One software
v3.0 (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
Data are presented as the mean ± standard deviation
of 3 independent experiments. Analysis was performed using SPSS
v17.0 software (SPSS, Inc., Chicago, IL, USA). Statistical
comparisons between groups were conducted using one-way analysis of
variance and the Tukey post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
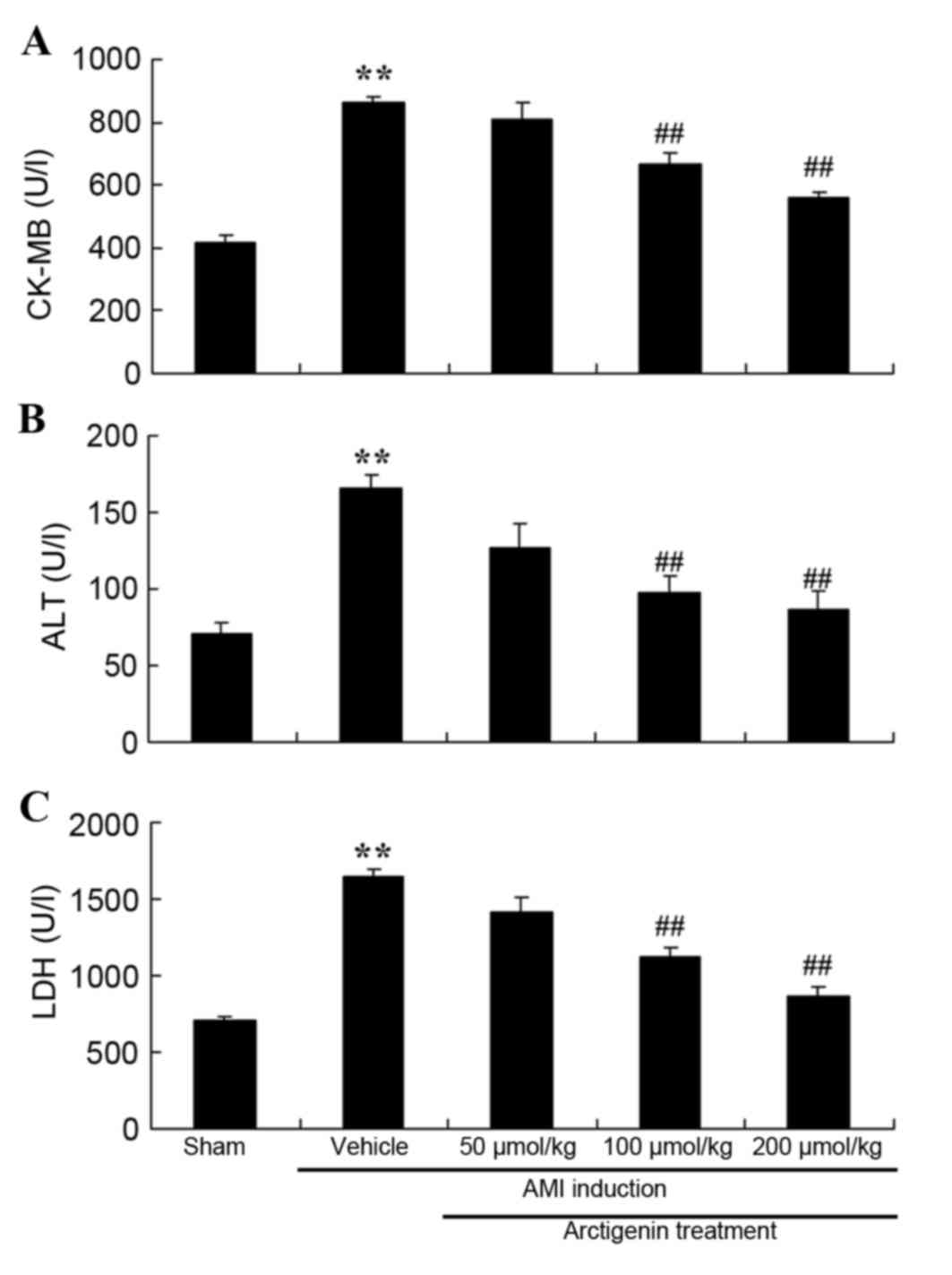
Arctigenin reduces CK-MB, ALT and LDH
levels in AMI rats
The present study evaluated the protective effects
of arctigenin by analyzing the levels of biochemical markers of AMI
in the rat model, including CK-MB, ALT and LDH (Fig. 2). The levels of CK-MB, ALT and LDH
were higher in the AMI model rats compared with the Sham group.
However, treatment with arctigenin (100 or 200 µmol/kg)
significantly reduced the levels of CK-MB, ALT and LDH compared
with in the untreated AMI control group (Fig. 2).
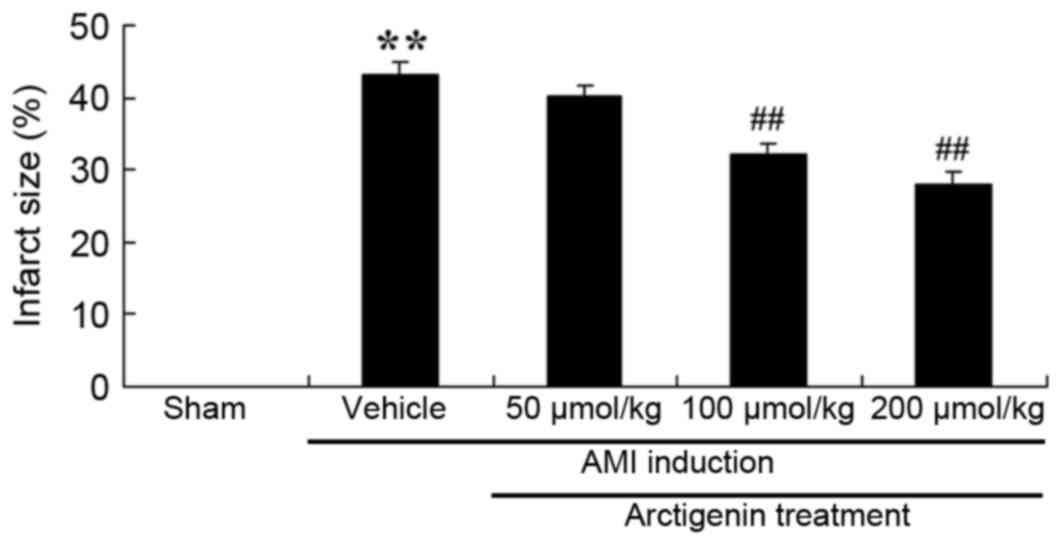
Arctigenin treatment reduces infarct
size in AMI rats
To confirm the protective effects of arctigenin on
AMI, the infarct size in AMI rats was examined. As presented in
Fig. 3, the infarct size in AMI
rats was significantly increased compared with the Sham group,
whereas treatment with arctigenin (100 or 200 µmol/kg) markedly
reduced the infarct size compared with untreated AMI rats (Fig. 3).
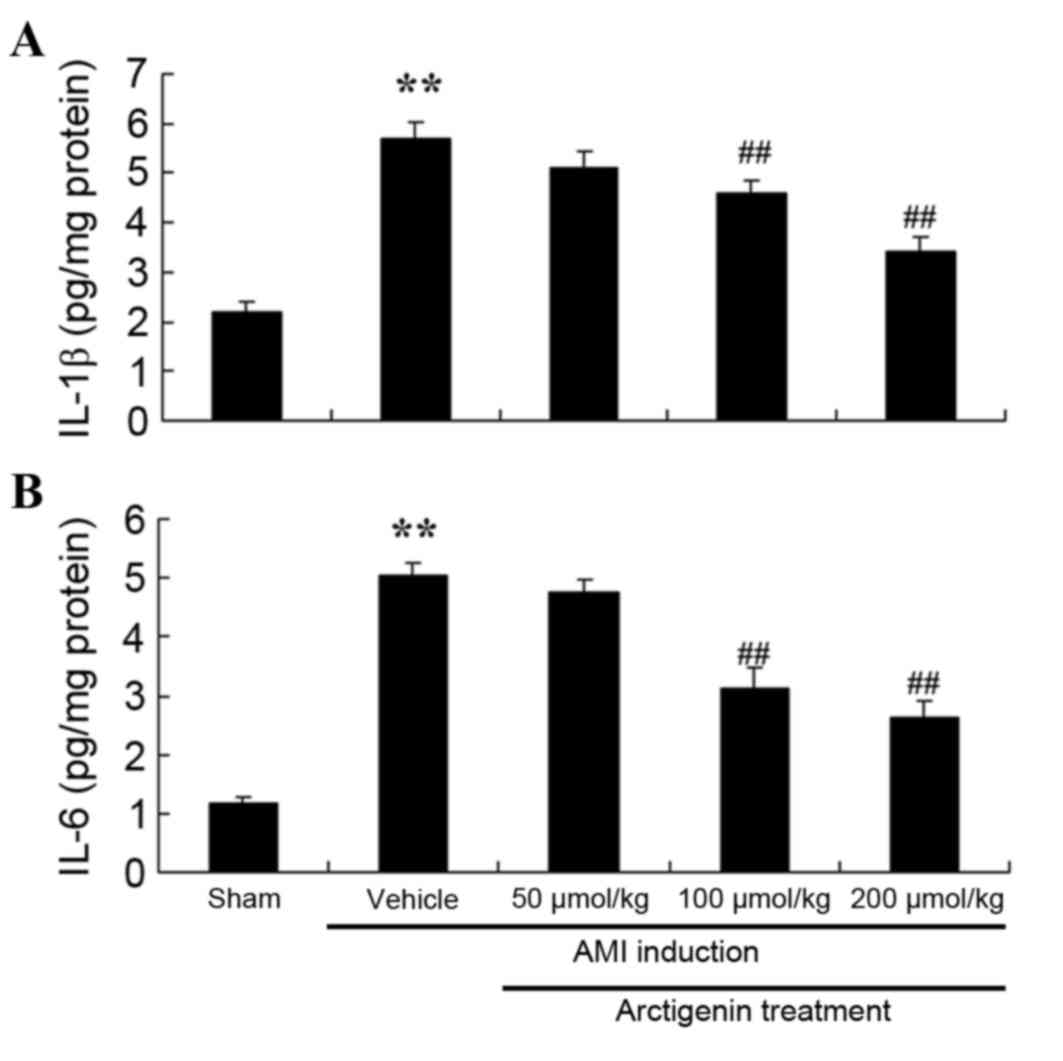
Arctigenin reduces inflammation in AMI
rats
The effects of arctigenin on AMI-induced
inflammation were investigated by examining the activity of IL-1β
and IL-6 (Fig. 4). Untreated AMI
model rats exhibited a significant increase in IL-1β and IL-6
activity compared with the Sham group. Conversely, treatment with
100 or 200 µmol/kg arctigenin significantly reduced the activity of
IL-1β and IL-6 compared with the untreated AMI group (Fig. 4).
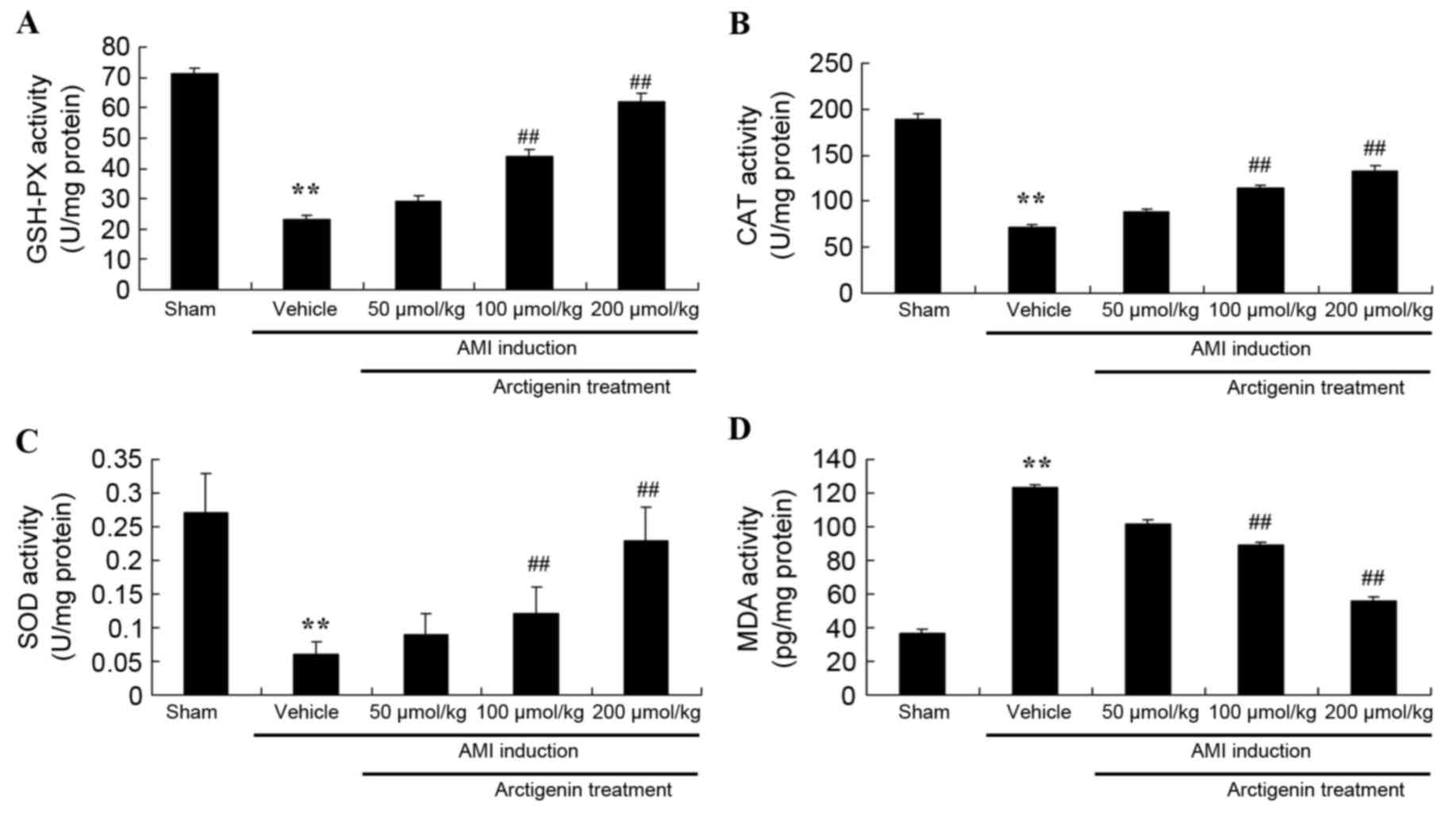
Arctigenin decreases oxidative stress
in AMI rats
The effects of arctigenin on AMI-induced oxidative
stress in AMI rats were examined. As shown in Fig. 5A-C, AMI resulted in decreased
GSH-PX, CAT and SOD activity compared with the Sham group. However,
treatment with arctigenin (100 or 200 µmol/kg) significantly
increased the activity of GSH-PX, CAT and SOD in AMI rats (Fig. 5A-C). Conversely, MDA activity was
higher in AMI rats compared with the Sham group (Fig. 5D); MDA activity was subsequently
reduced in AMI rats upon treatment with arctigenin (100 or 200
µmol/kg) (Fig. 5D). These results
demonstrated that arctigenin may possess antioxidative effects in
AMI, and thus may be effective as a clinical treatment for AMI.
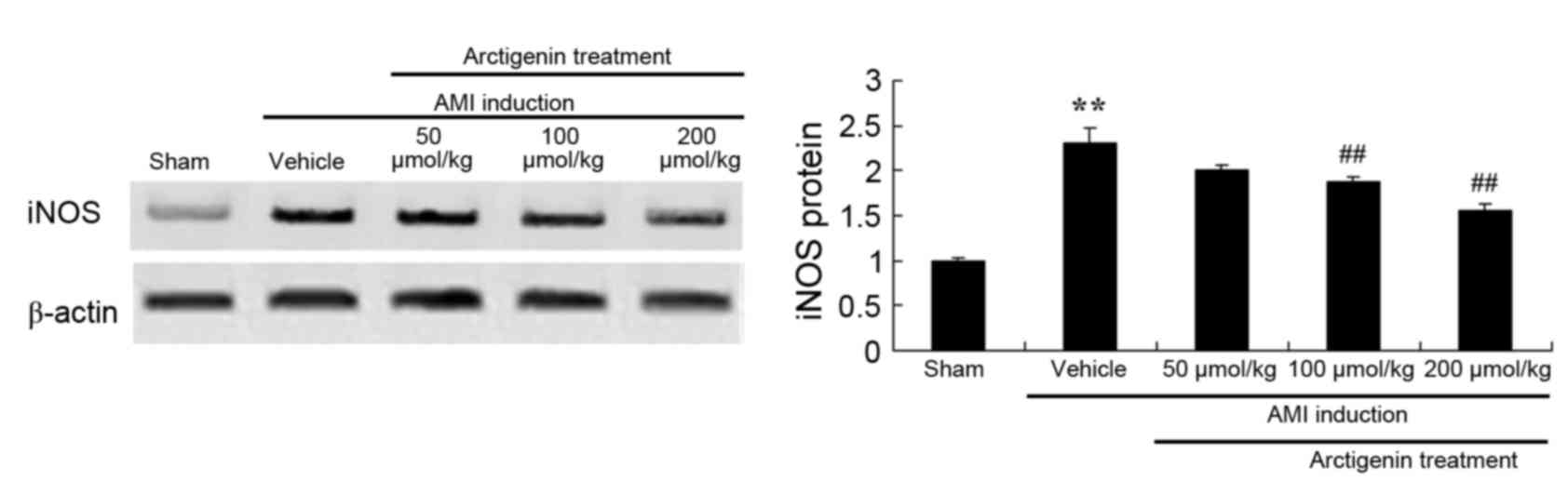
Arctigenin reduces iNOS protein
expression in AMI rats
Protein expression levels of iNOS were detected by
western blot analysis (Fig. 6).
iNOS protein expression was significantly increased in AMI rats,
compared with the Sham group, and was lowered in AMI rats treated
with arctigenin (100 or 200 µmol/kg).
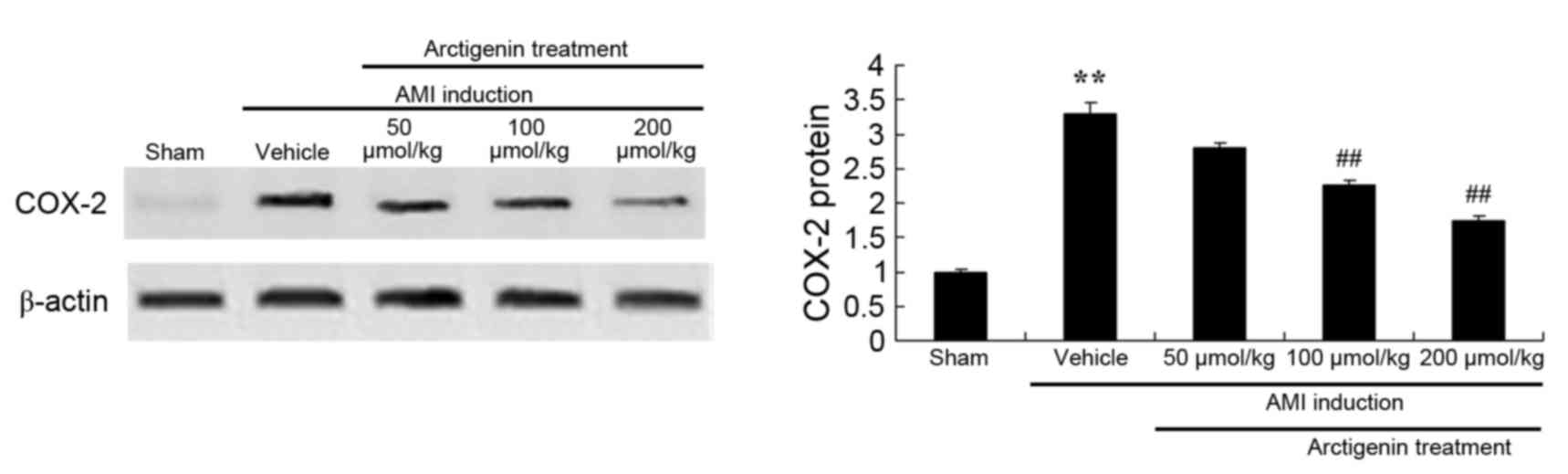
Arctigenin reduces the protein
expression levels of COX-2 in AMI rats
The effects of arctigenin on the protein expression
levels of COX-2 in AMI rats were determined by western blot
analysis (Fig. 7). COX-2 protein
expression was significantly higher in AMI model rats compared with
the Sham group. However, treatment with arctigenin (100 or 200
µmol/kg) significantly reduced the protein expression levels of
COX-2 in AMI rats.
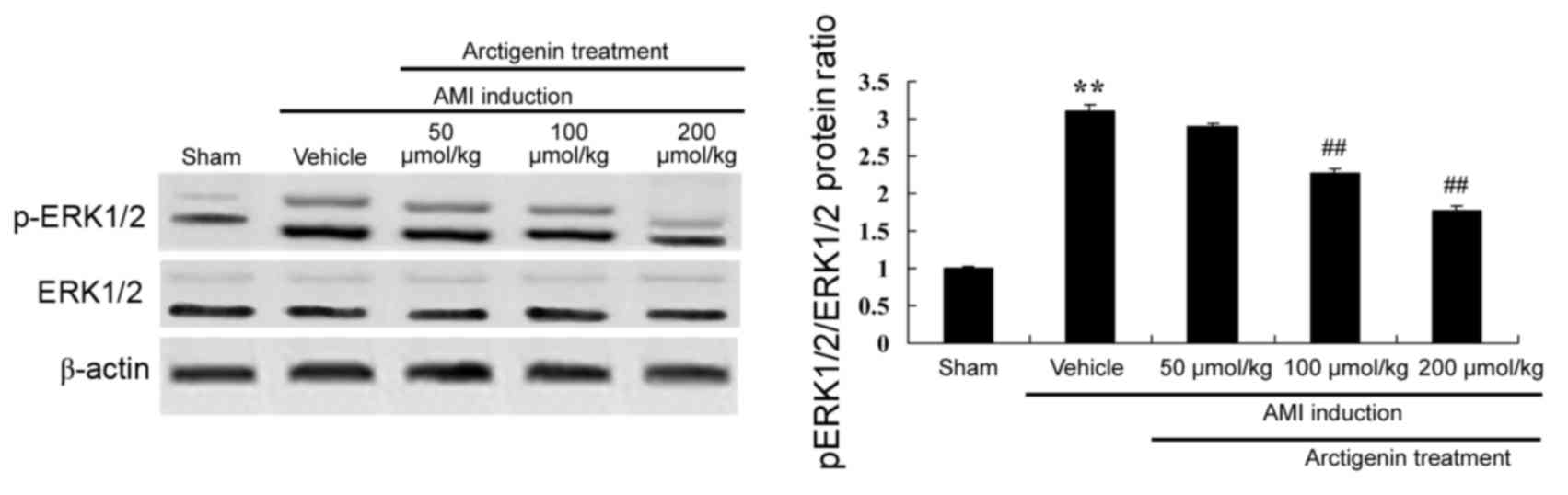
Arctigenin decreases p-ERK1/2 protein
expression in AMI rats
The protein expression levels of p-ERK1/2 were
analyzed by western blotting, in order to examine the underlying
mechanisms involved in AMI (Fig.
8). The protein expression of p-ERK1/2 was induced in the AMI
model group when compared with the Sham group (Fig. 8). Treatment with arctigenin (100 or
200 µmol/kg) significantly decreased the protein expression of
p-ERK1/2 AMI rats (Fig. 8).
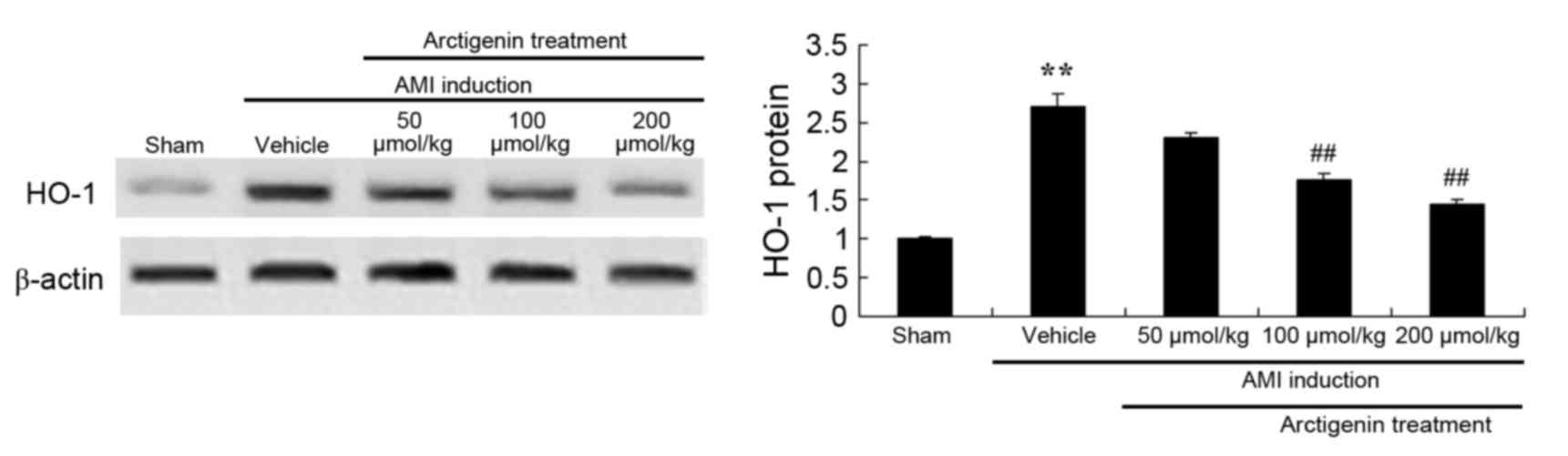
Arctigenin decreases the protein
expression levels of HO-1 in AMI rats
To confirm the underlying mechanisms of arctigenin
treatment in AMI rats, HO-1 protein expression was examined by
western blotting. As shown in Fig.
9, AMI induction led to an increase in the protein expression
levels of HO-1 in AMI rats compared with the Sham group. Elevated
HO-1 protein expression was significantly reduced in AMI rats
treated with arctigenin (100 or 200 µmol/kg) compared with
untreated AMI rats.
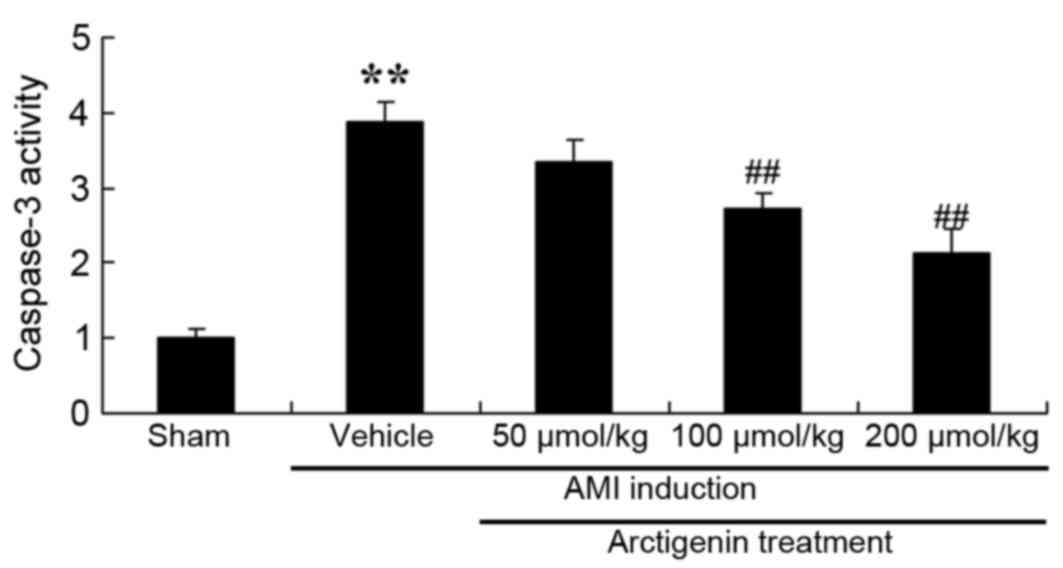
Arctigenin reduces caspase-3 activity
in AMI rats
The present study analyzed the effects of arctigenin
on AMI-induced apoptosis in rat heart cells by measuring caspase-3
activity by ELISA. As shown in Fig.
10, caspase-3 activity was significantly increased in AMI rats,
compared with the Sham group, whereas treatment with arctigenin
(100 or 200 µmol/kg) significantly reduced caspase-3 activity in
AMI rats (Fig. 10).
Discussion
AMI is an important pathological and lethal syndrome
worldwide. Following occurrence of AMI, dredging the blocked
coronary artery in a timely manner is the only effective
therapeutic strategy to save ischemic myocardial tissue and to
restore cardiac function (2).
However, reperfusion injury may lead to damage or death of ischemic
cardiac muscle cells (13).
Effective methods for the treatment of myocardial reperfusion
injury have been investigated; however, there is currently no
definite strategy or drug available (14). Despite the common therapeutic
strategy of timely ischemic myocardial perfusion recovery, 25% of
patients with AMI will develop chronic cardiac failure as a result
of reperfusion injury (15).
Reperfusion injury is a key factor for myocardial injury associated
with myocardial infarction recanalization; therefore, reducing
reperfusion injury is conducive to myocardial cell survival,
reducing the loss of myocardial function and reducing the
probability of developing chronic cardiac failure (16). In the present study, treatment with
arctigenin significantly lowered the AMI-induced levels of ALT,
CK-MB and LDH, and reduced the infarct size in AMI model rats.
Oxidative stress refers to an imbalance of oxidation
and antioxidation in the body (17). Oxidation may lead to granulocyte
inflammatory infiltration, increased protease secretion and the
production of a large amount of intermediate products (18). The degree of myocardial injury due
to the increased levels of oxygen radicals in peripheral
circulation can be evaluated through the change in activity of
endogenous antioxidant enzymes, such as SOD, in the peripheral
blood (17). Reduced glutathione
is a tripeptide that is composed of glutamic acid, cysteine and
glycine, and is expressed in various organs. It is a coenzyme that
is involved in the activation of numerous other enzymes,
participates in the Krebs cycle and sugar metabolism, and helps
maintain physiological functions of the cell (19). Results from the present study
revealed that the protective effects of arctigenin reduced
oxidative stress and iNOS expression in AMI model rats. Kou et
al (20) demonstrated that
arctigenin decreased COX-2 expression and inhibited STAT1/3
expression, which led to a decrease in iNOS expression. In
addition, Zhang et al (11)
reported that arctigenin exerts protective effects against
lipopolysaccharide (LPS)-induced oxidative stress and inflammation
by suppressing HO-1 and iNOS signaling in mice.
Tumor necrosis factor-α (TNF-α) is a multifunctional
protein that is mainly produced by the activated
scavenger/mononuclear cell system. Normal myocardial cells cannot
produce TNF-α (21); however, in
the case of AMI pump failure, TNF-α expression increases
significantly and becomes a reliable indicator by which to evaluate
the clinical prognosis of AMI (22). IL-6 is a proinflammatory cytokine
with numerous functions that is secreted by activated monocytes,
macrophages, T lymphocytes, endothelial cells and fibroblasts
(23). A previous study reported
that IL-6 may be the most powerful predictor of mortality caused by
cardiogenic shock in patients with AMI within 30 days (24). In the present study, AMI rats
treated with arctigenin exhibited significantly decreased IL-1β and
IL-6 activity.
Decreased coronary blood flow resulting from
arterial thrombosis or atherosclerosis may lead to myocardial
anoxia (25). In myocardial cells
of neonatal rats cultivated in vitro, hypoxia led to an
increase in the mRNA and protein expression levels of HO-1, whereas
under normoxic conditions, HO-1 expression was reduced (26). Environment-induced hypoxia may lead
to pulmonary hypertension and may induce right ventricular
hypertrophy (27). HO-1-knockout
mice under normoxic conditions exhibit normal behavior, whereas
under hypoxic conditions, despite having a similar degree of
pulmonary hypertension, HO-1 knockout mice were subject to more
serious right ventricular dilation and infarction accompanied by
atherothrombosis (28). In the
present study, treatment with arctigenin significantly reduced the
protein expression levels of HO-1 in AMI rats. Zhang et al
(11) reported that arctigenin
protects against LPS-induced oxidative stress and inflammation
through suppression of HO-1 and iNOS signaling in mice.
Ischemic preconditioning is a procedure that
prepares the myocardium to tolerate long-term ischemic damage by
subjecting myocardiocytes to transitory periods of ischemia
(29). A previous study
demonstrated that COX-2 may be involved in the preadaptation
process of myocardial ischemia and serves a key protective role
(30). Conversely, COX-2
inhibitors prevent the protective effects of the ischemia
preadaptation process by inhibiting the expression of prostaglandin
I2 receptor (30). In addition,
the delayed ischemic preconditioning protection mediated by
COX-2-regulated opioid-type receptors of rat myocardium may be
suppressed following treatment with the COX-2 inhibitor NS-398
(31). It has also been
demonstrated that activation of opioid receptors may induce the
delayed protective effects of ischemic preconditioning, which is
dependent on COX-2 expression, to mitigate myocardial suppression
and reduce myocardial infarct size (32). The present study demonstrated that
treatment with arctigenin significantly reduced the protein
expression levels of COX-2 in AMI model rats.
ERK1 and ERK2 are important members of the
mitogen-activated protein kinase signaling pathway superfamily
(33). They may be activated by
growth stimulating factors, such as growth factors, cytokines and
stretching, and are able to mediate various cellular responses,
including cell growth, differentiation and apoptosis (34). In cell culture experiments, ERK1/2
agonists significantly enhance hypertrophy and hyperplasia of
myocardiocytes (35). Furthermore,
hypertrophic myocardial cells also exhibit increased functional
ERK1/2 expression (36). In
vivo experiments also demonstrated that ventricular hypertrophy
due to acute or chronic pressure load may lead to varying degrees
of increased ERK activity, whereas ERK activity decreases during
periods of heart failure (36).
The results of the present study demonstrated that treatment with
arctigenin significantly decreased the protein expression levels of
p-ERK1/2 in AMI rats. Li et al (37) reported that arctigenin may suppress
transforming growth factor-β1 in renal tubular epithelial cells
through the reactive oxygen species-dependent ERK/nuclear factor-κB
signaling pathway.
In conclusion, the results of the present study
revealed that the protective effects of arctigenin reduced the
levels of ALT, CK-MB and LDH, and inhibited infarct size in AMI
rats. Arctigenin also exhibited antioxidative and anti-inflammatory
activities by suppressing the expression levels of iNOS, COX-2 and
HO-1, and activating ERK1/2 signaling. These findings suggested
that arctigenin may prove clinically useful in treating AMI.
References
|
1
|
Voors AA, von Haehling S, Anker SD,
Hillege HL, Struck J, Hartmann O, Bergmann A, Squire I, van
Veldhuisen DJ and Dickstein K; OPTIMAAL Investigators, : C-terminal
provasopressin (copeptin) is a strong prognostic marker in patients
with heart failure after an acute myocardial infarction: Results
from the OPTIMAAL study. Eur Heart J. 30:1187–1194. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wijnbergen I, Tijssen J, van't Veer M,
Michels R and Pijls NH: Gender differences in long-term outcome
after primary percutaneous intervention for ST-segment elevation
myocardial infarction. Catheter Cardiovasc Interv. 82:379–384.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Young JJ, Cox DA, Stuckey T, Babb J, Turco
M, Lansky AJ, Mehran R and Stone GW: Prospective, multicenter study
of thrombectomy in patients with acute myocardial infarction: The
X-Tract AMI registry. J Interv Cardiol. 20:44–50. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Baird SH, Menown IB, McBride SJ, Trouton
TG and Wilson C: Randomized comparison of enoxaparin with
unfractionated heparin following fibrinolytic therapy for acute
myocardial infarction. Eur Heart J. 23:627–632. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhu XY, Zhang ZL, Li P, Liang WY, Feng XR
and Liu ML: Shenyuan, an extract of American Ginseng and Corydalis
tuber formula, attenuates cardiomyocyte apoptosis via inhibition of
endoplasmic reticulum stress and oxidative stress in a porcine
model of acute myocardial infarction. J Ethnopharmacol.
150:672–681. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ghyasi R, Sepehri G, Mohammadi M,
Badalzadeh R and Ghyasi A: Effect of mebudipine on oxidative stress
and lipid peroxidation in myocardial ischemic-reperfusion injury in
male rat. J Res Med Sci. 17:1150–1155. 2012.PubMed/NCBI
|
|
7
|
Neri M, Fineschi V, Di Paolo M, Pomara C,
Riezzo I, Turillazzi E and Cerretani D: Cardiac oxidative stress
and inflammatory cytokines response after myocardial infarction.
Curr Vasc Pharmacol. 13:26–36. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xie N, Zhang W, Li J, Liang H, Zhou H,
Duan W, Xu X, Yu S, Zhang H and Yi D: alpha-Linolenic acid intake
attenuates myocardial ischemia/reperfusion injury through
anti-inflammatory and anti-oxidative stress effects in diabetic but
not normal rats. Arch Med Res. 42:171–181. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang Y, Wang Z, Chen H, Chen Z and Tian
Y: Antioxidants: Potential antiviral agents for Japanese
encephalitis virus infection. Int J Infect Dis. 24:30–36. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shin HS, Jung SY, Back SY, Do JR and Shon
DH: Arctigenin from fructus arctii (Seed of Burdock) reinforces
intestinal barrier function in Caco-2 cell monolayers. Evid Based
Complement Alternat Med. 2015:3681052015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang WZ, Jiang ZK, He BX and Liu XB:
Arctigenin protects against lipopolysaccharide-induced pulmonary
oxidative stress and inflammation in a mouse model via suppression
of MAPK, HO-1, and iNOS signaling. Inflammation. 38:1406–1414.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fan T, Jiang WL, Zhu J and Feng Zhang Y:
Arctigenin protects focal cerebral ischemia-reperfusion rats
through inhibiting neuroinflammation. Biol Pharm Bull.
35:2004–2009. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu F, Ming Q and Hou L: The effect of sex
counselling in the sexual activity of acute myocardial infarction
patients after primary percutaneous coronary intervention. Acta
Cardiol. 70:460–464. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Limalanathan S, Andersen GO, Klow NE,
Abdelnoor M, Hoffmann P and Eritsland J: Effect of ischemic
postconditioning on infarct size in patients with ST-elevation
myocardial infarction treated by primary PCI results of the POSTEMI
(POstconditioning in ST-Elevation Myocardial Infarction) randomized
trial. J Am Heart Assoc. 3:e0006792014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Palmerini T, Brener SJ, Mehran R, Dangas
G, Genereux P, Riva DD, Mariani A, Xu K and Stone GW: Leukocyte
count is a modulating factor for the mortality benefit of
bivalirudin in ST-segment-elevation acute myocardial infarction:
The HORIZONS-AMI trial. Circ Cardiovasc Interv. 6:518–526. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Palmerini T, Brener SJ, Genereux P,
Maehara A, Della Riva D, Mariani A, Witzenbichler B, Godlewski J,
Parise H, Dambrink JH, et al: Relation between white blood cell
count and final infarct size in patients with ST-segment elevation
acute myocardial infarction undergoing primary percutaneous
coronary intervention (from the INFUSE AMI trial). Am J Cardiol.
112:1860–1866. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang X, Wang Y, Jiang M, Zhu Y, Hu L, Fan
G, Wang Y, Li X and Gao X: Differential cardioprotective effects of
salvianolic acid and tanshinone on acute myocardial infarction are
mediated by unique signaling pathways. J Ethnopharmacol.
135:662–671. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bagatini MD, Martins CC, Battisti V,
Gasparetto D, da Rosa CS, Spanevello RM, Ahmed M, Schmatz R,
Schetinger MR and Morsch VM: Oxidative stress versus antioxidant
defenses in patients with acute myocardial infarction. Heart
Vessels. 26:55–63. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lorgis L, Zeller M, Dentan G, Sicard P,
Richard C, Buffet P, L'Huillier I, Beer JC, Cottin Y, Rochette L,
et al: The free oxygen radicals test (FORT) to assess circulating
oxidative stress in patients with acute myocardial infarction.
Atherosclerosis. 213:616–621. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kou X, Qi S, Dai W, Luo L and Yin Z:
Arctigenin inhibits lipopolysaccharide-induced iNOS expression in
RAW264.7 cells through suppressing JAK-STAT signal pathway. Int
Immunopharmacol. 11:1095–1102. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Abe Y, Ito K, Hao K, Shindo T, Ogata T,
Kagaya Y, Kurosawa R, Nishimiya K, Satoh K, Miyata S, et al:
Extracorporeal low-energy shock-wave therapy exerts
anti-inflammatory effects in a rat model of acute myocardial
infarction. Circ J. 78:2915–2925. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
White DA, Fang L, Chan W, Morand EF,
Kiriazis H, Duffy SJ, Taylor AJ, Dart AM, Du XJ and Gao XM:
Pro-inflammatory action of MIF in acute myocardial infarction via
activation of peripheral blood mononuclear cells. PLoS One.
8:e762062013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Oliveira NL, Ribeiro F, Silva G, Alves AJ,
Silva N, Guimaraes JT, Teixeira M and Oliveira J: Effect of
exercise-based cardiac rehabilitation on arterial stiffness and
inflammatory and endothelial dysfunction biomarkers: A randomized
controlled trial of myocardial infarction patients.
Atherosclerosis. 239:150–157. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ammirati E, Cannistraci CV, Cristell NA,
Vecchio V, Palini AG, Tornvall P, Paganoni AM, Miendlarzewska EA,
Sangalli LM, Monello A, et al: Identification and predictive value
of interleukin-6+ interleukin-10+ and interleukin-6-interleukin-10+
cytokine patterns in ST-elevation acute myocardial infarction. Circ
Res. 111:1336–1348. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Monu SR, Pesce P, Sodhi K, Boldrin M, Puri
N, Fedorova L, Sacerdoti D, Peterson SJ, Abraham NG and Kappas A:
HO-1 induction improves the type-1 cardiorenal syndrome in mice
with impaired angiotensin II-induced lymphocyte activation.
Hypertension. 62:310–316. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zeng B, Lin G, Ren X, Zhang Y and Chen H:
Over-expression of HO-1 on mesenchymal stem cells promotes
angiogenesis and improves myocardial function in infarcted
myocardium. J Biomed Sci. 17:802010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Czibik G, Derumeaux G, Sawaki D, Valen G
and Motterlini R: Heme oxygenase-1: An emerging therapeutic target
to curb cardiac pathology. Basic Res Cardiol. 109:4502014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shu T, Zeng B, Ren X and Li Y: HO-1
modified mesenchymal stem cells modulate MMPs/TIMPs system and
adverse remodeling in infarcted myocardium. Tissue Cell.
42:217–222. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Carnieto A Jr, Dourado PM, Luz PL and
Chagas AC: Selective cyclooxygenase-2 inhibition protects against
myocardial damage in experimental acute ischemia. Clinics (Sao
Paulo). 64:245–252. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vaithianathan R, Hockey PM, Moore TJ and
Bates DW: Iatrogenic effects of COX-2 inhibitors in the US
population: Findings from the medical expenditure panel survey.
Drug Saf. 32:335–343. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ma Y, Li H, Yue Z, Guo J, Xu S, Xu J, Jia
Y, Yu N, Zhang B, Liu S, et al: Cryptotanshinone attenuates cardiac
fibrosis via downregulation of COX-2, NOX-2, and NOX-4. J
Cardiovasc Pharmacol. 64:28–37. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Davies NM, Smith GD, Windmeijer F and
Martin RM: COX-2 selective nonsteroidal anti-inflammatory drugs and
risk of gastrointestinal tract complications and myocardial
infarction: An instrumental variable analysis. Epidemiology.
24:352–362. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Duan J, Yang Y, Liu H, Dou PC and Tan SY:
Osthole ameliorates acute myocardial infarction in rats by
decreasing the expression of inflammatory-related cytokines,
diminishing MMP-2 expression and activating p-ERK. Int J Mol Med.
37:207–216. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Peake BF, Nicholson CK, Lambert JP, Hood
RL, Amin H, Amin S and Calvert JW: Hydrogen sulfide preconditions
the db/db diabetic mouse heart against ischemia-reperfusion injury
by activating Nrf2 signaling in an Erk-dependent manner. Am J
Physiol Heart Circ Physiol. 304:H1215–H1224. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Koizumi K and Nakajima H: Serotonin
induces the migration of PC12 cells via the serotonin receptor
6/cAMP/ERK pathway. Biomed Rep. 2:29–33. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Reid EA, Kristo G, Yoshimura Y,
Ballard-Croft C, Keith BJ, Mentzer RM Jr and Lasley RD: In vivo
adenosine receptor preconditioning reduces myocardial infarct size
via subcellular ERK signaling. Am J Physiol Heart Circ Physiol.
288:H2253–H2259. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li A, Wang J, Zhu D, Zhang X, Pan R and
Wang R: Arctigenin suppresses transforming growth
factor-beta1-induced expression of monocyte chemoattractant
protein-1 and the subsequent epithelial-mesenchymal transition
through reactive oxygen species-dependent ERK/NF-kappaB signaling
pathway in renal tubular epithelial cells. Free Radic Res.
49:1095–1113. 2015. View Article : Google Scholar : PubMed/NCBI
|