Introduction
Leigh syndrome (also termed subacute necrotizing
encephalomyelopathy) is a rare, inherited, heterogeneous, and
progressive neurometabolic disorder (1). This syndrome is characterized by a
wide variety of neurologic abnormalities (2,3) with
degeneration of cognitive and motor functions, in most cases
resulting in death due to respiratory failure (4). Leigh syndrome normally begins between
the ages of three months and two years, and affects approximately 1
in 40,000 live births worldwide (5). Neurological symptoms are hallmarks of
Leigh syndrome and patients generally manifest a progressive
decline in central nervous system function because of necrotizing
lesions of the basal ganglia, cerebellum, or brainstem, as
visualized by magnetic resonance imaging (MRI) (3). Additional clinical manifestations and
ages of onset vary from case to case, encompassing a diverse array
(6,7). Leigh syndrome can result from
specific mutations in nuclear DNA (nDNA) or mitochondrial DNA
(mtDNA). Mutations in mtDNA are responsible for approximately 20%
of cases of Leigh syndrome (8,9). The
main mutations, which are both associated with the ATPase6
gene, are m.T8993G and m.T8993C (10). They result in the most frequent
cases of maternally inherited Leigh syndrome (MILS) (4,10).
The m.T8993C variant results in later onset, slower progression,
and lesser severity than the energy production-related m.T8993G
mutation (9,11). The T8993G or T8993C mutations
convert a leucine residue in the mitochondrial ATP synthase protein
into arginine or proline, respectively, reducing its activity, and
subsequently leading to ATP deficiency in the cells. T8993C-induced
reduction of ATP synthesis is also involved with deficiency of
complexes I and V in the mitochondrial oxidative phosphorylation
system (OXPHOS) (11,12), with consequent cell death.
Moreover, experimental data previously revealed that the m.T8993C
mutation leads to increased reactive oxygen species (ROS)
production, thus creating oxidative stress (13). Therefore, high-energy demanding
tissues, such as brain and heart, are most vulnerable to this
process, which in part explains the clinical features of Leigh
syndrome (4). The clinical
manifestations of these gene defects largely depend on the
abundance of mutated mtDNA molecules (12). High mutation loads (over 90%)
usually cause Leigh syndrome cases such as MILS, while lower levels
can cause less severe phenotypes such as neuropathy, ataxia, and
retinitis pigmentosa (NARP). Thus, a diverse array of clinical
features (10,14) presents a challenge to the
clinicians (7). Therefore, for
better diagnosis and treatment of this syndrome, molecular genetic
testing is required, in addition to clinical characterization.
Case report
Samples
Peripheral blood samples were collected from a
patient showing characteristics of Leigh syndrome and eight of his
family members at the Vietnam National Children's Hospital (VNCH;
Hanoi, Vietnam). The present study was approved by the ethics
committee of the VNCH and conformed to the guidelines and ethical
rules of the VNCH. All the subjects signed a consent form for
voluntary participation in the research.
Clinical description of patient
The patient was a 21-month-old male displaying
characteristics of Leigh syndrome (movement disorder, hypotonia,
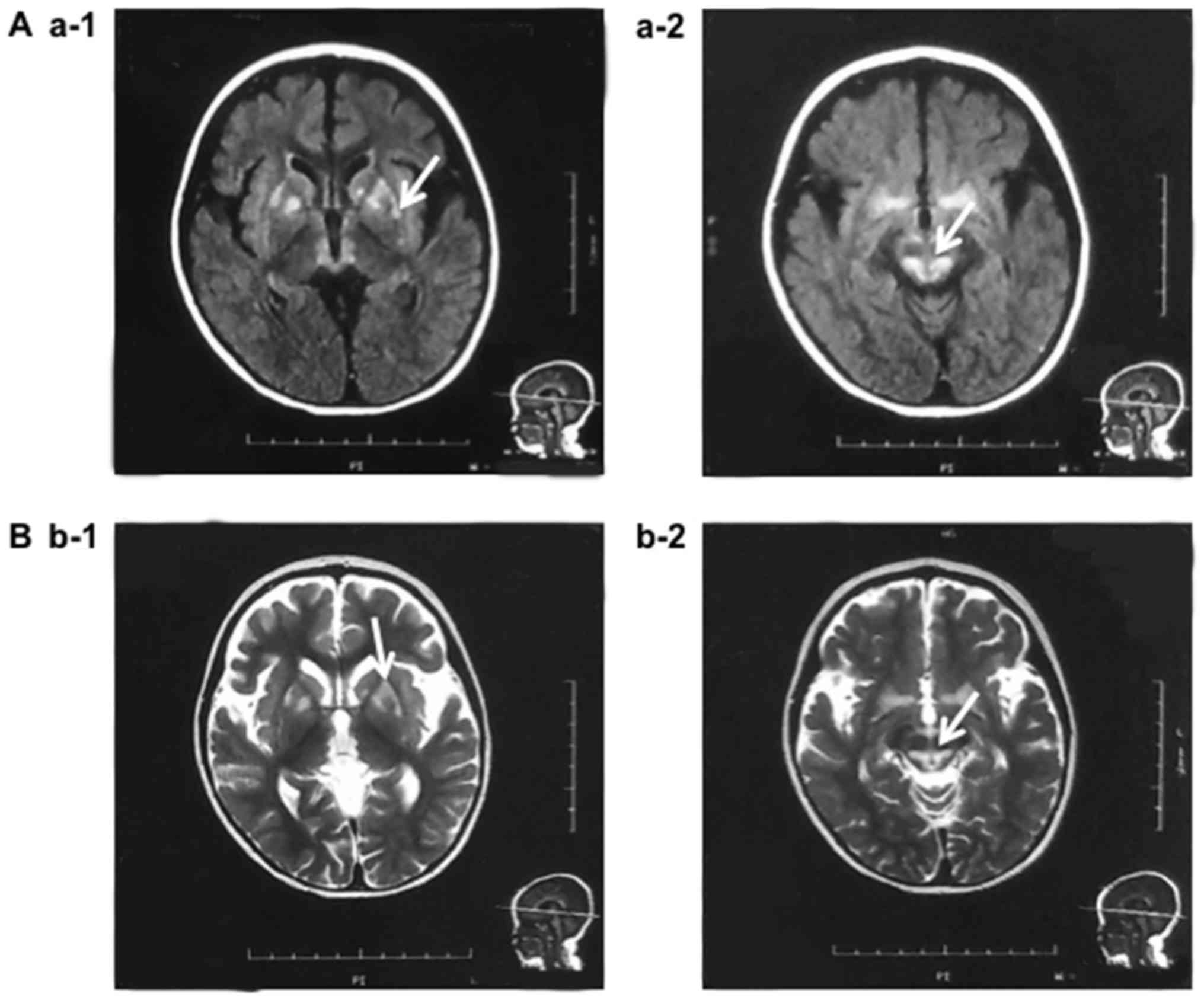
fever, intractable nausea and vomiting, and abnormal brain MRI as
shown in Fig. 1). Blood test
results of the patient showed lactatemia (8.2 mmol/l) and acidosis
associated with ketonuria. He was born after 40 weeks of gestation
to a 32-year-old mother and a 35-year-old father via normal
delivery. Only the patient showed clinical characteristics of Leigh
syndrome whereas the other eight members of his family were normal.
The patient died at 34 months of age.
MRI
Axial fluid-attenuated inversion recovery MRI (Axial
FLAIR MR) was conducted according to a previous study (15).
Detection of m.T8993C mutation using
PCR in combination with restriction fragment length polymorphism
(PCR-RFLP) and sequencing
Total DNA from peripheral blood samples was manually
extracted using the QIAamp DNA Mini kit (Qiagen, Valencia, CA, USA)
and spectrophotometrically quantified by NanoDrop ND-1000
(NanoDrop; Thermo Fisher Scientific, Inc., Wilmington, DE,
USA).
The m.T8993C mutation was detected using PCR-RFLP as
previously described (16).
Briefly, a mtDNA fragment of 402 bp length (spanning the 8768–9169
bp region of the mitochondrial genome) was amplified with the
following primers. Forward primer: T8993C Fw
5′-CAACTAACCTCCTCGGAC-3′ (corresponding to the 8768–8785 bp
region); reverse primer: T8993C Rv 5′-TGAAAACGTAGGCTTGGAT-3′
(9169–9151 bp region). Amplified PCR products were digested using
endonuclease HpaII, an enzyme that specifically recognizes
and cuts after the first nucleotide in the C/CGG sequence at the
base pair 8,993, and separated by 12% polyacrylamide gel
electrophoresis. Ethidium bromide-stained gels were then
photographed using Geldoc (Bio-Rad Laboratories, Inc., Hercules,
CA, USA). The amplified and digested mtDNA of affected individuals
showed three bands of lengths 402, 224, and 178 bp (heteroplasmic),
or two bands of lengths 224 and 178 bp (homoplasmic) in samples
taken from affected individuals, whereas the mtDNA of normal of
unaffected individuals showed only one band of length 402 bp. The
presence of the m.T8993C mutant was reconfirmed using DNA
sequencing (analyzed by the 1st Base Company, Singapore). The
analyzed DNA sequence was then compared to the reference sequence
(GenBank J01415.2).
Quantification of mtDNA copy number
using qPCR
The m.T8993C heteroplasmy level was quantitated by
qPCR using a Taqman Locked Nucleic Acid (LNA) probe (IDT Inc.,
Coralville, IA, USA) and iQ5 cycler (Biorad, USA) as previously
described for the A3243G mutation (17). In this case, a mtDNA fragment of 69
bp (8958–9026 bp region) was amplified with the following primers.
Forward primer: RT8993-Fw 5′-CGAAACCATCAGCCTACTCATTCAA-3′; reverse
primer: RT8993-Rv 5′-CCTGCAGTAATGTTAGCGGTTAGG-3′ (corresponding to
the 9026–9003 bp region). The LNA probes, Wt-8993T-HEX
5′-/5HEX/AAT+AGCCC+T+GGCC/3IABkFQ-3′ (8985–8997 bp region) and
Mt-8993C-FAM 5′-/5FAM/ATAGCCC+CGGCCGT/3IABkFQ/-3′ (8986–8999 bp
region), containing LNA nucleotides (nucleotides proceeded by the
‘+’ symbol in the given sequences) were designed for quantitating
the wild type and mutant genotypes, respectively. The copy numbers
of mutant and wild-type mtDNA in the samples were calculated from
standard curve, obtained by plotting the Ct values of
six standards on the X-axis vs. the logarithm of their copy numbers
on the Y-axis. The standards were 1:1 mixtures of the pJET1.2
plasmid with either the mutant m.T8993C or wild-type (T8993) DNA,
at concentrations increasing from 5.103 to
5.106 copies per µl The calculations were made by
averaging triplicate measurements. The level of heteroplasmy was
defined as the percentage of mutant copies obtained in total copies
of mutant and wild type DNA.
Statistical analysis
Data are presented as means ± standard error of the
mean (SEM). Differences between the generations were evaluated by
Pair sample t-test using Origin 8.0 (version 8.0; OriginLab,
Northampton, MA, USA). P<0.05 was considered to indicate a
statistically significant difference.
Clinical features and MRI of the Leigh
syndrome patient
MRI data of the patient revealed abnormal anatomical
characteristics. Axial fluid-attenuated inversion recovery MRI
(Axial FLAIR MR) showed a symmetric abnormal signal in the dorsal
medulla oblongata. Axial T2 weighted image (T2WI) MRI revealed
Sylvian fissure enlargement in association with abnormal signal in
the periventricular white matter and in the putamina, and caudate
heads (Fig. 1). The patient first
presented with characteristics of Leigh syndrome at 21 months of
age: muscular weakness (hypotonia), movement disorder, fever,
intractable nausea and vomiting. His blood test results indicated
lactatemia (8.2 mmol/l compared to the normal level of less than
2.8 mmol/l) and acidosis associated with ketonuria. During
treatment, the patient showed severe cognitive retardation,
episodes of seizures, pneumonia, lethargy, and fever. Severe
hypotonia associated with dystonia and dyspnea was also found. The
patient died at 34 months of age because of respiratory
failure.
Discovery of m.T8993C point mutation
in the patient and his family members
As T>C/G nucleotide changes at site 8,993 bp are
commonly diagnosed in Leigh syndrome patients, we carried out
PCR-RFLP using HpaII enzyme on the DNA samples. PCR-RFLP
results showed that three DNA bands (402, 224, and 178 bp) were
present in lanes 1, 2, 4–6, and 8 corresponding to DNA samples
taken from the patient, his mother, his cousin 1 (male), his cousin
2 (female), his aunt, and his grandmother, as shown in Fig. 2. This indicates that these family
members carried the m.T8993C/G point mutation and that the variant
was maternally inherited from the grandmother. The mutation was not
found in DNA samples taken from the rest of the family members
including the father, the husband of the aunt, and the grandfather
of the patient (Fig. 2; lanes 3,
7, and 9) as indicated by the presence of a single band of length
402 bp.
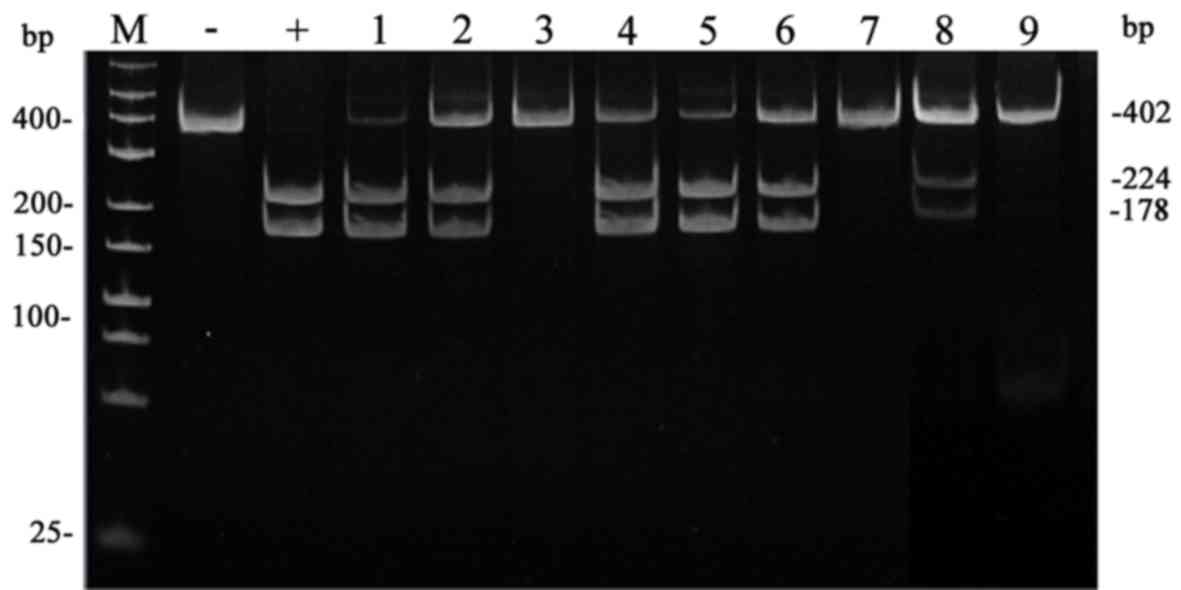 | Figure 2.PCR-RFLP analysis of the m.T8993C
mutation in the samples of the patient and his family members.
Polyacrylamide electrophoresis results of HpaII enzyme
digestions. Amplified mutant mtDNA shows bands of 402, 224, and 178
bp (heteroplasmic) or bands of 402 and 224 bp (homoplasmic);
wild-type mtDNA shows a band of 402 bp. Lane labels M, Marker (low
range DNA ladder); -, Negative control; +, Positive control; 1,
Patient; 2, Mother; 3, Father; 4, Cousin 1 (male); 5, Cousin 2
(female); 6, Aunt; 7, Husband of the aunt; 8, Grandmother; 9,
Grandfather. |
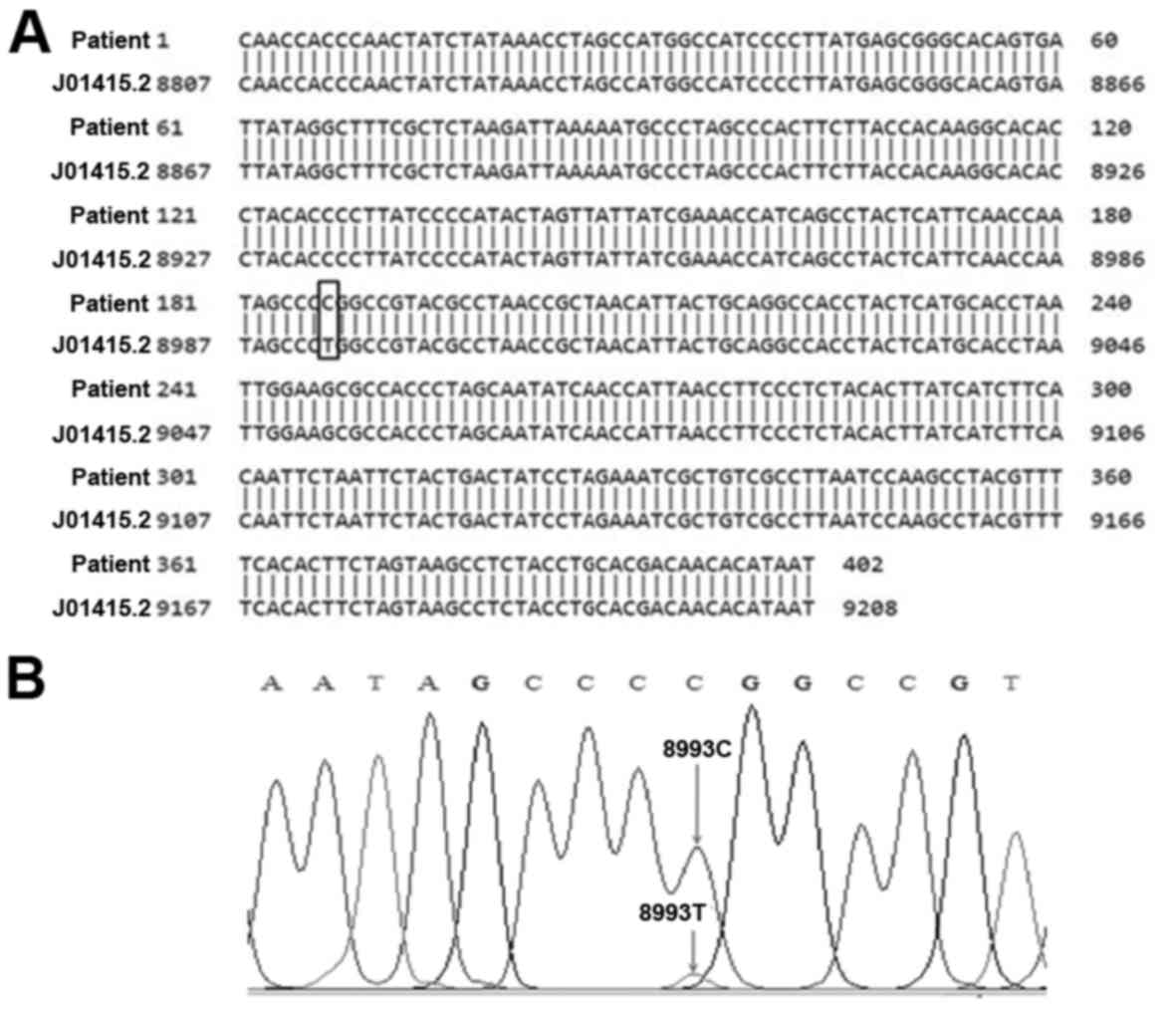
The DNA sequence obtained from the patient sample
was then compared to the GenBank reference sequence (J01415.2). The
presence of the T8993C mutation in the representative alignment
between the sequence of patient and the reference sequence is shown
in Fig. 3. Based on the DNA
sequencing data, we can conclude that the patient, his brothers and
mother have the T8993C mutation. Heterogeneity in the intensities
of the 224 and 178 bp bands among the samples containing the T8993C
mutation probably reflect different levels of heteroplasmy
(Fig. 2).
Quantification of m.T8993C
heteroplasmy levels in family members
As the level of heteroplasmy of m.T8993C is an
important factor affecting the phenotype and severity of Leigh
syndrome, we reconfirmed the presence of the mutation and
quantified heteroplasmy levels among the patient's family members
in triplicate by qPCR using LNA Taqman probes.
Amplification curves for the mutant genotype (FAM
channel, Fig. 4A) appeared only in
samples from the patient, his mother, cousin 1, his cousin 2, his
aunt, and his grandmother, but not in those from the patient's
father, his aunt's husband, or his grandfather, whereas the
amplification curves for the wild-type genotype (HEX channel,
Fig. 4B) appeared in all the
samples. The calculated heteroplasmy levels of the patient and his
family members (based on the standard curves) were significantly
different between the generations (Fig. 4 and Table I). The percentages of heteroplasmy
levels in peripheral blood samples taken from the patient, his
mother, cousin 1, cousin 2, his aunt, and his grandmother were
94.00±1.34, 71.66±3.22, 87.00±1.79, 91.24±2.50, 66.81±0.85, and
16.33±1.67%, respectively. The mutation was not detectable in
samples from his grandfather, his father, or his aunt's
husband.
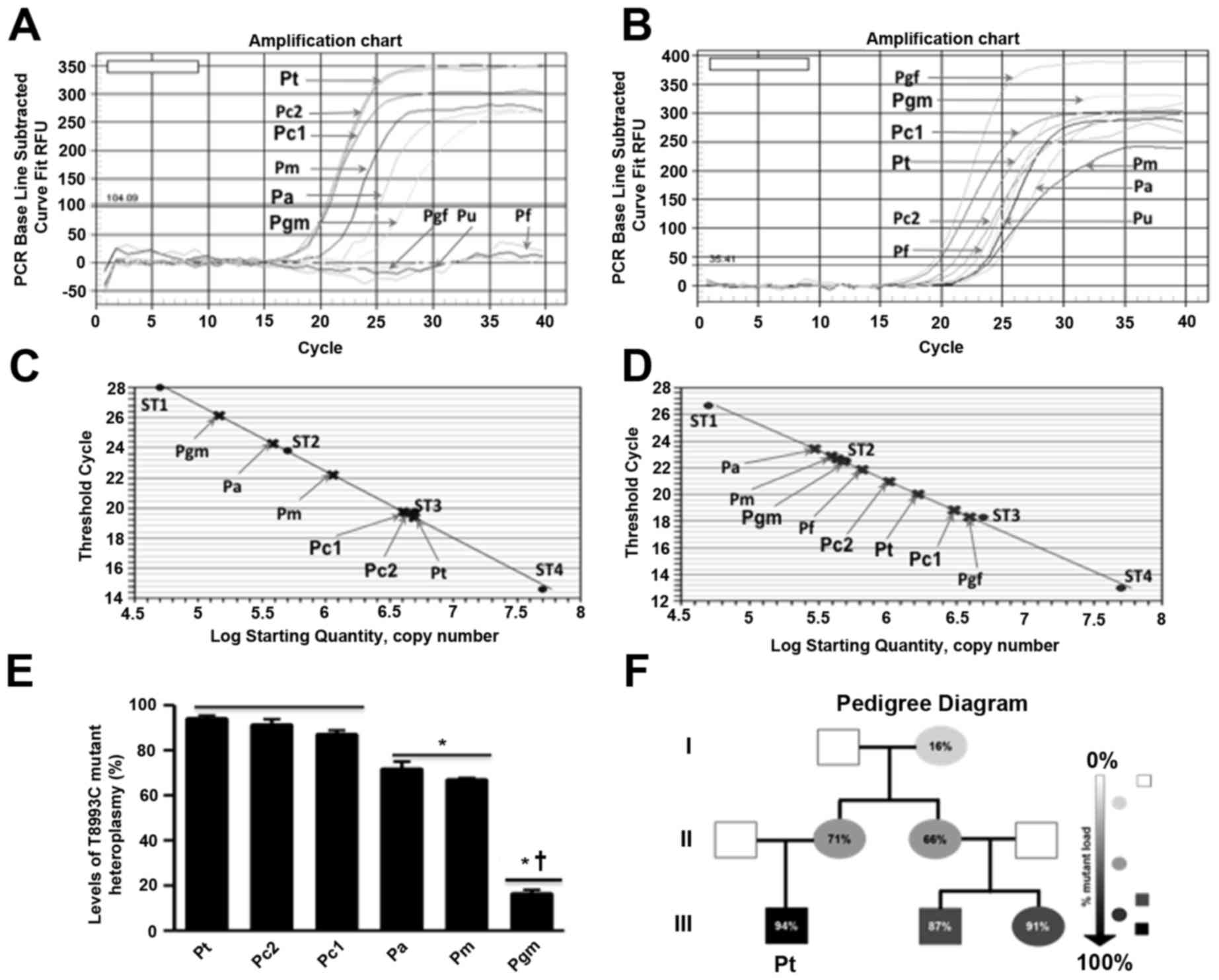 | Figure 4.Quantitative polymerase chain
reaction (qPCR) analysis of the T8993C mutation and pedigree
diagram of the patient's family. (A) qPCR amplification curves
quantifying the T8993C mutation in samples of the patient and his
family members using the (A) FAM and (B) HEX LNA probes. Standard
curves of Ct values generated by mixing plasmid DNA with
varying concentrations of (C) 100% mutant or (D) 100% wild-type
DNA, with overlaid Ct values of the patient and his family members.
ST1, ST2, ST3, and ST4 are the concentrations of standards
(5.103, 5.104, 5.105 and
5.106 molecules/µl, respectively). (E) Heteroplasmy
levels (%) in family members harboring the m.T8993C mutation.
*P<0.05 vs. the 3rd generation, †P<0.05 vs. the
2nd generation. (F) Pedigree diagram of the family. The gray or
black symbols show affected family members and the white symbol
indicates unaffected members. Numbers refer to percentage of
mutated mtDNA in the blood of family members. Circles and squares
denote females and males, respectively. Pt, Patient; Pm, Patient's
mother; Pf, Patient's father; Pc1, Patient's cousin 1; Pc2,
Patient's cousin 2; Pa, Patient's aunt; Pu, The husband of
patient's aunt; Pgm, Patient's grandmother; Pgf, Patient's
grandfather. |
 | Table I.Correlation between percentage
m.T8993C mutation heteroplasmy and clinical phenotype in patient's
family. |
Table I.
Correlation between percentage
m.T8993C mutation heteroplasmy and clinical phenotype in patient's
family.
| Family members | Age (years) | Clinical presence
of Leigh syndrome | Mutation load
(%) |
|---|
| Patient | 2 | Yes |
94.00±1.34 |
| Mother | 35 | No |
71.66±3.22 |
| Father | 37 | No |
0.00±0.00 |
| Cousin 1 | 6 | No |
87.00±1.79 |
| Cousin 2 | 3 | No |
91.24±2.50 |
| Aunt | 32 | No |
66.81±0.85 |
| Aunt's husband | 34 | No |
0.00±0.00 |
| Grandmother | 60 | No |
16.33±1.67 |
| Grandfather | 62 | No |
0.00±0.00 |
The qPCR results presented in Fig. 4 and Table I also correlate well with the
change from low to high intensity of the 224 and 178 bp bands
obtained by PCR-RFLP (Fig. 2). The
results reconfirmed that among mutation bearers, the grandmother
harbored the level of heteroplasmy (16.33±1.67%), while the patient
bore the level of heteroplasmy (94.00±1.34%). Both of his cousins
harbored high levels of heteroplasmy, while his mother and his aunt
bore milder levels of mutation loads.
Discussion
Leigh syndrome is a rare, heritable, heterogeneous,
and progressive neurodegenerative disorder, appearing mostly during
infancy or early childhood (1–3,7,18)
and it affects approximately 1 in 40,000 live births worldwide
(5). In Vietnam, no patients have
been found in a previous screening (16). Here, we report the first case of a
young Vietnamese patient clinically presenting the symptoms of
Leigh syndrome who was genetically confirmed to harbor T8993C mtDNA
mutation. In contrast to the patient, other patient family members
carrying the T8993C mutation were neurologically asymptomatic and
in them the percentage of heteroplasmy was lower than in the
patient. The asymptomatic range was found to be between 16 and 91%
approximately.
In this study, the patient presented clinical
characteristics common to Leigh syndrome, as reported previously
(1,2,11,13,19).
Clinical features of Leigh syndrome such as epilepsy, intractable
nausea and vomiting accompanied by abnormal brain imaging were
observed in the patient (Fig. 1).
Similar to a previous study (2),
the clinical features of our patient also included muscular
hypotonia. The patient was also found to have lactatemia (8.2
mmol/l) and ketonuria. He lost the ability to speak and,
eventually, could no longer walk at two years of age.
MRI of his brain revealed a symmetric abnormal
signal in the dorsal medulla oblongata and Sylvian fissure
enlargement in association with an abnormal signal in the
periventricular white matter and in the putamina, and caudate heads
(Fig. 1), which were pathognomonic
for Leigh syndrome as demonstrated previously (2,20).
He also exhibited deterioration of cognitive and motor functions.
In agreement with earlier reports (21), the patient was ultimately diagnosed
with Leigh syndrome after targeted mutation analysis revealed high
heteroplasmy (94%) of mtDNA bearing the m.T8993C mutation.
Although, m.T8993C heteroplasmy has also been associated with
neurogenic weakness, ataxia and retinitis pigmentosa [NARP syndrome
(22)], it's correlation with
disease characteristics is not clearly understood (6), particularly for explaining the normal
health of other family members of the patient. The patient was
treated with coenzyme Q10 and vitamins and showed some progress.
Unfortunately, he kept continuing to deteriorate and died of
respiratory failure at 34 months of age during an episode of chest
infection, as is commonly reported for such patients (14).
Mutation at the mtDNA nucleotide 8993 site is a
relatively common cause of Leigh syndrome (8) with a correlation between the amount
of mutant mtDNA and clinical severity (11,20,22).
The change of nucleotide (either T>G or T>C) at this site is
hypothesized to impair OXPHOS, particularly I and V complexes,
leading to ROS over-generation and mitochondrial energy metabolic
malfunction, with subsequent cell death (11). It is reported that the mtDNA 8993
mutation site is in heteroplasmy, with cells having both wild-type
and mutant mtDNA and the heteroplasmy level is an important factor
determining the phenotype and severity of the disease (12). The tissues demanding high energy
are most vulnerable to this process and, in part, are responsible
for the clinical characteristics of Leigh syndrome patients. The
mtDNA T8993C mutation is generally considered to be clinically
milder than the T8993G mutation (11). However, in cases where the levels
of heteroplasmy exceed approximately 90–95%, progressive
neurodegeneration characterized by bilateral lesions of the basal
ganglia, which may extend to the brainstem, lead to the eventual
death of the patient (13,22). In our study, the patient's second
cousin carried the T8993C mutation at 91.24±2.50% levels of
heteroplasmy but showed no clinical symptoms of Leigh syndrome.
This level of heteroplasmy is still very close to the normal range
of reported cases (11,22); even a case of ≥95% of heteroplasmy
has been reported not to show most symptoms of Leigh syndrome
(22). In concordance with the
literature (11,22), we found that our Vietnamese patient
presenting with clinical Leigh syndrome harbored the highest
heteroplasmy level of the T8993C mutation in his family. The other
family members carrying T8993C mutation were neurologically
asymptomatic, with lower percentages of heteroplasmy compared to
that of the patient (Fig. 4 and
Table I). Prenatal diagnosis of
the T8993C mutation is technically possible and there is a
correlation between the percentage of heteroplasmy of the T8993C
mutation and the clinical phenotype (12). Similar to a previous report
regarding Leigh patients harboring high T8993C heteroplasmy levels
in the blood (10), our patient
showed worsening clinical progression, was unable to speak,
hypotonic, and also showed motor disability. However, variability
in the phenotypic expression of patients carrying the T8993C
mutation has also been observed (3,11,22).
As the level of heteroplasmy of mtDNA in the patient's blood was at
approximately 95%, he showed early muscular hypotonia and abnormal
brain MRI (11). Another case
report described the long-term follow-up of a Leigh syndrome
patient who presented at four years of age with the typical T8993C
mutation (95% heteroplasmy), but showed an unexpected resolution of
symptoms and a favorable outcome. This suggests the need for
caution in predictive counseling of patients suspected to have
Leigh syndrome and their parents (22). No strong correlation between the
clinical features and heteroplasmy has been reported (3). Therefore, co-variation in
heteroplasmy among tissues and/or over time, and the clinical
presentation of Leigh syndrome with brainstem or spinal cord
involvement should be tracked in patients with this syndrome
(6,14). In our study, except for typical
clinical presentation by the patient, all the family members
carrying the mutation were clinically normal. A pedigree analysis
confirmed maternal inheritance of the T8993C mutation with
heteroplasmy. Genetic testing revealed that the proportion of the
mtDNA carrying the T8993C mutation was 16.33±1.67 in the patient's
maternal grandmother, 71.66±3.22% in the patient's mother, and
66.81±0.85% in the patient's maternal aunt. The grandmother,
mother, and the aunt were all clinically normal. The patient also
had two symptomatic maternal first cousins, the aunt's children,
who had approximately 87 to 91% mtDNA with the m.T8993C mutation,
but they were neurologically asymptomatic. This finding can be
explained by the tight correlation between the heteroplasmy and the
development of the phenotype in T8993C carriers (11,23).
That is, the threshold of heteroplasmy of this mutation for the
appearance of effects is high. Moreover, the mutant load can change
over time and the clinical characteristics of individuals could be
dependent on other factors, such as the frequency of environmental
stressors like illness (6,9). As mentioned above, nucleotide change
at mtDNA 8,993 bp affects ATPase activity and thus be associated
with MILS or NARP when the mutational load is greater than 90%, or
between 50–60%, respectively (13,22).
Greater than 95% proportion of m.T8993C mutation has also been
revealed in an adult diagnosed with NARP syndrome (19,22).
The varying degrees of heteroplasmy produce a wide
range of clinical symptoms and most T8993C carrying patients
present one or more abnormal neurologic characteristics, including
brainstem or basal ganglia dysfunction, seizures, hypotonia,
periodic illness, and progressive neurodegeneration (2,19,23).
As a bioenergetic defect is unlikely to be the main reason for
explaining all the Leigh syndrome-associated phenotypes and their
pathogenesis (13,24), more study is required, especially
to address the higher frequency of ataxia in T8993C-based Leigh
syndrome cases (23). Predictably,
ATP synthesis rate was only mildly reduced by the m.T8993C mutation
at levels of 94% heteroplasmy (13), especially because only a 22%
reduction of ATP synthesis has been previously reported in
homoplasmic m.T8993C cybrids (24). So far, increased oxidative stress
may be the major cause of the occurrences of MILS cases associated
with the T8993C mutation (13). At
the molecular level, the difference in severity between T8993C and
T8993G carrying patients is that the T8993C mutation results in
substitution of leucine with proline (p.Leu156Pro) in ATP synthase
subunit 6 (Atp6p), while the T8993G mutation results in
substitution of leucine with arginine (p.Leu156Arg) in Atp6p
(11). Leu and Pro both are
hydrophobic amino acids and their side residues have similar sizes.
Thus the Leu vs. Pro changes should not be very different with
respect to their effect on activity of Atp6p protein. However,
replacement of Leu by Arg does induce a structural defect in human
F1F0-ATPase that causes a severe impairment
in ATP synthesis (25).
It is well known that mtDNA mutations are randomly
subjected to a bottleneck effect and segregate to the oocytes
forming in the female fetus (26).
Differential amplification could lead to a rapid diversification of
mtDNA genotypes in a single generation. Heteroplasmy and mutant
types (either m.T8993G or m.T8993C) play important roles in
determining the age of disease onset as well as in levels of
clinical severity. As pointed out in Fig. 4 and Table I, the heteroplasmic state of the
m.T8993C mutation was significantly increased over successive
generations in this Vietnamese family. Interestingly, in the two
preceding generations, none of the paternal members harbor the
mutation; only mothers and their children carry the variant (as
seen in the pedigree diagram in Fig.
4F). Perhaps, it is a result of a bottleneck effect (9,24).
If this effect takes place during or after early embryonic
development, there may be variable mutational loads in tissues and
between ages. We suppose that the high heteroplasmic presentation
in our patient was probably due to random segregation and that a
bottleneck effect occurred during his mother's oogenesis. In
contrast to this patient, the mtDNA genetic bottleneck effect and
genetic drift in other family members might have occurred during
gastrulation because their genetic profiles did not match their
clinical records (27). However,
severity of Leigh syndrome occasionally depends on the tissue
distribution and on the mutant load of the pathogenic variant
(6,24). In this family, all children of
females with the mtDNA pathogenic variant remained at risk of
developing Leigh symptoms. In agreement with the previous studies
about this disease, mothers had much lower mutation loads than
children and displayed normal health (9), suggesting that the m.T8993C mutation
is likely to have a higher tendency of being selected for during
early oogenesis. Here, the patient was found to have the highest
heteroplasmy of m.T8993C with typical clinical features of Leigh
syndrome while other family member remained asymptomatic. In the
first generation, the patient's grandmother bore the lowest level
of mutational load (approximately 16%). His mother and aunt, the
members of the second generation, exhibited a lower heteroplasmy
percentage (between 67 and 71%). The patient and his two younger
cousins, representative of the third generation, harbored the
highest mutational burden (approximately 91%). it is possible that
the T8993C mutation could aggregate rapidly towards homoplasmy
through generations of this family, and their clinical
manifestations become more severe, as has been demonstrated in
other cases (9).
To date, there is no cure for mtDNA T8993C
mutation-affected patients, and treatment options are mostly
unsatisfactory (18). Furthermore,
mitochondrial genetics, ATP production, ROS levels, and other
undefined factors likely differentiate the onset and severity of
the disorder (19). In order to
understand correspondence between the genetic profiles and clinical
manifestation more investigation is needed.
In conclusion, this is the first case of an m.T8993C
mutation-associated Leigh syndrome case identified in Vietnam. We
also described the correlation between the percentage of
heteroplasmy of this mutation and the clinical phenotype of the
patient's family. We hope that these data lead to the improvement
of clinical diagnosis and treatment of Leigh syndrome patients.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Vietnam
National University, Hanoi (Project code: KLEPT.16.03 to TNP).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CALW performed qPCR analysis of the patient and his
family members, BHTB performed PCR-RFLP analysis of the patient and
his family, TTV isolated DNA from the samples and contributed to
writing the manuscript, HLTN analyzed qPCR data, prepared the
figures and contributed to writing the manuscript, BKP checked the
PCR-RFLP data, VMN checked the qPCR data, VAP contributed to
assessing the clinical data of the patient and his family members,
VHC analyzed NMR data, diagnosed and treated the patient and
contributed to writing the manuscript. TNP coordinated the study
with TTV, HLTN and VHC and finalized the manuscript. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the VNCH and the patient's representative and his
family members signed consent forms to participate in the
study.
Consent for publication
The authors declare that identifying information,
including names, initials, date of birth or hospital numbers,
images of the patient and his family members were not included in
the manuscript and publication of the data presented in the
manuscript does not compromise anonymity or confidentiality.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Leigh PN, Al-Sarraj S and DiMauro S:
Impact commentaries. Subacute necrotising encephalomyelopathy
(Leigh's disease; Leigh syndrome). J Neurol Neurosurg Psychiatry.
86:363–365. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Piao YS, Tang GC, Yang H and Lu DH:
Clinico-neuropathological study of a Chinese case of familial adult
Leigh syndrome. Neuropathology. 26:218–221. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Finsterer J: Leigh and leigh-like syndrome
in children and adults. Pediatr Neurol. 39:223–235. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Thorburn DR, Rahman J and Rahman S:
Mitochondrial DNA-associated Leigh syndrome and NARPAdam MP,
Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K and Amemiya
A: Source GeneReviews® [Internet]. University of
Washington; Seattle, WA: pp. 1993–2018
|
|
5
|
Piekutowska-Abramczuk D: The molecular
background of Leigh syndrome. Neurol Neurochir Pol. 42:238–250.
2008.(In Polish). PubMed/NCBI
|
|
6
|
White SL, Shanske S, McGill JJ, Mountain
H, Geraghty MT, DiMauro S, Dahl HH and Thorburn DR: Mitochondrial
DNA mutations at nucleotide 8993 show a lack of tissue- or
age-related variation. J Inherit Metab Dis. 22:899–914. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bonfante E, Koenig MK, Adejumo RB,
Perinjelil V and Riascos RF: The neuroimaging of Leigh syndrome:
Case series and review of the literature. Pediatr Radiol.
46:443–451. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Santorelli FM, Shanske S, Macaya A, DeVivo
DC and DiMauro S: The mutation at nt 8993 of mitochondrial DNA is a
common cause of Leigh's syndrome. Ann Neurol. 34:827–834. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Makino M, Horai S, Goto Y and Nonaka I:
Mitochondrial DNA mutations in Leigh syndrome and their
phylogenetic implications. J Hum Genet. 45:69–75. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Santorelli FM, Shanske S, Jain KD, Tick D,
Schon EA and DiMauro S: A T->C mutation at nt 8993 of
mitochondrial DNA in a child with Leigh syndrome. Neurology.
44:972–974. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Morava E, Rodenburg RJ, Hol F, de Vries M,
Janssen A, van den Heuvel L, Nijtmans L and Smeitink J: Clinical
and biochemical characteristics in patients with a high mutant load
of the mitochondrial T8993G/C mutations. Am J Med Genet A.
140A:863–868. 2006. View Article : Google Scholar
|
|
12
|
Tatuch Y, Pagon RA, Vlcek B, Roberts R,
Korson M and Robinson BH: The 8993 mtDNA mutation: Heteroplasmy and
clinical presentation in three families. Eur J Hum Genet. 2:35–43.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Baracca A, Sgarbi G, Mattiazzi M, Casalena
G, Pagnotta E, Valentino ML, Moggio M, Lenaz G, Carelli V and
Solaini G: Biochemical phenotypes associated with the mitochondrial
ATP6 gene mutations at nt8993. Biochim Biophys Acta. 1767:913–919.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huntsman RJ, Sinclair DB, Bhargava R and
Chan A: Atypical presentations of Leigh syndrome: A case series and
review. Pediatr Radiol. 32:334–340. 2004.
|
|
15
|
Bakshi R, Ariyaratana S, Benedict RH and
Jacobs L: Fluid-attenuated inversion recovery magnetic resonance
imaging detects cortical and juxtacortical multiple sclerosis
lesions. Arch Neurol. 58:742–748. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Truong HT, Nguyen TVA, Nguyen LV, Pham VA
and Phan TN: Screening of common point-mutations and discovery of
new T14727C change in mitochondrial genome of Vietnamese
encephalomyopathy patients. Mitochondrial DNA A DNA Mapp Seq Anal.
27:441–448. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Truong HT, Nguyen TVA, Nguyen THL, Pham VA
and Phan TN: Sensitive quantification of mitochondrial mutation
using new Taqman probes. Cent Eur J Med. 9:839–848. 2014.
|
|
18
|
Kumagai R, Ichikawa K, Yasui T, Kageyama Y
and Miyabayashi S: Adult Leigh syndrome: Treatment with intravenous
soybean oil for acute central respiratory failure. Eur J Neurol.
6:613–615. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ruhoy IS and Saneto RP: The genetics of
Leigh syndrome and its implications for clinical practice and risk
management. Appl Clin Genet. 7:221–234. 2014.PubMed/NCBI
|
|
20
|
Makino M, Horai S, Goto Y and Nonaka I:
Confirmation that a T-to-C mutation at 9176 in mitochondrial DNA is
an additional candidate mutation for Leigh's syndrome. Neuromuscul
Disord. 8:149–151. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Enns GM, Bai RK, Beck AE and Wong LJ:
Clinical correlations in a family with variable tissue
mitochondrial DNA T8993G mutant load. Mol Genet Metab. 88:364–371.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Debray FG, Lambert M, Lortie A, Vanasse M
and Mitchell GA: Long-term outcome of Leigh syndrome caused by the
NARP-T8993C mtDNA mutation. Am J Med Genet A. 143A:2046–2051. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fujii T, Hattori H, Higuchi Y and Tsuji M:
Phenotypic differences between T->C and T->G mutations at nt
8993 of mitochondrial DNA in Leigh syndrome. Pediatr Neurol.
18:275–277. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pallotti F, Baracca A, Hernandez-Rosa E,
Walker WF, Solaini G, Lenaz G, Melzi D'Eril GV, Dimauro S, Schon EA
and Davidson MM: Biochemical analysis of respiratory function in
cybrid cell lines harbouring mitochondrial DNA mutations. Biochem
J. 384:287–293. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Baracca A, Barogi S, Carelli V, Lenaz G
and Solaini G: Catalytic activities of mitochondrial ATP synthase
in patients with mitochondrial DNA T8993G mutation in the ATPase 6
gene encoding subunit a. J Biol Chem. 275:4177–4182. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dahl HH, Thorburn DR and White SL: Towards
reliable prenatal diagnosis of mtDNA point mutations: Studies of
nt8993 mutations in oocytes, fetal tissues, children and adults.
Hum Reprod. 15 Suppl 2:S246–S255. 2000. View Article : Google Scholar
|
|
27
|
St John JC, Facucho-Oliveira J, Jiang Y,
Kelly R and Salah R: Mitochondrial DNA transmission, replication
and inheritance: A journey from the gamete through the embryo and
into offspring and embryonic stem cells. Hum Reprod Update.
16:488–509. 2010. View Article : Google Scholar : PubMed/NCBI
|