Introduction
Synovial sarcoma is a high-grade soft tissue sarcoma
that may occur in various regions of the body, particularly in
para-articular regions; it is the fourth most common tumor of soft
tissues, accounting for 5–10% of all soft tissue sarcomas (1). Although the available treatments have
improved, 25% of patients will succumb to synovial sarcoma within
the first 5 years following diagnosis (2). Surgery is the main treatment for
synovial sarcoma (3,4); however, a previous study reported
that postoperative chemotherapy for synovial sarcoma may
effectively inhibit tumor growth (5), and thus postoperative chemotherapy
for survival is necessary. The combination of surgery and
chemotherapy has resulted in an ~60% 5-year survival rate, yet the
10-year survival rate remains low (1). As a result, it is essential for
additional studies to identify novel chemotherapeutic drugs for
synovial sarcoma therapy.
Selenium is an essential trace element that serves
important roles in different physiological functions in the human
body (6). Numerous epidemiological
and clinical studies have suggested that physiological levels of
selenium may have a wide range of biological effects, including
strong protective effects against heart disease and age-related
diseases (7,8). Additional studies have indicated that
physiological and supranutritional selenium exhibit chemopreventive
or therapeutic activities on human solid cancers, such as lung
cancer, colorectal cancer and leukemia, by inducing apoptosis in
cancer cells with minimal side effects to normal cells, within a
proper dose range (9–12). Pharmacokinetics and toxicity of
sodium selenite have been reported in the treatment of patients
with carcinoma in a phase I clinical trial (13). Therefore, selenium may serve as a
potential auxiliary chemotherapeutic drug for synovial sarcoma in
the future by initiating cancer cell apoptosis.
Apoptosis is an important biological process that
leads to programmed cell death through the regulation of
apoptosis-related gene expression, and serves an important role in
a variety of tumor cells (14).
Apoptosis is mediated by the regulation of numerous proteins, such
as the apoptosis regulator Bcl-2 (Bcl-2) protein family and
caspases. Bcl-2 inhibits the induction of apoptosis, and
Bcl-2-associated X protein (Bax) induces apoptosis. High expression
of Bcl-2 affects the susceptibility of cells to the induction of
apoptosis and is often associated with low expression of Bax
(15). Bcl-2 and other Bcl-2
protein family members target intracellular organelles, including
the endoplasmic reticulum as well as outer mitochondrial and
nuclear membranes, where they modulate responses to a number of
cell death stimuli (15). Damage
to the outer mitochondrial membrane subsequently results in
increased permeability and the release of apoptosis-associated
molecules (16). These molecules
subsequently activate apoptotic factors, such as Caspase (Casp)-9
and its downstream factors, Casp-3 and poly (ADP-ribose) polymerase
(PARP) (16). However, the
mechanisms by which selenium activates apoptotic machinery are not
well understood. Autophagy is a survival strategy that is used by
cells experiencing nutrient deprivation or other stresses, and is
widely involved in the pathogenesis of many diseases, cancer in
particular (17,18). During autophagy, cytoplasmic
material is sequestered into double-membrane vesicles,
autophagosomes, which fuse with lysosomes and their contents are
subsequently degraded and recycled. Beclin-1 and
microtubule-associated protein 1 light chain 3 (LC3) are markers of
autophagy: Beclin-1 is associated with the trafficking of lysosomal
enzymes to lysosomes as well as autophagic vesicle nucleation, and
LC3, particularly LC3-II, is a marker of autophagosome formation
(19). In addition, autophagy is
responsible for the degradation of p62, which is a selective
autophagy receptor for the degradation of ubiquitinated substrates
(19). However, different studies
report different results regarding the role of selenium in tumor
cell autophagy. Selenium was reported to inhibit autophagy in
leukemia cells (20), whereas in
lung cancer and certain other tumors, selenium was revealed to
promote tumor cell autophagy (10,21).
Therefore, it is difficult to draw a conclusion regarding the
effects of selenium on tumor cell autophagy, particularly in
synovial sarcoma cells. The individual roles of apoptosis and
autophagy, and the interplay between these processes are complex
and may be different in different cells as well as in relation to
various stressors. On the one hand, a previous study indicated that
autophagy may protect certain tumor cells from chemotherapy
drug-induced apoptosis in vivo and in vitro (22). On the other hand, extensive or
persistent autophagy may also induce cell death; this autophagic
cell death is termed type II programmed cell death (23,24).
Thus, autophagy may serve as an adapter between cell death and
survival (25). Limited evidence
is available regarding the roles of selenium on the underlying
mechanisms in human tumor cells, and the effects of sodium
selenite, an inorganic selenium compound, in synovial sarcoma cells
have not been reported.
The aims of the present study were to determine the
mode of action of sodium selenite in the context of its antitumor
activity on synovial sarcoma, to investigate the relationship
between apoptosis and autophagy, and to examine the molecular
mechanisms underlying sodium selenite treatment in cancer cells.
Results from the present study may provide the first evidence to
suggest that sodium selenite induces apoptosis and inhibits
autophagy in SW982 cells in vitro. These observations
demonstrated that sodium selenite may serve as a novel adjuvant
therapy in the treatment of synovial sarcoma.
Materials and methods
Cells and main reagents
The SW982 human synovial sarcoma cell line was
obtained from The Cell Bank of Type Culture Collection of the
Chinese Academy of Sciences (Shanghai, China). Sodium selenite was
purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). LC3
(cat. no. L7541) and p62 (cat. no. p0067) antibodies were purchased
from Sigma-Aldrich; Merck KGaA. The β-actin (cat. no. bs-0061R)
antibody was purchased from Biosen Biotech Company (Beijing,
China). Pro-Caspase (Casp)-3 (cat. no. ab32351), cleaved-Casp-3
(cat. no. ab207612), PARPp85 (cat. no. ab32561) and Bcl-2 (cat. no.
ab32124) antibodies were purchased from Abcam (Cambridge, UK).
Beclin-1 (cat. no. 3738) and Bax (cat. no. 2772) antibodies were
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA).
3-methyladenine (3-MA) was purchased from Selleck Chemicals
(Shanghai, China). Rapamycin was purchased from Sigma-Aldrich;
Merck KGaA. Fluorescein isothiocyanate-conjugated immunoglobulin G
(IgG) was purchased from Thermo Fisher Scientific, Inc. (Waltham,
MA, USA). Bovine serum albumin was purchased from Sigma-Aldrich;
Merck KGaA.
Cell culture and treatment
SW982 cells were cultured in Dulbecco's modified
Eagle medium/Ham's Nutrient Mixture F12 medium (Thermo Fisher
Scientific, Inc.) containing 10% fetal bovine serum (Gibco; Thermo
Fisher Scientific, Inc.), penicillin G (100 U/ml), and streptomycin
(100 µg/ml) in a 5% CO2-humidified atmosphere at 37°C. A
previous study suggested that the reference range for blood
selenium level was 80–150 µg/l (1.02–1.91 µmol/l) (26); blood selenium concentrations of
1,400 ng/ml (equivalent to 17.7 µM) are toxic and may cause
symptoms of selenium poisoning, including hair and nail loss,
disorders of the respiratory system and paralysis (27,28).
Therefore, the present study used non-toxic sodium selenite
concentrations (0, 5, 10 and 15 µM) and toxic sodium selenite
concentrations (20 and 30 µM) for investigation. The culture
periods ranged between 0 and 72 h of continuous exposure to sodium
selenite.
Inhibition of autophagy
SW982 cells (5×104 cells/ml) were seeded
into 6-well plates and then incubated in a 5%
CO2-humidified atmosphere at 37°C. Following overnight
incubation, SW982 cells were pretreated with an autophagy
inhibitor, 3-MA (5 mM) for 2 h and subsequently incubated with
sodium selenite (10 µM) for 24 h at 37°C.
Induction of autophagy
SW982 cells (5×104 cells/ml) were seeded
into 6-well plates and then incubated in a 5%
CO2-humidified atmosphere at 37°C. Following overnight
incubation, SW982 cells were pretreated with 100 nM rapamycin for 2
h at 37°C and subsequently incubated at 37°C with sodium selenite
(10 µM) for 24 h.
Cell viability experiments
Cell viability was measured using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. Briefly, SW982 cells (5×104 cells/ml) were seeded
into 96-well plates, incubated in a 5% CO2-humidified
atmosphere at 37°C overnight and exposed to concentrations of
sodium selenite ranging between 0 and 30 µM for 0–72 h at 37°C.
Following incubations, MTT solution (20 µl; 5 mg/ml) was added to
each well, and the cells were incubated for an additional 4 h at
37°C. Following removal of the remaining medium, dimethyl sulfoxide
(150 µl) was added to each well to solubilize the precipitate. The
optical density (OD) was measured at 570 nm with a microplate
reader (Thermo Fisher Scientific, Inc.), and the following formula
was used to calculate viability: Cell viability (%)=(OD of the
experimental sample/OD of the control group) ×100. The half maximal
inhibitory concentrations (IC50) were determined using
GraphPad Prism 5.0 statistical software (GraphPad Software, Inc.,
La Jolla, CA, USA).
Morphological changes in SW982 cells
assay
SW982 cells (5×104 cells/ml) were
incubated in 96-well plates with either various concentrations (0,
5, 10 and 15 µM) of sodium selenite for 24 h or with 10 µM sodium
selenite for various lengths of time (0, 6, 12 and 24 h).
Morphological changes of SW982 cells were determined using an
inverted microscope (Nikon Corporation, Tokyo, Japan). The
morphological changes of SW982 cells include cell density, cell
deformation and cell pycnosis.
Apoptosis detection
Following the treatments with various concentration
or length of exposure to sodium selenite at 37°C, SW982 cells
(5×104 cells/ml) were detached with trypsin, washed
twice with 1X PBS and resuspended in annexin V binding buffer (200
µl; 7SeaPharmTech, Shanghai, China). Subsequently, cells were
incubated with annexin V-fluorescein isothiocyanate (7SeaPharmTech)
for 15 min at room temperature in the dark, followed by propidium
iodide (7SeaPharmTech) for 5 min at 4°C in the dark. Apoptotic
cells were analyzed using a Guava EasyCyteHT flow cytometer (Merck
KGaA).
Transmission electron microscopy (TEM)
analysis of autophagy
Following the various treatments, SW982 cells
(5×104 cells/ml) were detached with trypsin, washed
twice with PBS and fixed in ice-cold 2% glutaraldehyde/0.1 M
phosphate buffer (pH 7.2) for 2 h at 4°C, post-fixed in 1% osmium
tetroxide for 2 h at 4°C, washed with PBS, dehydrated in a graded
ethanol series (30, 50, 70, 90 and 100%) at 4°C (each concentration
for 10 min) and embedded in 1:1 propylene oxide/embedding resin at
37°C for 24 h. The resin blocks cut with a LKB-V microtome (LKB
Bromma, Sollentuna, Sweden), and thin (60 nm) sections were picked
up on 200-mesh copper grids and stained with uranyl acetate and
lead citrate for 10 min at room temperature. The sections were
examined with a H-7650 transmission electron microscope (Hitachi,
Ltd., Tokyo, Japan).
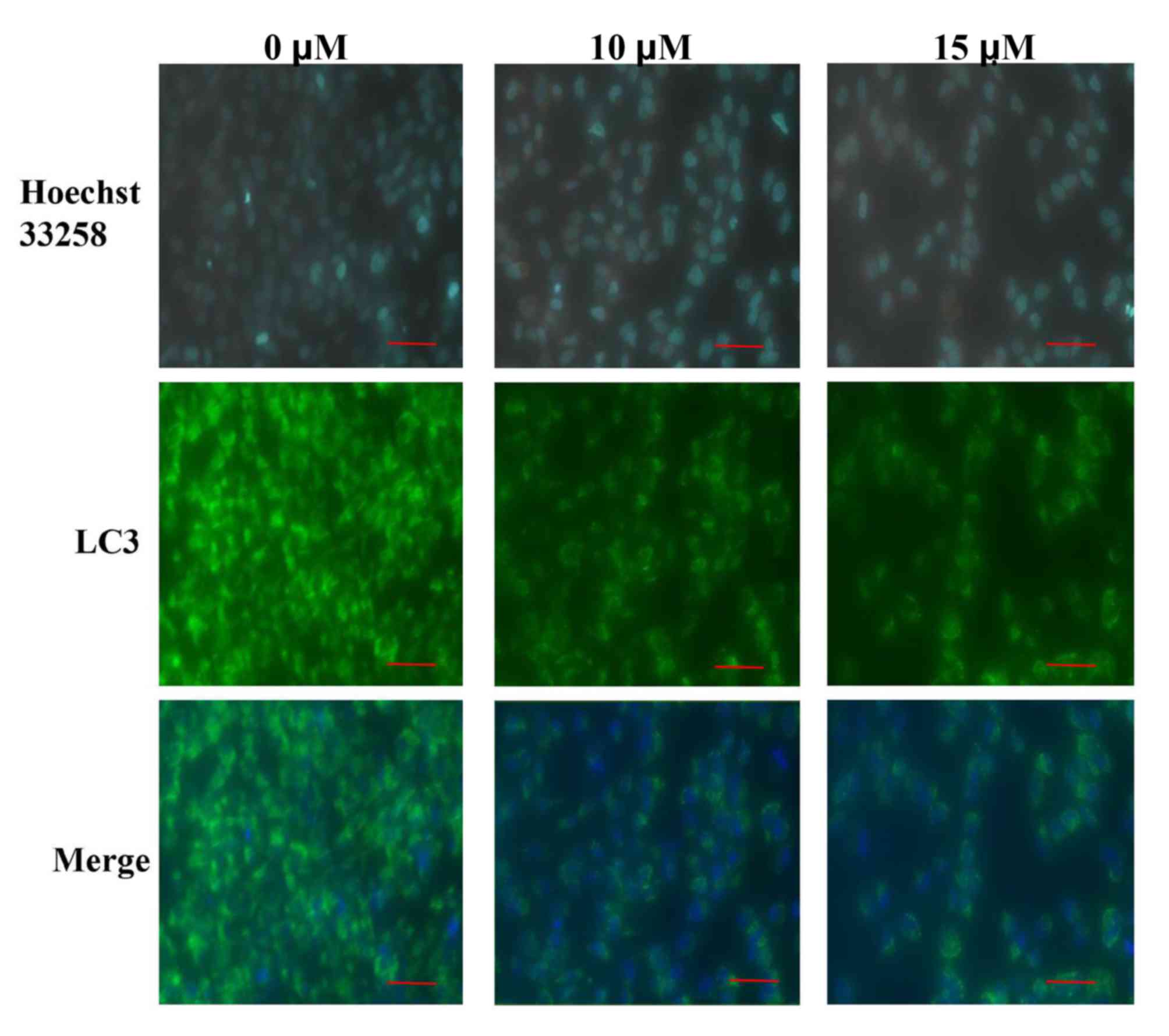
LC3 immunofluorescence
SW982 cells (5×104 cells/ml) were seeded
into 24-well plates and then incubated in a 5%
CO2-humidified atmosphere at 37°C overnight. Following
overnight incubation, cells were treated with 0, 10 and 15 µM
sodium selenite for 24 h at 37°C. Subsequently, the medium was
removed, cells were washed twice with PBS and then fixed with 3.7%
paraformaldehyde and treated with 0.2% Triton X-100 to permeabilize
for 30 min on ice. Following blocking with 2% bovine serum albumin
for 1 h at room temperature, cells were subsequently incubated with
LC3 antibodies [1:200 dilution with phosphate buffered saline with
0.1% Tween-20 (PBST)] for 2 h, and then incubated withfluorescein
isothiocyanate-conjugated immunoglobulin G (IgG; 1:100) with PBST
for 1 h at room temperature. Following this, cells were incubated
with Hoechst 33258 solution (10 µg/ml) for 15 min at room
temperature. A fluorescence microscope (Nikon Corporation) was used
to determine LC3 immunofluorescence (magnification, ×200). A total
of 5 fields of vision/per well were investigated.
Western blotting analysis
Following the various treatments and incubation in a
5% CO2-humidified atmosphere at 37°C, the medium was
removed and SW982 cells were washed twice with cold PBS and
solubilized in Triton lysis buffer (50 mM Tris-HCl, pH 7.4; 150 mM
NaCl; 0.2 mM EDTA; 1% Triton X-100; 1% sodium deoxycholate and 0.1%
SDS) and protease inhibitor cocktail (Beyotime Institute of
Biotechnology, Shanghai, China) on ice. Protein concentrations were
determined using the Bicinchoninic Acid assay (Thermo Fisher
Scientific, Inc.). The amount of supernatant loaded into each well
was calculated according to the protein concentrations. Either 10%
or 12% SDS-PAGE was prepared for western blotting analysis, and
each well was loaded with 20 µg of protein. Proteins were
transferred onto an Immoblin-P nitrocellulose membrane (EMD
Millipore, Billerica, MA, USA), blocked in 10% non-fat milk in
Tris-buffered saline + 0.1% Tween-20 (TBST) for 2 h at room
temperature and then incubated overnight at 4°C with the following
primary antibodies: Anti-Bax, 1:600; anti-Bcl-2, 1:100;
anti-pro-caspase-3, 1:200; anti-cleaved-caspase-3, 1:200;
anti-PARPp85, 1:200; anti-P62, 1:2,000; anti-Beclin-1, 1:500;
anti-LC3-II, 1:500; and anti-β-actin, 1:750. Membranes were washed
with TBST buffer and reacted with the appropriate horseradish
peroxidase-conjugated secondary antibodies (goat anti-rabbit IgG;
1:10,000; Thermo Fisher Scientific, Inc.) for 1 h at room
temperature. Following incubation with the secondary antibodies,
the membranes were washed thrice with TBST and once with TBS and
developed using an enhanced chemiluminescence kit (Thermo Fisher
Scientific, Inc.) and a GeneGnome 5 western blotting detection
system (Synoptics Ltd., Cambridge, UK). β-actin was used as the
internal control and for the normalization of protein expression,
and densitometric analysis was performed using Image J2 software
(National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Data are presented as the mean ± standard deviation;
experiments were repeated thrice (n=3). All statistical analyses of
the experimental data were performed using GraphPad Prism 5.0
statistical software (GraphPad Software, La Jolla, CA, USA).
Two-tailed Student's t-test was used to determine significant
differences in the normal distribution of the data of the two
groups. Data for multiple variable comparisons were analyzed by
one-way analysis of variance or the Kruskal-Wallis test, which were
then followed by the Bonferroni post-hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
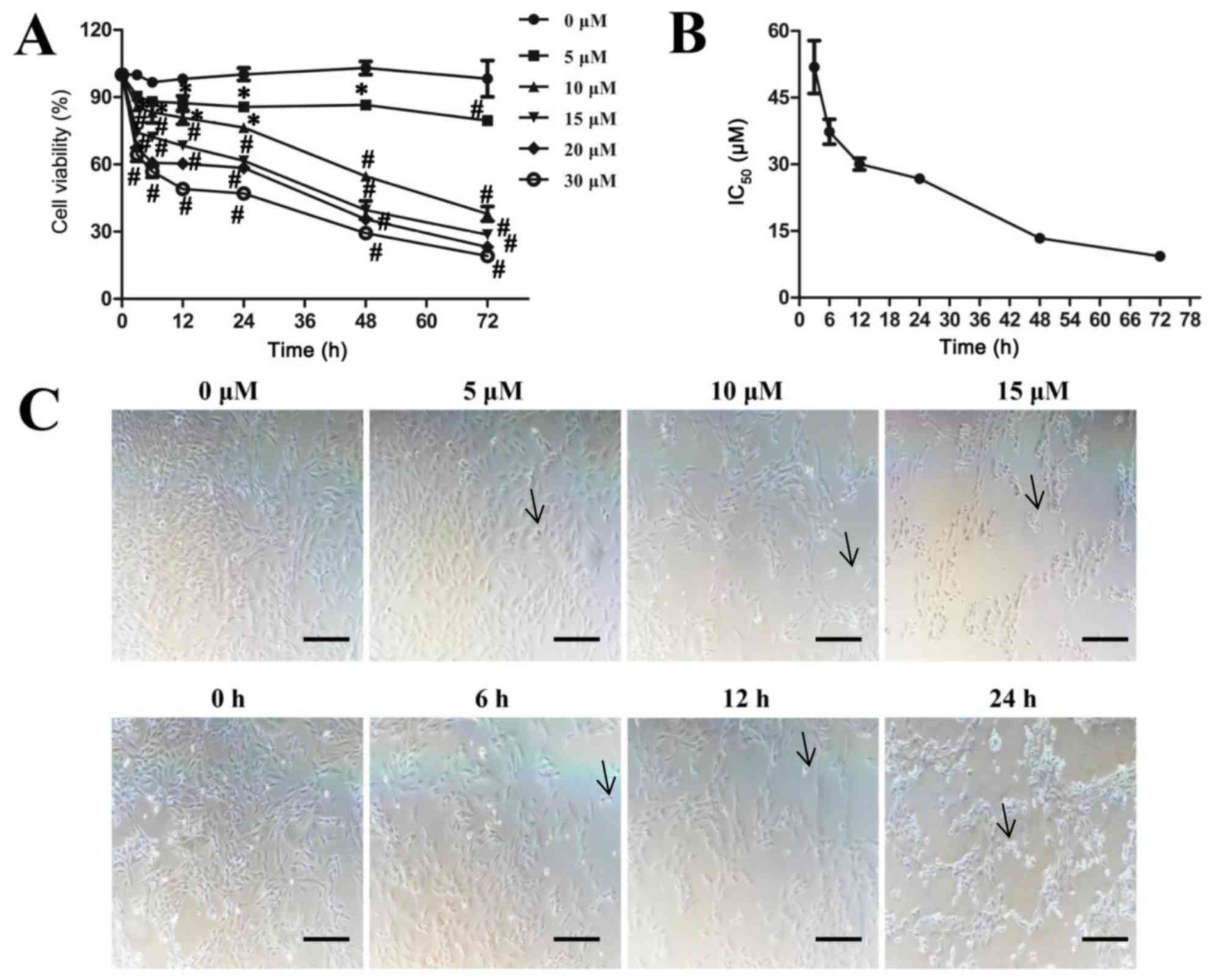
Sodium selenite reduces cell viability
in SW982 human synovial sarcoma cells
The effects of sodium selenite on SW982 cell
viability were examined with the MTT assay. SW982 cells were
treated with sodium selenite at concentrations ranging between 0
and 30 µM, which included both nutritious and toxic doses, for 0 to
72 h. The results indicated that sodium selenite significantly
inhibited SW982 cell viability in a time- and dose-dependent manner
(Fig. 1A). The IC50
values of 51.9±5.9 µM at 3 h, IC50 values of 37.3±2.8 µM
at 6 h, IC50 values of 30.1±1.3 µM at 12 h,
IC50 values of 26.8±1.0 µM at 24 h, IC50
values of 13.4±0.4 µM at 48 h and IC50 values of 9.3±0.4
µM at 72 h of treatment (Fig. 1B).
Significant inhibitory effects were observed with 5 µM or greater
sodium selenite treatment for 12 h; similarly, ≥10 µM sodium
selenite treatment for 3 h significantly inhibited cell viability.
Furthermore, toxic doses (>17.7 µM) of sodium selenite (24) also significantly inhibited cell
viability (Fig. 1A). In addition,
morphological changes in SW982 cells exposed to sodium selenite
were observed: The cells shrunk, retracted from neighboring cells,
lost their flat and polygonal shape and ultimately detached from
the culture dish (arrow); thus suggesting that cell death was
induced by sodium selenite (Fig.
1C). Following treatment with sodium selenite, the cell density
was significantly decreased in a dose and time-dependent manner.
These results indicated that sodium selenite inhibits cell
viability in SW982 cells.
Sodium selenite induces apoptosis in
SW982 cells by regulating expression of Casp-3, PARPp85 and
Bcl-2/Bax proteins
The effects of sodium selenite on apoptosis in SW982
cells were examined by flow cytometry. The results indicated that
sodium selenite treatment induced apoptosis in a time- and
dose-dependent manner (Fig. 2A-C).
To determine the mechanisms by which sodium selenite induced
apoptosis, western blotting assays were used to analyze the
expression levels of apoptosis-related proteins. The results
demonstrated that sodium selenite treatment significantly reduced
the expression of the anti-apoptotic protein Bcl-2 and pro-Casp-3
protein, and increased the expression of the pro-apoptotic protein
Bax, cleaved-Casp-3 and PARPp85 in a time- and dose-dependent
manner (Fig. 2D-F). These results
revealed that sodium selenite induced apoptosis in SW982 cells
through the activation of Casp-3, PARPp85, Bcl-2 and Bax
proteins.
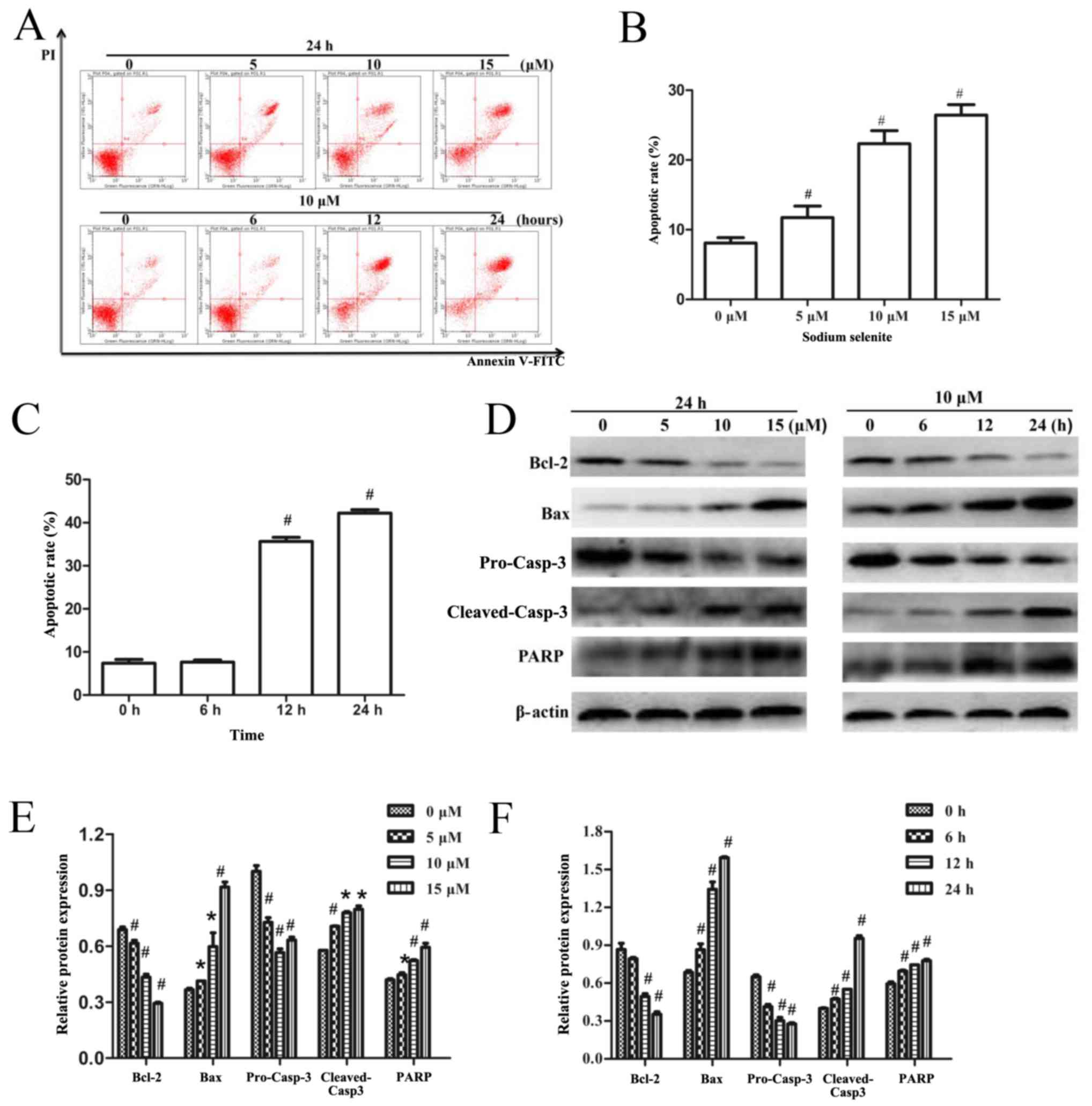 | Figure 2.Sodium selenite induces apoptosis in
SW982 cells via regulation of Casp-3, PARPp85, Bcl-2 and Bax
protein expression. (A) Cells were treated with various
concentrations (0, 5, 10 and 15 µM) of sodium selenite for 24 h or
with 10 µM sodium selenite for 0, 6, 12 and 24 h. Apoptotic rates
were examined using an Annexin V-FITC/PI double-stain assay.
Annexin V-FITC-positive and PI-negative stained cells indicate
early apoptosis (lower right quadrant), Annexin V-FITC-positive and
PI-positive double-stained cells indicate late apoptosis (upper
right quadrant), and Annexin V-FITC-negative and PI-positive
stained cells represent dead cells (upper left quadrant). (B and C)
Apoptotic rates of SW982 cells analyzed by flow cytometry following
treatment with either (B) various concentrations of sodium selenite
or (C) various incubation times with 10 µM sodium selenite. (D)
Cell lysates were subjected to western blotting to determine the
levels of apoptosis-related proteins Bcl-2, Bax, pro-Casp-3,
cleaved-Casp-3 and PARP; β-actin was used as an internal control
and to normalize the expression data. (E and F) Densitometric
analysis of the apoptosis-related proteins from Part D. Data are
presented as the mean ± standard deviation of at least three
independent experiments; *P<0.05 and #P<0.01 vs.
untreated control (0 µM or 0 h). Bax, Bcl-2-associated X protein;
Casp, caspase; FITC, fluorescein isothiocyanate; PARP, poly
(ADP-ribose) polymerase; PI, propidium iodide. |
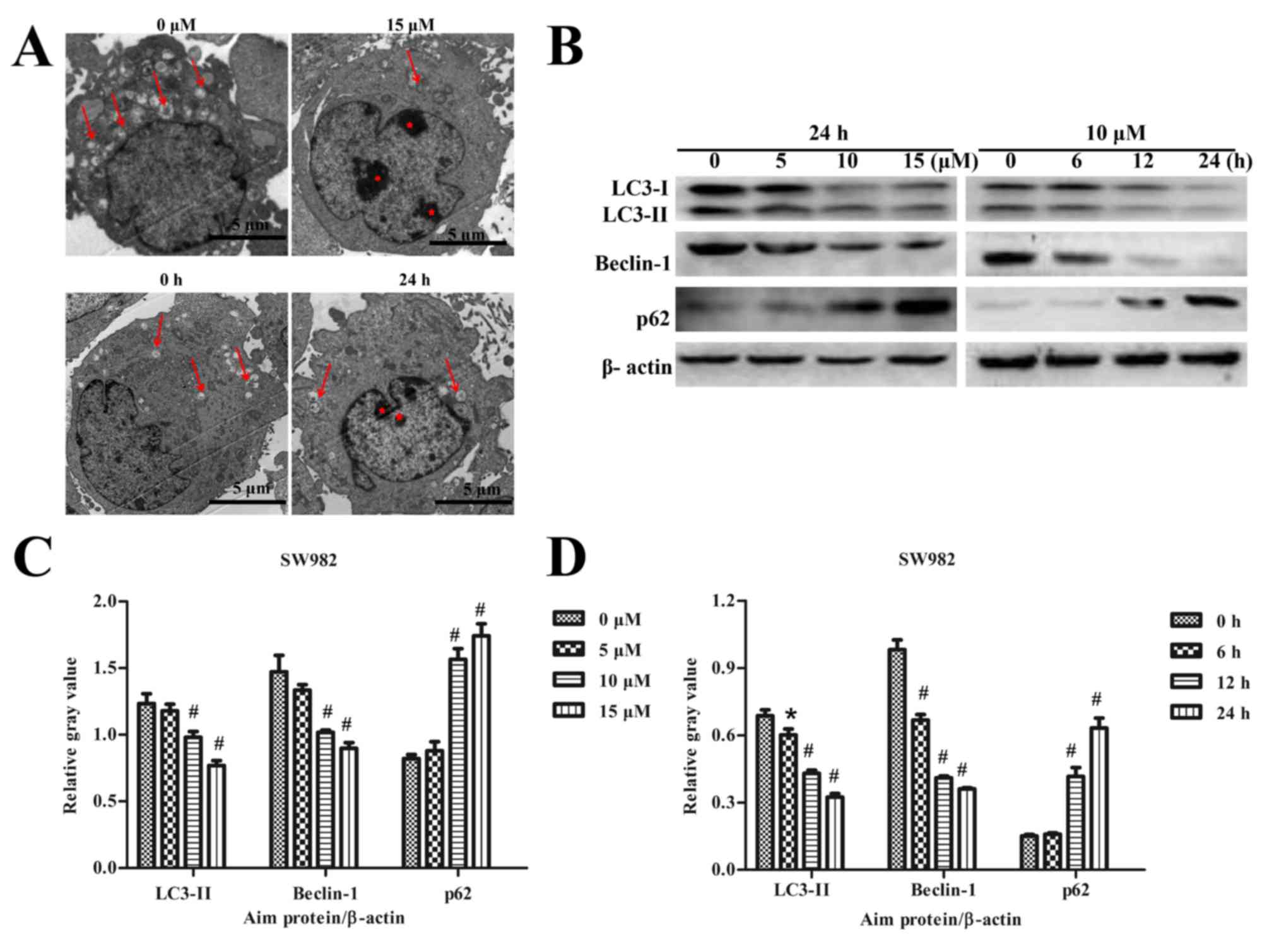
Sodium selenite inhibits autophagy in
SW982 cells
The effects of sodium selenite on autophagy in SW982
cells were also examined. TEM results revealed that a higher number
of autophagic vesicles formed in the untreated (0 µM) control group
compared with the number of autophagy vesicles that formed
following treatment with 15 µM sodium selenite (Fig. 1A). Cells treated with 10 µM sodium
selenite for 24 h also exhibited reduced formation of autophagic
vesicles compared with the control group at 0 h. The TEM results
suggested that sodium selenite inhibited autophagy in SW982 cells
(Fig. 3A).
Expression of the autophagy-related proteins LC3,
Beclin-1 and p62 were examined by western blotting. The results
revealed a significant decrease in the protein expression levels of
LC3-II and Beclin-1, whereas the level of p62 expression was
significantly increased in a dose- and time-dependent manner,
compared with the respective untreated controls (Fig. 3B-D). Furthermore, LC3
immunofluorescence experiments were conducted to further verify the
effects of sodium selenite on SW982 cell autophagy. The results
demonstrated an enhanced expression of LC3 immunofluorescence
visible in the untreated (0 µM) control group compared with the
groups treated with 10 and 15 µM sodium selenite (Fig. 4). LC3 fluorescence demonstrated
that the expression of LC3 was decreased following treatment with
sodium selenite (Fig. 4). These
results confirmed that sodium selenite treatment inhibited
autophagy in SW982 cells.
Inhibition of autophagy enhances sodium
selenite-induced apoptosis in SW982 cells. Based on the
aforementioned results, the present study aimed to verify that
sodium selenite induces apoptosis and inhibits autophagy in SW982
cells following treatment with sodium selenite and to determine the
inter-relationship between apoptosis and autophagy. Therefore,
cells were incubated with 3-MA, an inhibitor of autophagy. TEM
results indicated that 3-MA inhibited autophagy in SW982 cells
(Fig. 5A). Western blotting assays
also confirmed that 3-MA inhibited autophagy by altering the
expression of LC3-II (Fig. 5B and
C). Thus, we clearly demonstrated that autophagy was inhibited
by 3-MA. In addition, western blotting demonstrated that sodium
selenite treatment in combination with 3-MA resulted in a
significant decrease in the expression of anti-apoptotic protein
Bcl-2 and significant increase in the expression of pro-apoptotic
protein Bax compared with cells treated with sodium selenite alone
(Fig. 5B, D and E).
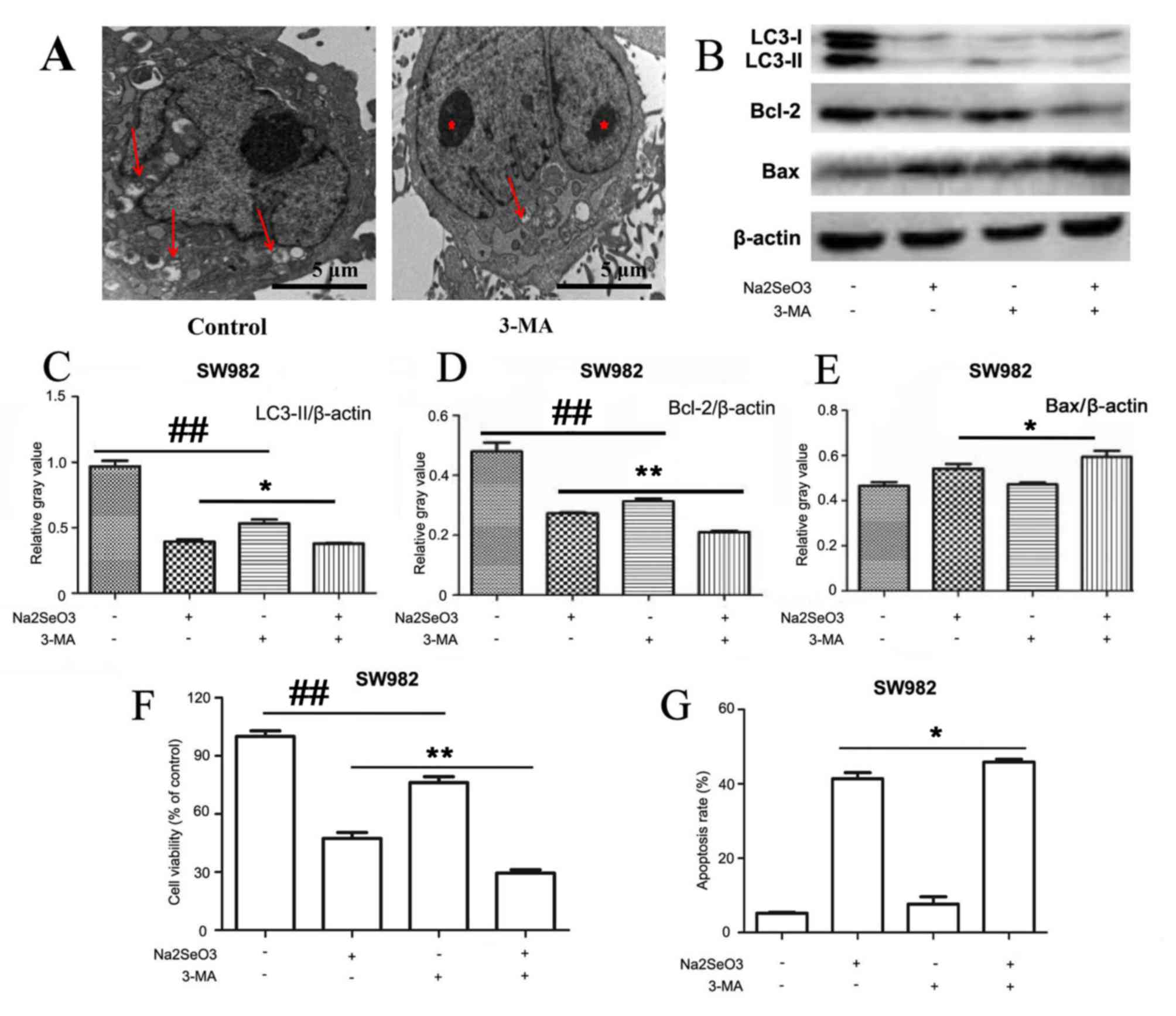 | Figure 5.Inhibition of autophagy enhances
sodium selenite-induced apoptosis in SW982 cells. SW982 cells were
pretreated, 3-MA (5 mM), for 2 h and subsequently incubated with
sodium selenite (10 µM) for 24 h. (A) Autophagosomes (arrows) and
nuclear condensation (red asterisks) were examined by transmission
electron microscopy; scale bar, 5 µm. (B) Cell lysates were
subjected to western blotting to examine the expression levels of
autophagy-related protein LC3 and apoptosis-related proteins Bcl-2
and Bax; β-actin was used as an internal control. Densitometric
analysis of (C) LC3-II, (D) Bcl-2 and (E) Bax expression in SW982
cells treated with sodium selenite (10 µM) in the absence or
presence of 3-MA (5 mM). (F) Cell viability was determined by using
the MTT assay. (G) Apoptotic rates were measured by flow cytometry.
Data are presented as the mean ± standard deviation of at least
three independent experiments; *P<0.05 and **P<0.01,
Na2SeO3 + 3-MA treated cells vs.
Na2SeO3 treated cells;
##P<0.01, untreated cells vs. 3-MA treated cells.
3-MA, 3-methyladenine; Bax, Bcl-2-associated X protein; LC3,
microtubule-associated protein 1 light chain 3;
Na2SeO3, sodium selenite. |
MTT experiments revealed that cell viability was
significantly reduced when cells were co-treated with sodium
selenite and 3-MA compared with treatment with sodium selenite
alone (Fig. 5F). Furthermore, flow
cytometric analyses revealed that cells treated with sodium
selenite combined with 3-MA exhibited a significantly increased
apoptotic rate compared with treatment with sodium selenite alone
(Fig. 5G).
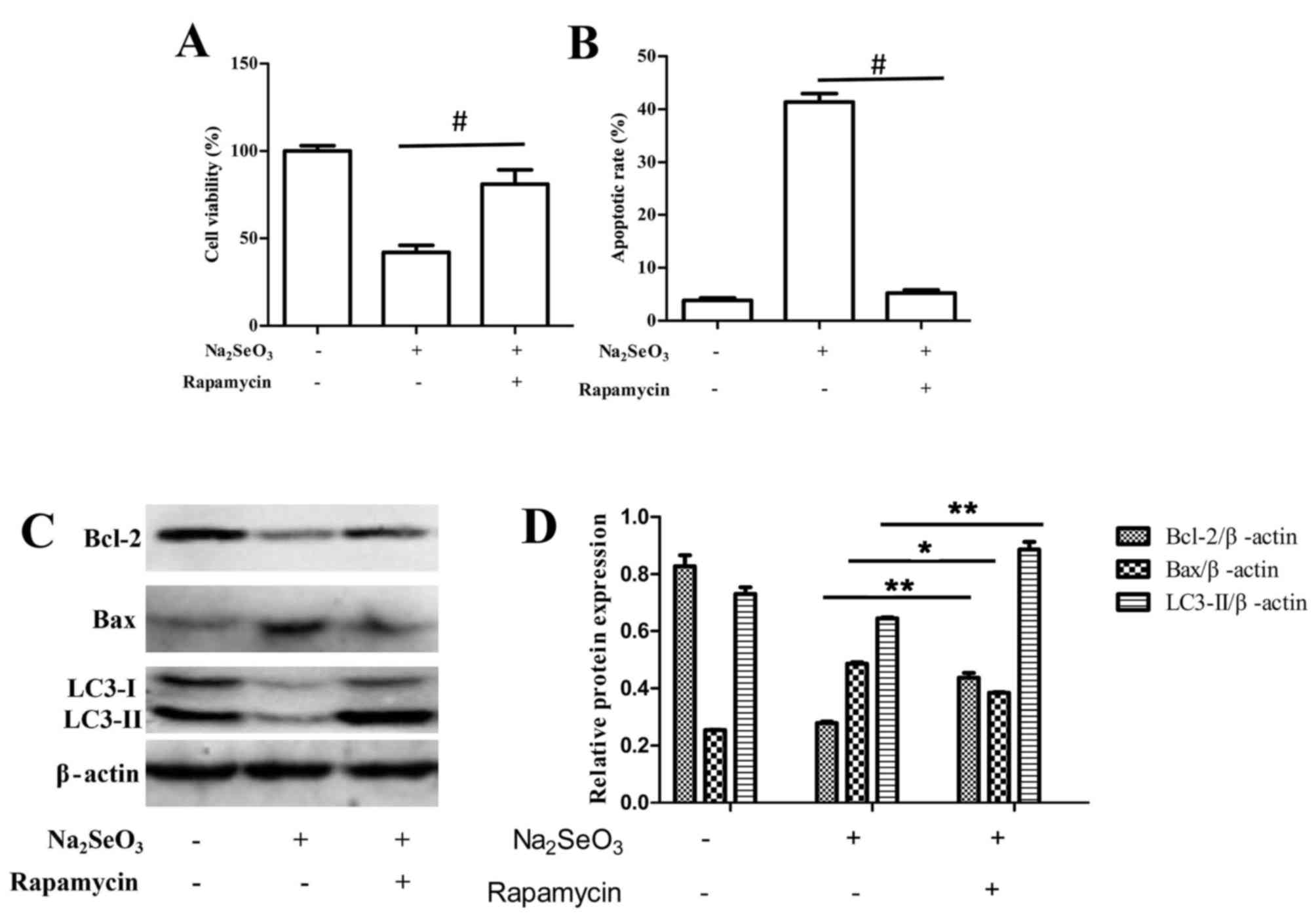
It was also demonstrated that upon stimulation of
autophagy by rapamycin treatment, selenite-mediated cytotoxicity
and apoptosis was significantly reduced compared with untreated
cells (Fig. 6). Following
treatment with sodium selenite+ 100 nM rapamycin, the induction of
autophagy significantly promoted cell viability and decreased the
rate of apoptosis compared with sodium selenite treatment alone
(Fig. 6A and B, respectively).
Western blotting results also indicated that sodium selenite+ 100
nM rapamycin notably increased the expression of anti-apoptotic
protein Bcl-2 and decreased the expression of pro-apoptotic protein
Bax compared with cells treated with sodium selenite alone
(Fig. 6C and D).
Overall, these results indicated that suppression of
autophagy may enhance sodium selenite-induced apoptosis in SW982
cells; therefore, autophagy may protect cells from death by
antagonizing sodium selenite-induced apoptosis in SW982 cells.
Discussion
The effects of selenium on human synovial sarcoma
have not been clearly elucidated. Given the antitumor effects of
selenium, the present study was particularly interested in the
effects of sodium selenite, an inorganic selenium compound, on the
human synovial sarcoma cell line SW982. It was hypothesized that
sodium selenite inhibited cell viability and induced apoptosis in
SW982 cells; therefore the effects of sodium selenite exposure on
cell viability were analyzed. The results demonstrated that sodium
selenite significantly inhibited SW982 cell viability. Apoptosis
serves a crucial role in a number of pathophysiological processes
(15). Flow cytometry was used to
observe that sodium selenite treatment significantly increased
SW982 cell apoptosis via activation of Casp-3, PARPp85, Bcl-2 and
Bax proteins. However, further studies are needed to determine the
underlying mechanisms behind apoptosis. Autophagy is a cellular
degradation pathway for the clearance of damaged or superfluous
protein and organelles, and occurs in many tumor cells (19). Autophagy serves as a mechanism of
cell survival by favoring stress adaptation over cell death
(19). Excessive self-digestion
and degradation of essential cellular components may induce what is
known as autophagic cell death (19). The results of the present study
demonstrated that sodium selenite inhibited autophagy in SW982
cells in vitro. The respective roles and interplay between
apoptosis and autophagy are complex and may vary in different cells
and in the context of various stress types. Previous observations
indicated that autophagy serves a role in preventing apoptosis
(22), whereas other studies
reported that active autophagy increased apoptosis (23,24).
Several previous studies indicated that sodium selenite enhances,
not suppresses, autophagic activity in certain types of cancer
cells (10,21). Thus, it is difficult to draw a
conclusion regarding the effects of selenium on tumor cell
autophagy. The mechanisms behind this observation are complex and
require further investigation. The respective roles and the
interplay between apoptosis and autophagy are complicated and are
likely to vary in different cells and in response to different
stressors. In the present study, autophagy was suppressed in sodium
selenite-treated SW982 cells by co-treating cells with 3-MA, which
significantly increased the apoptotic rates. In addition, apoptosis
was downregulated when sodium selenite was combined with rapamycin,
an inducer of autophagy. In summary, the data suggested that
autophagy may protect SW982 cells from death by antagonizing sodium
selenite-induced apoptosis; therefore, autophagy may play a
protective role in SW982 cells.
The Akt/mammalian target of rapamycin (mTOR)
signaling pathway is initiated by ligand-activated receptor
tyrosine kinases on the plasma membrane, such as insulin-like
growth factor 1 receptor and platelet-derived growth factor
receptors, which recruit phosphatidylinositol 3-kinase (PI3K)
directly to the receptor (29,30).
PI3K converts phosphatidylinositol 4,5-bisphosphate to
phosphatidylinositol 3,4,5-triphosphate, which subsequently
recruits Akt to the membrane, where it is activated and facilitates
the downstream activation of mTOR (29). Akt/mTOR activation suppresses
autophagy in mammalian cells, which suggested that inactivation of
Akt/mTOR may promote autophagy (31). In addition, a previous study
reported that the Akt/mTOR pathway positively regulates autophagy
(32). A number of other previous
studies indicated that sodium selenite exposure enhances autophagic
activity in certain types of cancer cells (10,21).
Given the dual function of Akt/mTOR pathway in autophagy, the
present study hypothesizes that the Akt/mTOR pathway may be
involved in the inhibition of sodium selenite-induced autophagy in
SW982 synovial sarcoma cells. A previous study using animal models
with activated Akt demonstrated that oncogenesis through this
pathway was dependent upon downstream activation of mTOR (33). Akt/mTOR pathway activation is
associated with sarcoma oncogenesis through a number of mechanisms,
such as: Mast/stem cell growth factor receptor Kit and
platelet-derived growth receptor-α mutations in gastrointestinal
stromal tumours; phosphatidylinositol 4,5-bisphosphate 3-kinase
catalytic subunit-α isoform mutations in myxoid/round-cell
liposarcomas; or other pathognomonic alterations that promote
reliance upon the pathway, such as the dependence of RNA binding
protein EWS-friend leukemia integration 1 transcription factor gene
fusion-driven oncogenesis upon the IGF-1 receptor in Ewing sarcoma
(29). Faghiri and Bazan (34) demonstrated that in single-dose
oxidative stress-induced apoptosis, phosphorylation of Akt, mTOR,
and p70S6K is both time- and dose-dependent. Furthermore, it has
been demonstrated that wortmannin (a PI3K inhibitor that functions
upstream of Akt) and rapamycin (mTOR inhibitor that functions
upstream in the mTOR/p70S6K pathway) inhibit the PI3K and
mTOR/p70S6K pathways, respectively, increase the apoptosis of human
retinal pigment epithelial cells, and inhibits the phosphorylation
of Akt and p70S6K otherwise induced by single-dose oxidative stress
(34). Thus, the Akt/mTOR pathway
maybe associated with both apoptosis and autophagy and may serve an
important role in sodium selenite-induced apoptosis and autophagy
suppression in SW982 cells. However, the underlying mechanisms
require further research.
The characteristics of synovial sarcoma cells
significantly differ from one cell line to another. Therefore, the
results obtained from a single cell line may not always be
interpreted as representing the biology of synovial sarcoma. In
this regard, at least 3–4 different cell lines should be used to
verify that sodium selenite may serve as a potential adjuvant agent
for the treatment of synovial sarcoma. Over the course of this
study, additional cell lines could not be obtained, as only one
synovial sarcoma cell line exists in China. Future studies should
use an alternative cell line of synovial sarcoma to verify the
present study results.
Acknowledgements
Not applicable.
Funding
This study was supported by The National Natural
Science Foundations of China (grant nos. 81271948 and
81601877).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
LY, YsC, KX and PX conceived and designed the
experiments. LY and YsC performed the experiments. LY, JlZ, YL, XW,
JS and SmL analyzed the data. LY wrote the manuscript. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Reference
|
1
|
Minami Y, Kohsaka S, Tsuda M, Yachi K,
Hatori N, Tanino M, Kimura T, Nishihara H, Minami A, Iwasaki N and
Tanaka S: SS18-SSX-regulated miR-17 promotes tumor growth of
synovial sarcoma by inhibiting p21WAF1/CIP1. Cancer Sci.
105:1152–1159. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ito T, Ouchida M, Morimoto Y, Yoshida A,
Jitsumori Y, Ozaki T, Sonobe H, Inoue H and Shimizu K: Significant
growth suppression of synovial sarcomas by the histone deacetylase
inhibitor FK228 in vitro and in vivo. Cancer Lett.
224:311–319. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sun Y, Wang H, Lin F, Hua J and Zhou G:
Inhibition of proliferation and gene expression regulation by
(−)-epigallocatechin-3-gallate in human synovial sarcoma cells. Med
Oncol. 28:1463–1468. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Haldar M, Randall RL and Capecchi MR:
Synovial sarcoma: From genetics to genetic-based animal modeling.
Clin Orthop Relat Res. 466:2156–2167. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Eilber FC, Brennan MF, Eilber FR, Eckardt
JJ, Grobmyer SR, Riedel E, Forscher C, Maki RG and Singer S:
Chemotherapy is associated with improved survival in adult patients
with primary extremity synovial sarcoma. Ann Surg. 246:105–113.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cui Z, Li C, Li X, Zhang Q, Zhang Y, Shao
J and Zhou K: Sodium selenite (Na2SeO3)
induces apoptosis through the mitochondrial pathway in CNE-2
nasopharyngeal carcinoma cells. Int J Oncol. 46:2506–2514. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rayman MP: Selenium and human health.
Lancet. 379:1256–1268. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fairweather-Tait SJ, Bao Y, Broadley MR,
Collings R, Ford D, Hesketh JE and Hurst R: Selenium in human
health and disease. Antioxid Redox Signal. 14:1337–1383. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Klein EA: Selenium: Epidemiology and basic
science. J Urol. 171:S50–S53. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Park SH, Kim JH, Chi GY, Kim GY, Chang YC,
Moon SK, Nam SW, Kim WJ, Yoo YH and Choi YH: Induction of apoptosis
and autophagy by sodium selenite in A549 human lung carcinoma cells
through generation of reactive oxygen species. Toxicol Lett.
212:252–261. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li Z, Meng J, Xu TJ, Qin XY and Zhou XD:
Sodium selenite induces apoptosis in colon cancer cells via
Bax-dependent mitochondrial pathway. Eur Rev Med Pharmacol Sci.
17:2166–2171. 2013.PubMed/NCBI
|
|
12
|
Li Z, Shi K, Guan L, Jiang Q, Yang Y and
Xu C: Activation of p53 by sodium selenite switched human leukemia
NB4 cells from autophagy to apoptosis. Oncol Res. 21:325–331. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Brodin O, Eksborg S, Wallenberg M,
Asker-Hagelberg C, Larsen EH, Mohlkert D, Lenneby-Helleday C,
Jacobsson H, Linder S, Misra S and Björnstedt M: Pharmacokinetics
and toxicity of sodium selenite in the treatment of patients with
carcinoma in a phase I clinical trial: The SECAR study. Nutrients.
7:4978–4994. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gao H, Sun W, Zhao W, Hao W, Leung CH, Lu
J and Chen X: Total tanshinones-induced apoptosis and autophagy via
reactive oxygen species in lung cancer 95D cells. Am J Chin Med.
43:1265–1279. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kontos CK, Christodoulou MI and Scorilas
A: Apoptosis-related BCL2-family members: Key players in
chemotherapy. Anticancer Agents Med Chem. 14:353–374. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hirchaud F, Hermetet F, Ablise M,
Fauconnet S, Vuitton DA, Prétet JL and Mougin C: Isoliquiritigenin
induces caspase-dependent apoptosis via downregulation of HPV16 E6
expression in cervical cancer Ca Ski cells. Planta Med.
79:1628–1635. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
You L, Shou J, Deng D, Jiang L, Jing Z,
Yao J, Li H, Xie J, Wang Z, Pan Q, et al: Crizotinib induces
autophagy through inhibition of the STAT3 pathway in multiple lung
cancer cell lines. Oncotarget. 6:40268–40282. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu K, Cai YS, Lu SM, Li XL, Liu L, Li Z,
Liu H and Xu P: Autophagy induction contributes to the resistance
to methotrexate treatment in rheumatoid arthritis fibroblast-like
synovial cells through high mobility group box chromosomal protein
1. Arthritis Res Ther. 17:3742015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Klionsky DJ, Abeliovich H, Agostinis P,
Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA,
Ballabio A, et al: Guidelines for the use and interpretation of
assays for monitoring autophagy in higher eukaryotes. Autophagy.
4:151–175. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ren Y, Huang F, Liu Y, Yang Y, Jiang Q and
Xu C: Autophagy inhibition through PI3K/Akt increases apoptosis by
sodium selenite in NB4 cells. BMB Rep. 42:599–604. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Králová V, Benešová S, Cervinka M and
Rudolf E: Selenite-induced apoptosis and autophagy in colon cancer
cells. Toxicol In Vitro. 26:258–268. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tsai JP, Lee CH, Ying TH, Lin CL, Lin CL,
Hsueh JT and Hsieh YH: Licochalcone A induces autophagy through
PI3K/Akt/mTOR inactivation and autophagy suppression enhances
Licochalcone A-induced apoptosis of human cervical cancer cells.
Oncotarget. 6:28851–28866. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Amaravadi RK, Yu D, Lum JJ, Bui T,
Christophorou MA, Evan GI, Thomas-Tikhonenko A and Thompson CB:
Autophagy inhibition enhances therapy-induced apoptosis in a
Myc-induced model of lymphoma. J Clin Invest. 117:326–336. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Singh BN, Kumar D, Shankar S and
Srivastava RK: Rottlerin induces autophagy which leads to apoptotic
cell death through inhibition of PI3K/Akt/mTOR pathway in human
pancreatic cancer stem cells. Biochem Pharmacol. 84:1154–1163.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Allan LA and Clarke PR: Apoptosis and
autophagy: Regulation of caspase-9 by phosphorylation. FEBS J.
276:6063–6073. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sutter ME, Thomas JD, Brown J and Morgan
B: Selenium toxicity: A case of selenosis caused by a nutritional
supplement. Ann Intern Med. 148:970–971. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nuttall KL: Evaluating selenium poisoning.
Ann Clin Lab Sci. 36:409–420. 2006.PubMed/NCBI
|
|
28
|
Johnson CC, Fordyce FM and Rayman MP:
Symposium on ‘Geographical and geological influences on nutrition’:
Factors controlling the distribution of selenium in the environment
and their impact on health and nutrition. Proc Nutr Soc. 69:pp.
119–132. 2010; View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ho AL, Vasudeva SD, Laé M, Saito T,
Barbashina V, Antonescu CR, Ladanyi M and Schwartz GK: PDGF
receptor alpha is an alternative mediator of rapamycin-induced Akt
activation: Implications for combination targeted therapy of
synovial sarcoma. Cancer Res. 72:4515–4525. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hay N and Sonenberg N: Upstream and
downstream of mTOR. Genes Dev. 18:1926–1945. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang J, Zhao D, Xie Z and Qi Y:
Down-regulation of AKT combined with radiation-induced autophagy
and apoptosis roles in MCF-7 cells. Biomed Mater Eng. 26 Suppl
1:S2259–S2265. 2015.PubMed/NCBI
|
|
32
|
Zeng X and Kinsella TJ: Mammalian target
of rapamycin and S6 kinase 1 positively regulate
6-thioguanine-induced autophagy. Cancer Res. 68:2384–2390. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Majumder PK, Febbo PG, Bikoff R, Berger R,
Xue Q, McMahon LM, Manola J, Brugarolas J, McDonnell TJ, Golub TR,
et al: mTOR inhibition reverses Akt-dependent prostate
intraepithelial neoplasia through regulation of apoptotic and
HIF-1-dependent pathways. Nat Med. 10:594–601. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Faghiri Z and Bazan NG: PI3K/Akt and
mTOR/p70S6K pathways mediate neuroprotectin D1-induced retinal
pigment epithelial cell survival during oxidative stress-induced
apoptosis. Exp Eye Res. 90:718–725. 2010. View Article : Google Scholar : PubMed/NCBI
|