Introduction
Hepatocellular carcinoma (HCC) is the fifth most
common cancer in men and the seventh in women, and the second or
third most common cause of cancer-associated mortalities worldwide
(1–3). However, the molecular pathogenesis of
HCC is not fully understood. It is well known that during the
process of tumor development, there are numerous levels of gene
transcriptional regulation initiate and promote the process of
tumor development and progression. Hence, improved understanding of
the underlying molecular processes of tumor initiation and the
identification of key molecular events for transcriptional
regulation that promotes tumor initiation and growth are of great
significance in developing therapeutic strategies.
Genome sequencing research has revealed that the
human genome is comprised of <2% protein-coding genes and
>90% of the genome is transcribed as non-coding RNA (ncRNA)
(4). Noncoding RNAs, an example of
which includes microRNAs (miRNAs), are non-coding RNAs which are
18–25 nucleotides in length, and long non-coding RNAs (lncRNAs)
which are defined as transcripts containing >200 nucleotides
that have a critical role in tumor occurrence and progression. It
has been demonstrated that miRNAs are involved in every type of
cancer examined to date, and the effects of miRNAs are mediated by
binding to target mRNAs or lncRNAs, either to suppress mRNA
translation or to degrade the miRNA-bound mRNA or lncRNA (5). Emerging evidence suggests that
lncRNAs exhibit various critical roles in global gene regulation,
including roles as the decoy, guide and in scaffolding (6,7). It
has previously been demonstrated that miRNAs and lncRNAs are
involved in tumorigenesis, acting either as oncogenes or tumor
suppressors in HCC (8). However,
multiple avenues for feedback and interconnectivity among miRNAs,
lncRNAs and mRNAs in HCC may potentially engender emergent
cooperative behavior.
With the emergence of microarray technologies
(9), the characterization of the
mammal transcriptome may occur at an unprecedented resolution
(10). The National Center for
Biotechnology Information (NCBI) Gene Expression Omnibus (GEO;
www.ncbi.nlm.nih.gov/geo/) provides the
largest public repository of microarray data in existence (11,12).
Therefore, effective integration of HCC GEO datasets may lead to
identification of differentially expressed lncRNAs, miRNAs and
mRNAs at transcription level, which provide a better research means
for tumor diagnosis, treatment and prognosis. There are numerous
HCC-associated lncRNA, miRNA and mRNA microarray data; however,
there are few studies on the integrative analysis of GEO datasets
in transcriptional regulation associated with HCC. Therefore,
understanding the potential regulation of lncRNAs, miRNAs and mRNAs
expression is critical for revealing novel therapeutic targets and
prognostic factors in management of HCC (13,14).
In the present study, differentially expressed
lncRNAs, miRNAs and mRNAs that promote tumor initiation and growth
were identified. The regulatory networks, between mRNA, miRNA and
lncRNA were constructed, in addition to the co-expression network
of mRNA-lncRNA. The key features of miRNAs regulatory effects on
lncRNAs were annotated and validated in HCC specimens.
Materials and methods
Cell culture
The normal human hepatocyte cell line (L02) and
human hepatocellular carcinoma cell lines (SMMC7721, Bel7404, Huh7
and PLC/PRF/5) were purchased from Cell Bank of Chinese Academy of
Sciences (Shanghai, China) and maintained in Dulbecco's modified
Eagle's medium (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 10% fetal bovine serum
(Invitrogen; Thermo Fisher Scientific, Inc.), 100 U/ml penicillin,
100 µg/ml streptomycin, and then cultured at 37°C in a humidified
atmosphere containing 5% CO2.
Patients and specimens
The experimental protocol was approved by the
Institutional Ethics Review Board of Daping Hospital, the Third
Military Medical University. Written informed consent was obtained
from all participants. A total of 10 patients with HCC were
recruited randomly at inpatient service of the Department of
Hepatobiliary Surgery, the Third Military Medical University
between 2013 and 2014. Patients underwent surgical HCC resection;
HCC and corresponding non-tumor liver tissues were collected. All
specimens were snap-frozen in liquid nitrogen or stored at −80°C.
The crystal HCC and non-tumor tissue sections (4 µm) were stained
with H&E: Sections were mounted on glass slides, fixed with
fixative (4% paraformaldehyde) for 20 min at room temperature,
air-dried for 15 min, stained in hematoxylin solution (Beyotime
Institute of Biotechnology, Haimen, China) for 2 min at 42°C, then
washed in running distilled water for 10 min. Subsequently,
sections were stained in 0.5% eosin solution (Beyotime Institute of
Biotechnology) for 1 min at 42°C, and then washed again,
dehydrated, and mounted at room temperature to ensure homogenous
cell population of tissues. Individual patients were excluded if
they received any chemotherapy and radiotherapy. Among those 10
patients, 7 were male and 3 were female. The age ranged from 35–72
years; 8 patients were serum positive for hepatitis B surface
antigen.
Selection of GEO datasets, processing
and construction of networks
The NCBI GEO (www.ncbi.nlm.nih.gov/geo) (11) was searched for expression profiling
studies on mRNAs, miRNAs and lncRNAs in HCC. Explicit search
strategies for three types of GEO datasets were applied to
preliminary selection. The strategy for mRNA datasets, [((((liver
cancer (Title)) OR Liver Neoplasms (MeSH Terms)) OR hepatocellular
carcinoma (Title)) AND Homo sapiens (Organism))] AND [(mRNA) OR
gene expression); for miRNA, (liver cancer (Title)) OR Liver
Neoplasms (MeSH Terms)) OR hepatocellular carcinoma (Title)) AND
Homo sapiens (Organism))] AND miRNA; for lncRNA, [((((liver cancer
(Title)) OR Liver Neoplasms (MeSH Terms)) OR hepatocellular
carcinoma (Title)) AND Homo sapiens (Organism))] AND lncRNA, with a
time limit until August 2015. The inclusion and exclusion criteria
included GEO datasets of RNA transcriptome analysis of human HCC
tissues from microarray in published articles; GEO datasets from
non-HCC tissue samples were excluded. A total of nine sets of GEO
microarray data were identified from the initial literature search
and manual search (Table I).
According to NCBI probe annotation files, gene expression levels
(average levels of the corresponding probes) were calculated. For
lncRNA genes, expression levels were calculated according to human
transcript sequences (refseq version) downloaded from UCSC
(www.genome.ucsc.edu/) (15) and lncRNAs probe sequence annotation
from NONCODE V4 (16).
Differentially expressed genes were screened through a matrix of
gene expression levels with the bioconductor limma R package
(17). The threshold of
differentially expressed genes screened was P<0.05, |fold
change|>1.5. Data were integrated according to the results of
the differentially expressed genes in the final screening. The
standards of screening differentially expressed genes suggest that
genes must be present and differentially expressed in at least two
datasets, and that the trend of expression alteration is
consistent. The target mRNA of miRNA was screened from microT-CDS
(18), miRanda (19), miRDB (20), PITA (21), TargetScan 6.2 (22), miRWalk (23) and miRecords (24), where miRWalk and miRecords are
included in the validated target genes. Screening for the miRNA
target gene involved the miRNA and the target gene appearing in the
validation database or in at least three prediction databases, and
the miRNAs and the target genes exhibiting differing expression
levels, with opposite expression alteration trends. The regulatory
network of miRNAs-mRNAs was constructed using Cytoscape software
(25). In addition, the target
genes for each miRNA were analyzed by Kyoto Encyclopedia of Genes
and Genomes (KEGG) (26)
enrichment (P<0.05, and at least two genes in the pathway). The
expression levels of lncRNA and mRNA in the three GEO datasets
(GSE58043, GSE55191, GSE27462) (27–29)
were first integrated, and then screened according to their
expression correlation (Pearson correlation), and screened with
|Pearson correlation coefficient| ≥0.7. The co-expression network
of mRNAs-lncRNAs was constructed using Cytoscape software (30) and the co-expressed mRNAs were
analyzed for KEGG enrichment analysis of each lncRNA. According to
the miRNA sequence and lncRNA sequence of NONCODE V4, miRanda
software was used to predict the interaction between miRNAs and
lncRNAs. The associated pairs were screened according to the miRNA
and lncRNA differing expressions and opposite trends, and the
networks were constructed using Cytoscape software.
 | Table I.Selected GEO datasets. |
Table I.
Selected GEO datasets.
| Microarray
types | GEO accession | Number of tumor
samples | Number of adjacent
non-tumor samples |
|---|
| mRNA
microarray | GSE25097 | 268 | 243 |
|
| GSE57957 | 39 | 39 |
|
| GSE22405 | 24 | 24 |
| miRNA
microarray | GSE31384 | 166 | 166 |
|
| GSE36915 | 68 | 21 |
|
| GSE10694 | 78 | 78 |
| lncRNA
microarray | GSE58043 |
7 |
7 |
|
| GSE55191 |
3 |
3 |
|
| GSE27462 |
5 |
5 |
Transfection of miRNA mimics
Cells were seeded in 6-well plates at a
concentration of 2×105 cells/well. When cells reached
40–60% confluence, 150 nM miRNA mimics (Guangzhou RiboBio Co.,
Ltd., Guangzhou, China), and negative control (NC) miRNA mimics
were transfected using Lipofectamine® 2000 reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol, for 24 h. The miRNA mimics and NC mimics
were synthesized by Guangzhou RiboBio Co. Ltd. and sequences are
listed in Table II.
| Table II.Mimics of miRNAs. |
Table II.
Mimics of miRNAs.
| miRNA mimic | Sense (5′-3′) | Anti-sense
(5′-3′) |
|---|
| Hsa-miR-101-3p
mimic (miR-101) |
UACAGUACUGUGAUAACUGAA |
CAGUUAUCACAGUACUGUAUU |
| Hsa-miR-125b-5p
mimic (miR-125b) |
UCCCUGAGACCCUAACUUGUGA |
ACAAGUUAGGGUCUCAGGGAUU |
| Hsa-miR-130a-3p
mimic (miR-130a) |
CAGUGCAAUGUUAAAAGGGCAU |
CCCUUUUAACAUUGCACUGUU |
| Hsa-miR-195-5p
mimic (miR-195) |
UAGCAGCACAGAAAUAUUGGC |
CAAUAUUUCUGUGCUGCUAUU |
| Hsa-miR-145-3p
mimic (miR-145) |
GGAUUCCUGGAAAUACUGUUCU |
AACAGUAUUUCCAGGAAUCCUU |
| Negative control
mimic |
UCACAACCUCCUAGAAAGAGUAGA |
UACUCUUUCUAGGAGGUUGUGAUU |
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from tissue samples and
cells using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocols. The
total RNA was quantified by using a Nanodrop ND-1000
spectrophotometer (NanoDrop Technologies; Thermo Fisher Scientific,
Inc.), and one microgram of RNA was used for cDNA synthesis using
GeneAmp RNA PCR kit (Applied Biosystems; Thermo Fisher Scientific,
Inc.). Gene expression was examined by SYBR-Green PCR Master mix
(Applied Biosystems; Thermo Fisher Scientific, Inc.) using the
primers (as listed in Table III)
with Bio-Rad CFX96 qPCR system. The detection run started at 50°C
for 2 min, 95°C for 2 min, followed by 45 cycles at 95°C for 15 sec
and at 60°C for 1 min. The data were analyzed using the
2ΔCt method or 2−ΔΔCq method (31). The lncRNA expression levels were
normalized to the GAPDH level.
| Table III.Sequences of long non-coding RNA
primers for reverse transcription-quantitative polymerase chain
reaction assays. |
Table III.
Sequences of long non-coding RNA
primers for reverse transcription-quantitative polymerase chain
reaction assays.
| Name | Primer
sequence | Annealing
temperature | Product length
(bp) |
|---|
| NONHSAG048960
(NON960)a |
|
|
|
| F |
TTCAGGAGACACGCGGACTA | 59 | 294 |
| R |
GCTAATCTGGGTCAGGAGCG |
|
|
| NONHSAG041512
(NON512) |
|
|
|
| F |
TTAGGATAGGATGGGCTTTTTCTGT | 60 | 101 |
| R |
AATTGCCCTAGACCCAGTGGT |
|
|
| NONHSAG016418
(NON418) |
|
|
|
| F |
AACTGGGCTTCCGTAGAACG | 60 | 242 |
| R |
GAAGGGGTGTAACGGGCAAA |
|
|
| NONHSAG028411
(NON411) |
|
|
|
| F |
CTCAATGGCCTGGGAGGTTT | 59 | 160 |
| R |
ACAAGTTCTGTGAGGGCAGG |
|
|
| NONHSAG020621
(NON621) |
|
|
|
| F |
TCTGGTGGACCCAACTCTGT | 60 | 153 |
| R |
CTTTGTCTTAGGCCAGCGGT |
|
|
| NONHSAG012658
(NON658) |
|
|
|
| F |
AGCTTAGTCGCTCATCTGGC | 61 | 226 |
| R |
AGTCAGCCAGTTCGGAAACC |
|
|
| NONHSAG019946
(NON946) |
|
|
|
| F |
CCCATACTTCCCCTTCCAGC | 60 | 122 |
| R |
ATTGCAGTTGGGCAGAGTGA |
|
|
| NONHSAG034094
(NON094) |
|
|
|
| F |
GTCGTGTCTCCTTCTTGGGG | 60 | 109 |
| R |
AGCGGTCATTATCTAGCGCC |
|
|
| NONHSAG048615
(NON615) |
|
|
|
| F |
CCCTACAAGTGGCTTTCGTG | 60 | 327 |
| R |
CGGACCCCAGAATACACCAC |
|
|
| NONHSAG006679
(NON679) |
|
|
|
| F |
TGTCTGATTCTGTCTGCTCCA | 61 | 172 |
| R |
CCGCATTTTCCCCATTCCAG |
|
|
| NONHSAG051177
(NON679) |
|
|
|
| F |
CTGCAAGTTTTGACCACGTCC | 60 | 94 |
| R |
AGACAATGAACAGGGCACAGAT |
|
|
| NONHSAG001301
(NON301) |
|
|
|
| F |
CCACAGTCCCGCTTACTTGT | 60 | 263 |
| R |
TTAAACCCGAGGGGGAGGAT |
|
|
| GAPDH |
|
|
|
| F |
TGCACCACCAACTGCTTAGC | 58 | 87 |
| R |
GGCATGGACTGTGGTCATGAG |
|
|
Statistical analysis
Data are present as the mean ± standard error of the
mean. The statistical significance between the experimental groups
was assessed using one-way analysis of variance, unpaired Student's
t-test or non-parametric test. Statistical analysis and curve
fitting were performed using GraphPad Prism software 5.1 (GraphPad
Software, Inc., San Diego, CA, USA) and SPSS 19.0 for Windows
(IBM-SPSS, Chicago, IL, USA). P<0.05 was considered to indicate
a statistically significant difference.
Results
Identification of differentially
expressed mRNAs, miRNAs and lncRNAs
Microarray expression data of HCC associated mRNAs,
miRNAs, and lncRNAs were collected from GEO: This consisted of a
total of 3 mRNA expression datasets, including 331 samples of tumor
tissue and 306 samples of adjacent non-tumor tissues, 3 miRNA
expression datasets, including 312 samples of tumor tissue and 265
samples of adjacent non-tumor tissue, and 3 lncRNA expression
datasets, including 15 samples of tumor tissue and 15 samples of
adjacent non-tumor tissue (Table
I). The results demonstrated that 628 mRNAs, 15 miRNAs, and 49
lncRNAs were significantly differentially expressed in HCC.
Construction of regulatory networks
and co-expression network
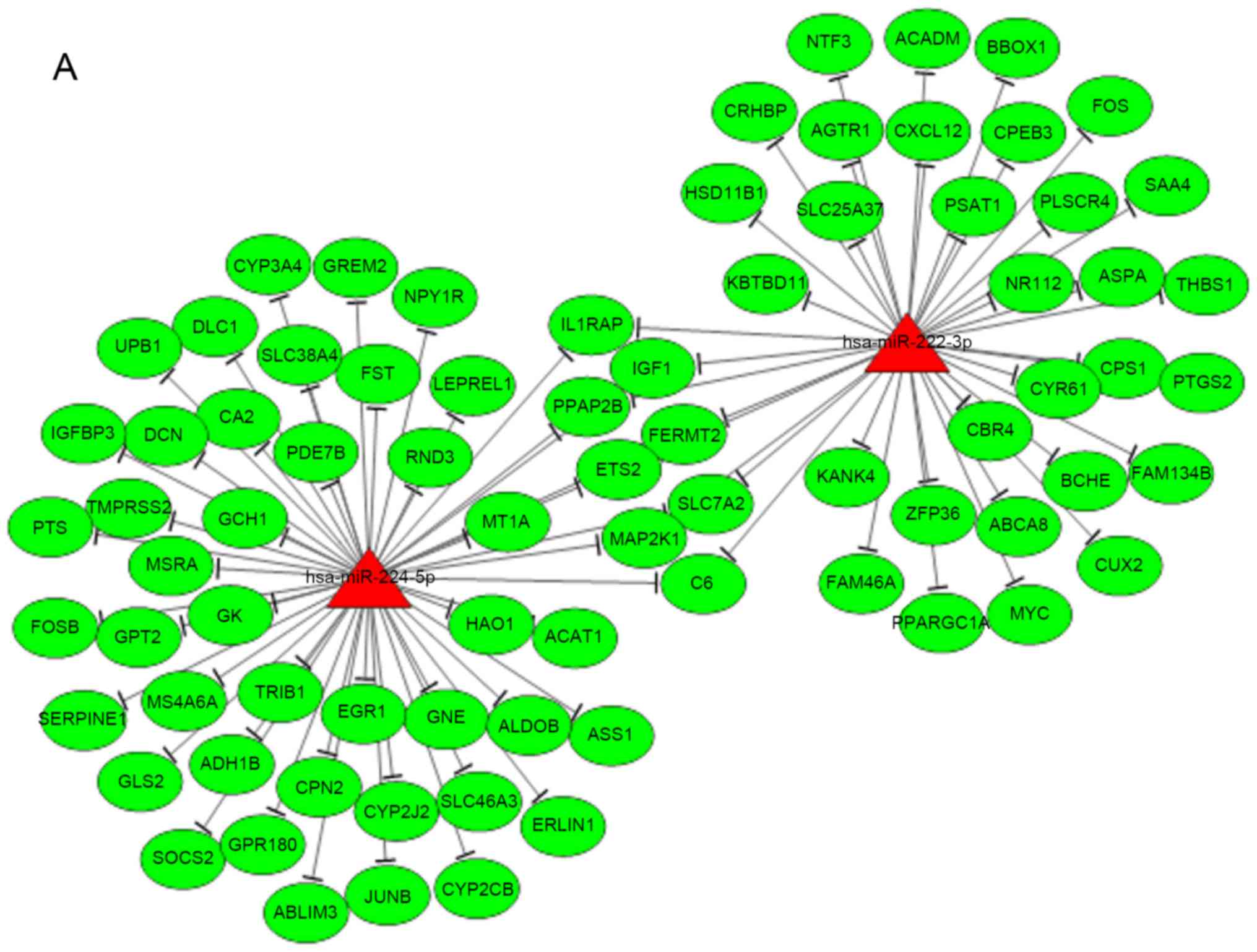
The miRNA-mRNA regulatory network was constructed
with a total of 87 pairs of upregulated miRNAs and downregulated
target mRNAs (Fig. 1A), 255
association pairs of downregulated miRNAs and upregulated target
mRNAs (Fig. 1B). According to the
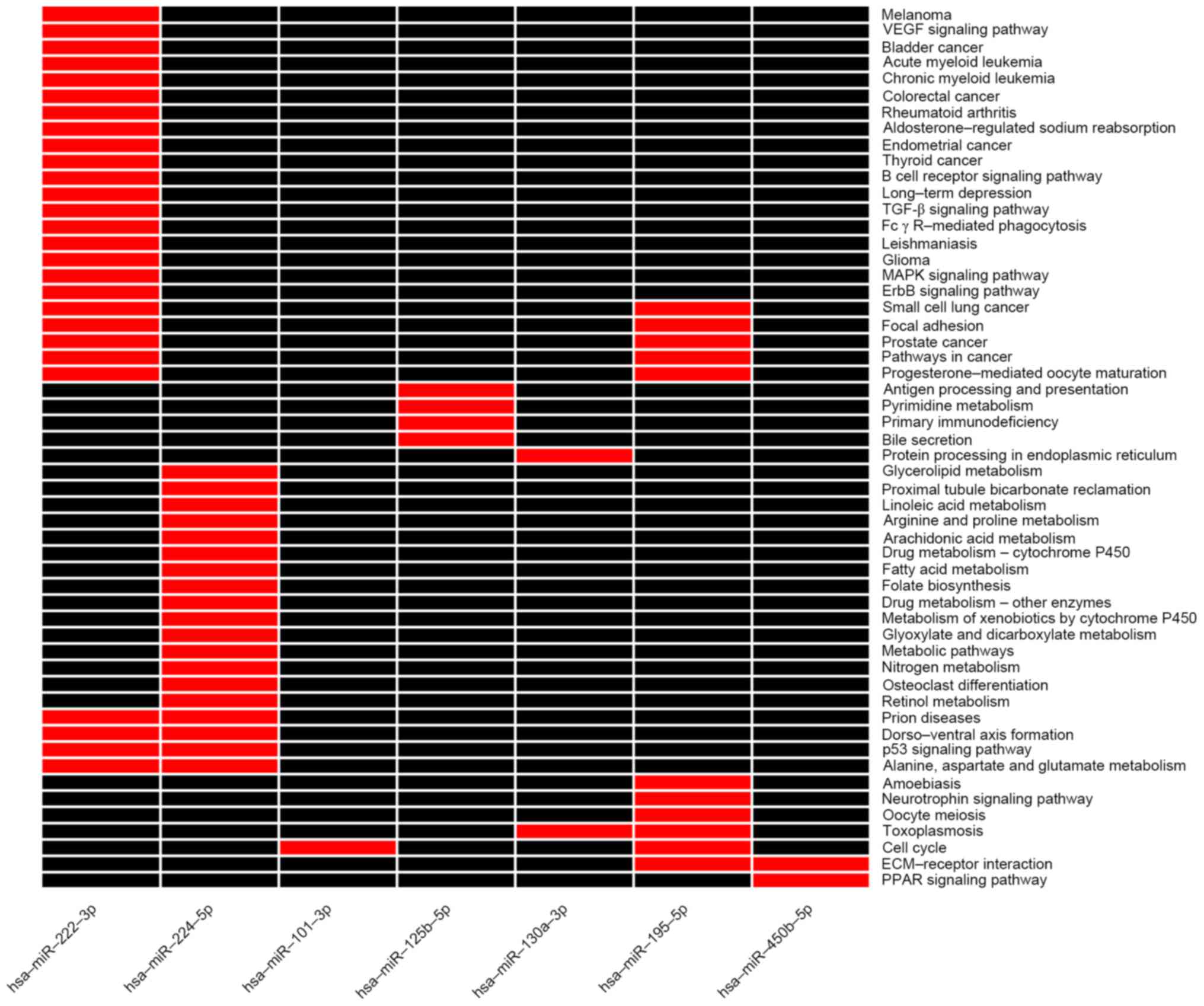
results of the enrichment analysis (Fig. 2), the target genes of
hsa-miR-222-3p and hsa-miR-195-5p were enriched in cancer
associated pathways, hsa-miR-224-5p in p53 associated pathways, and
hsa-miR-125b-5p in the pathway of antigen presentation and immune
correlation. Previous studies additionally demonstrated that
hsa-miR-222-3p (32–34), hsa-miR-125-5p (35), hsa-miR-224-5p (34) and hsa-miR-195 (36,37)
are involved in the critical processes of tumorigenesis. In
addition, differentially expressed mRNAs exhibit an important role
in HCC tumorigenesis, including a-fetoprotein (38,39),
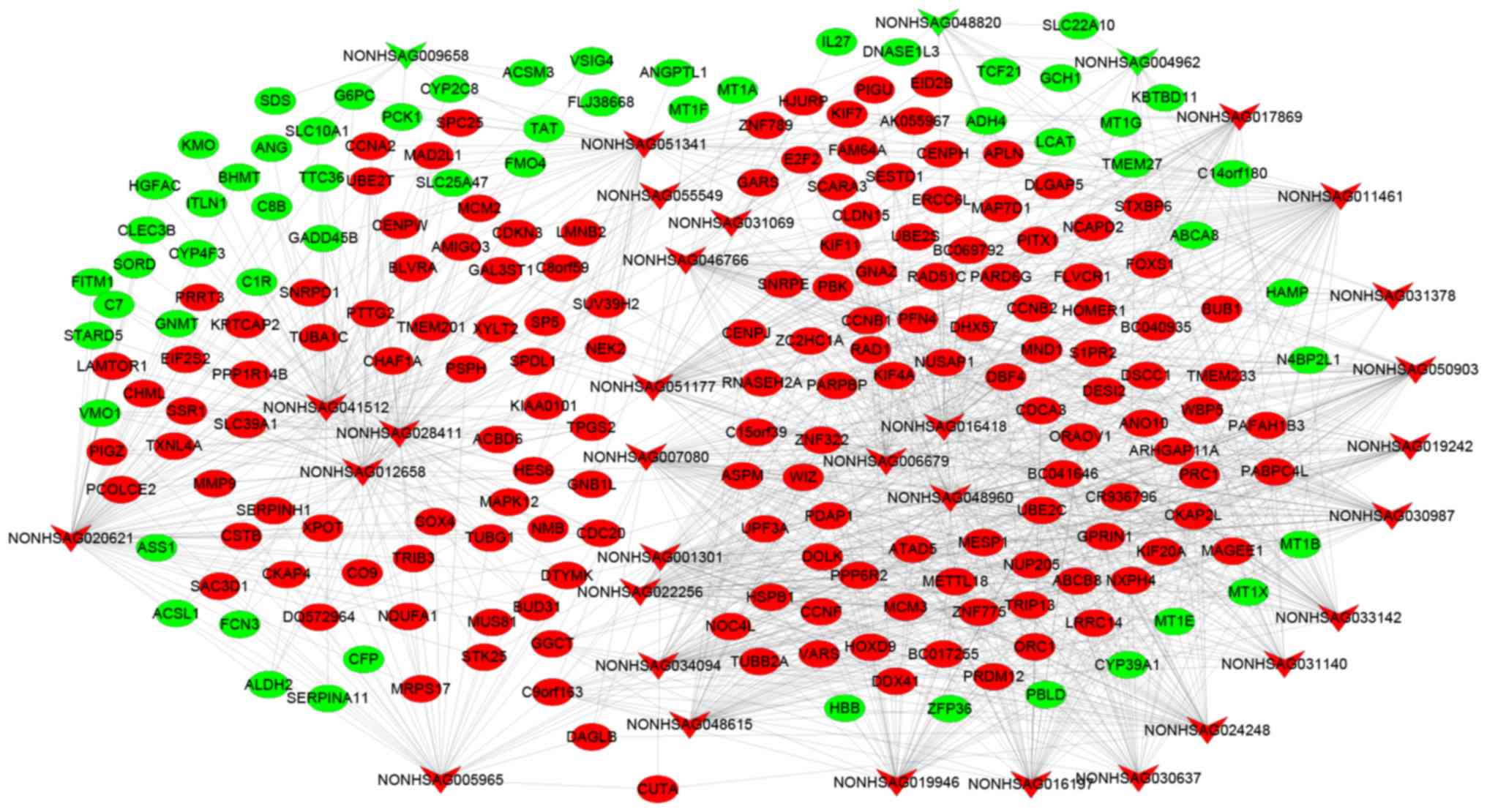
glypican 3 (40,41), and forkhead box M1 (42). Fig.
3 presents the co-expression network of mRNAs-lncRNAs. The
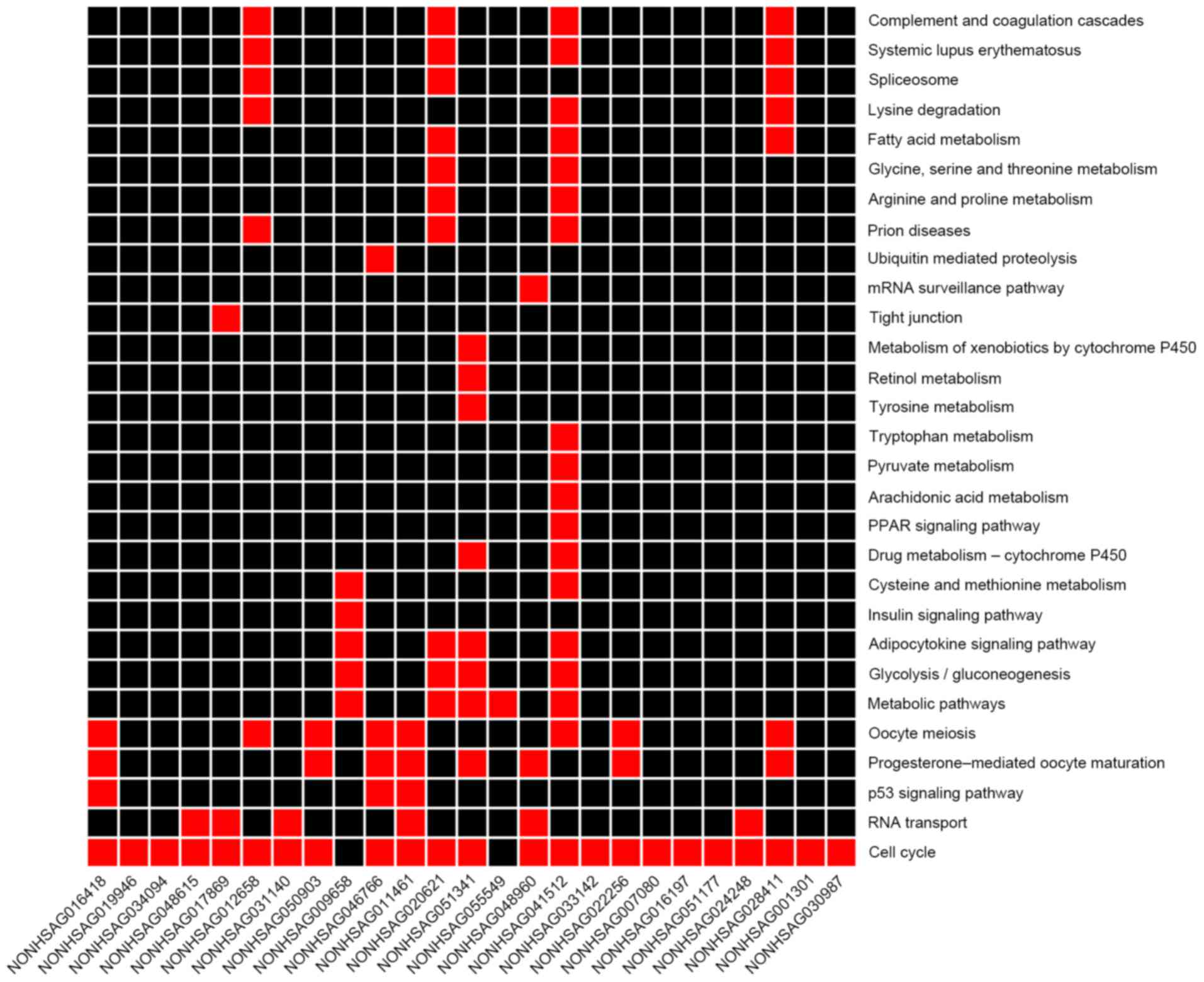
enrichment results of mRNA KEGG co-expressed with lncRNA are
presented in Fig. 4, in which the
majority of lncRNAs co-expressed with mRNAs were enriched in the
cell cycle pathway. It was revealed that NONHSAG046766,
NONHSAG011461 and NONHSAG016418 were enriched in the p53 pathway.
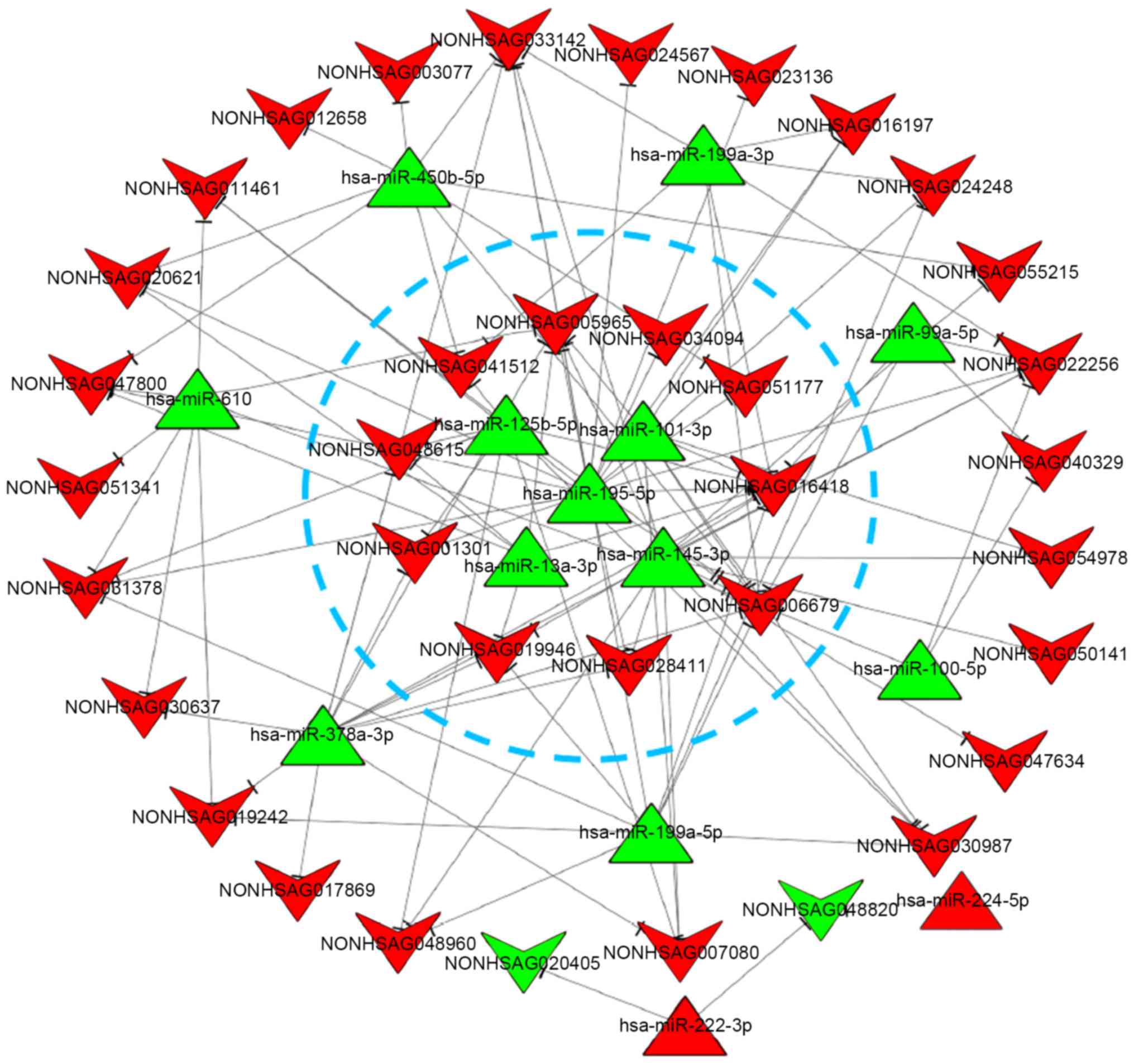
According to the results of miRNA-regulated lncRNAs predicted by
miRanda, combined with the significantly differentially expressed
miRNAs and lncRNAs, the miRNA-lncRNA regulatory network was
constructed with a total of 110 miRNA-lncRNA association pairs
screened (Fig. 5). According to
the number of connections per miRNA and lncRNA (as node) and the
KEGG results, 5 miRNAs and 10 lncRNAs were selected as key
differentially expressed noncoding RNAs in integrative analysis
(presented in the blue circle).
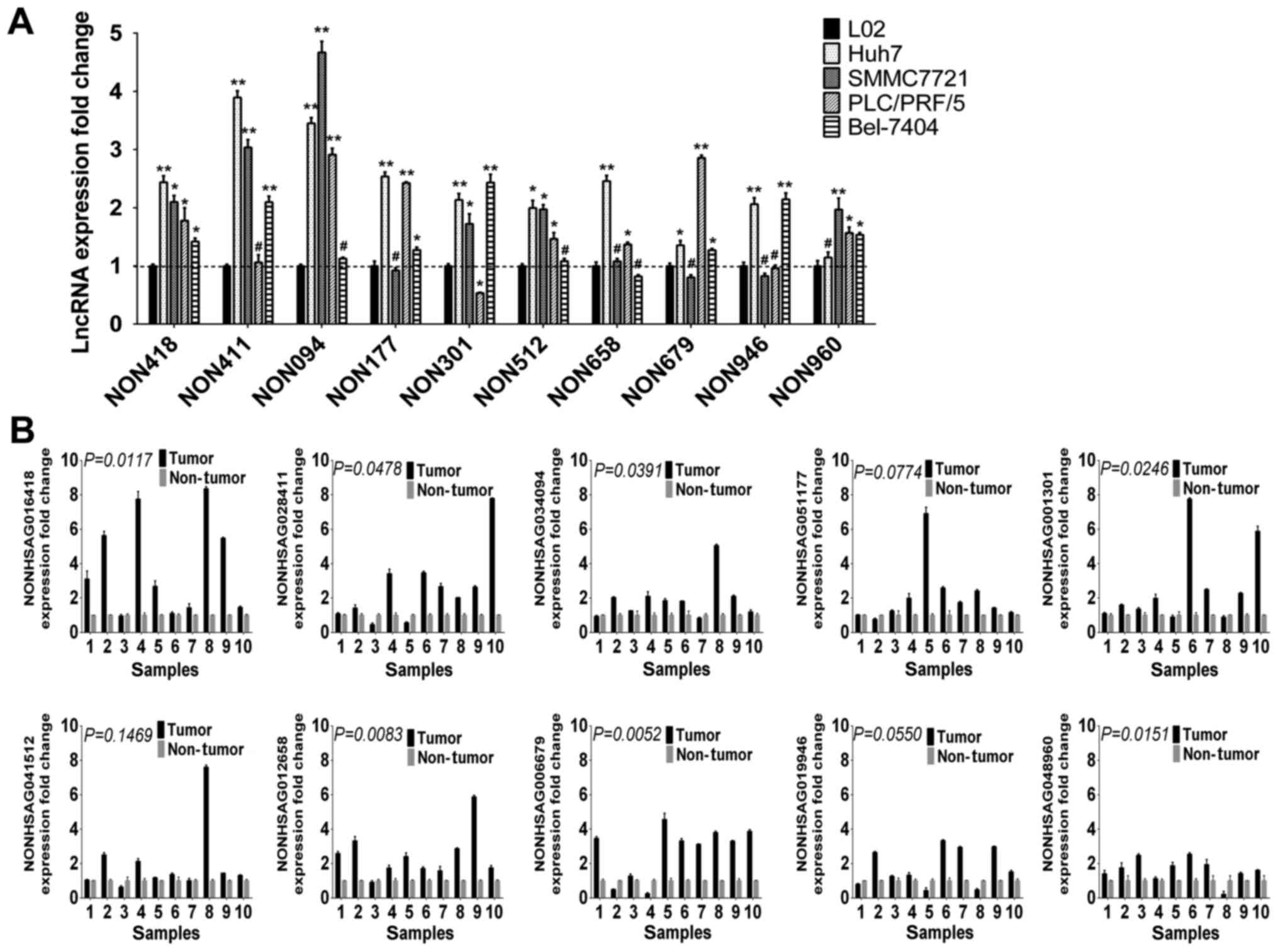
Expression of lncRNAs in cell lines
and HCC tissues
To validate the findings of the integrated
microarray analysis, transcripts of 10 key differentially expressed
lncRNAs, which were upregulated in integrated analysis, were
analyzed by RT-qPCR in 5 cell lines and 10 pairs of randomly
selected, paired tumor and non-tumor liver tissues from 10 HCC
patients. The present study primarily focused on predicted
upregulated lncRNAs, as lncRNAs may be more readily used as early
diagnosis markers or therapeutic targets, compared with
downregulated lncRNAs. The RT-qPCR analysis verified the findings
of integrated microarray analysis. Compared with the human normal
liver cell line (L02) and human non-tumor adjacent tissues, the 10
lncRNA expression levels were increased in cancer cell lines
(Fig. 6A) and tumor tissues
(Fig. 6B).
Validation of miRNA-lncRNA regulatory
associations in vitro
Interactions between lncRNAs and miRNAs, which are
important classes of noncoding RNAs in eukaryotes, provide an
additional layer of control in gene regulation. The 5 miRNAs and 10
lncRNAs in core miRNAs-lncRNAs regulatory network were selected to
validate miRNAs regulatory effect on lncRNAs. Following
transfection of miRNAs, the majority of the upregulated lncRNAs
altered their expression levels as predicted, and were
downregulated compared with negative control (NC) (Fig. 7).
Discussion
Hepatocellular carcinoma remains a primary challenge
due to its high morbidity and mortality without early diagnosis and
lack of effective treatment. A few molecular biomarkers have been
successfully used in clinical diagnostics, particularly as
prognostic or diagnostic tools, and even as therapeutic targets for
HCC. LncRNAs were once considered as the transcription noise,
however, in-depth studies, along with a large number of clinical
observations and experimental studies, have revealed that lncRNAs
exhibit an important role in tumorigenesis and cancer development
by interacting with miRNAs, mRNAs, and even proteins. Increasing
evidence suggests that dysregulated lncRNAs are closely associated
with the initiation and progression of HCC. Long intergenic
non-coding RNA LINC00152 functions in gastric cancer (43); highly expressed long intergenic
noncoding RNA UFC1 interacts with the mRNA stabilizing protein HuR
to increase levels of β-catenin in HCC cells (44); growth arrest-specific 5 regulates
apoptosis in prostate cancer (45). The results of these studies suggest
that there are numerous novel lncRNAs that remain to be identified
and investigated.
The present study systematically analyzed the
complex effects of interrelated mRNAs, miRNAs and lncRNAs to
provide networks for revealing the dysregulated lncRNAs. Currently,
there are numerous investigations regarding mRNAs and miRNAs,
however, research on lncRNAs remains limited, and the functions and
mechanism of numerous lncRNAs remain to be elucidated. The present
study integrated GEO expression microarrays to identify
differentially expressed mRNAs, miRNAs and lncRNAs, and further
constructed networks to reveal the potential function and
regulation mechanisms of the identified dysregulated lncRNAs.
Consistent with the predicted results, the majority of lncRNAs were
significantly differentially expressed in hepatocellular carcinoma
cells and tissues. The results revealed that a particular set of
miRNAs and lncRNAs were potentially involved in regulative
mechanisms in HCC development at the transcription level. Previous
molecular biology research conducted on the interaction between
miRNAs and lncRNAs demonstrates that lncRNAs may be regulated by
miRNAs. Cao et al (27)
discovered that miR-34a targets and regulates linRNA UFC1 in HCC
cells, Xu et al (45)
reported that lncRNA-AC130710 is targeted by miR-129-5p in gastric
cancer, and in addition, lncRNA MEG3 may be regulated by miR-29 in
HCC (46). These studies indicated
that lncRNAs may be targeted and regulated by miRNAs in the process
of tumor genesis and evolution.
However, the present study had various limitations
that should be acknowledged. The first is the shortage of
expression microarray data for HCC in public GEO datasets. Results
in the present study were primarily obtained through integrative
analysis of the GEO database, and numerous cases included in the
GEO microarray and analysis platform were not uniform. The
integrative analysis was primarily based on the differentially
expressed genes of the GEO microarray. Secondly, it should be
emphasized that the regulatory networks or mechanisms analyzed in
the study were only bioinformatically predicted, and expressions of
a few lncRNAs were verified in cell lines and patients. In the
future, further validation and functional examination of.
miRNAs-lncRNAs may be conducted in vivo and in
vitro.
In conclusion, the present integrative analysis of
the GEO transcriptomic data provided a comprehensive meaningful
insight into the tumorigenesis of HCC and an understanding of the
underlying mRNA-miRNA-lncRNA molecular mechanisms involved. The
present study demonstrated a method to identify a novel class of
potential biomarkers in HCC development. These findings indicated
that upregulated lncRNAs, downregulated by miRNAs, may serve as
potential molecular targets for the development of specific
therapies for HCC.
Acknowledgements
The authors would like to thank Miss Jia Wang
(research assistant) of the Department of Environmental Hygiene,
College of Preventive Medicine, Third Military Medical University
(Chongqing, China) for the critical reading of the manuscript. The
present study was supported by the National Natural Science
Foundation of China (grant no. 81270523).
Glossary
Abbreviations
Abbreviations:
|
HCC
|
hepatocellular carcinoma
|
|
GEO
|
Gene Expression Omnibus
|
|
miRNA
|
microRNA
|
|
lncRNA
|
long non-coding RNA
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
genomes
|
References
|
1
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: Epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bosetti C, Turati F and La Vecchia C:
Hepatocellular carcinoma epidemiology. Best Pract Res Clin
Gastroenterol. 28:753–770. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wallace MC, Preen D, Jeffrey GP and Adams
LA: The evolving epidemiology of hepatocellular carcinoma: A global
perspective. Expert Revi Gastroenterol Hepatol. 9:765–779.
2015.
|
|
4
|
Shi X, Sun M, Liu H, Yao Y and Song Y:
Long non-coding RNAs: A new frontier in the study of human
diseases. Cancer Lett. 339:159–166. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhao T, Xu J, Liu L, Bai J, Wang L, Xiao
Y, Li X and Zhang L: Computational identification of epigenetically
regulated lncRNAs and their associated genes based on integrating
genomic data. FEBS Lett. 589:521–531. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shahandeh A: Molecular mechanisms of
oncogenic long non-coding RNAs. Biosci Res. 10:38–54. 2013.
|
|
7
|
Marchese FP and Huarte M: Long non-coding
RNAs and chromatin modifiers: Their place in the epigenetic code.
Epigenetics. 9:21–26. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang G, Lu X and Yuan L: LncRNA: A link
between RNA and cancer. Biochim Biophys Acta. 1839:1097–1109. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee NH and Saeed AI: MicroarraysProtocols
for Nucleic Acid Analysis by Nonradioactive Probes. Hilario E and
Mackay JF: 353. Humana Press; New York, NY: pp. 265–300. 2007,
View Article : Google Scholar
|
|
10
|
Butte A: The use and analysis of
microarray data. Nat Rev Drug Discov. 1:951–960. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Edgar R, Domrachev M and Lash AE: Gene
expression omnibus: NCBI gene expression and hybridization array
data repository. Nucleic Acids Res. 30:207–210. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Barrett T, Troup DB, Wilhite SE, Ledoux P,
Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M and
Edgar R: NCBI GEO: Mining tens of millions of expression
profiles-database and tools update. Nucleic Acids Res.
35:D760–D765. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Salmena L, Poliseno L, Tay Y, Kats L and
Pandolfi PP: A ceRNA hypothesis: The rosetta stone of a hidden RNA
language? Cell. 146:353–358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tay Y, Rinn J and Pandolfi PP: The
multilayered complexity of ceRNA crosstalk and competition. Nature.
505:344–352. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rosenbloom KR, Armstrong J, Barber GP,
Casper J, Clawson H, Diekhans M, Dreszer TR, Fujita PA, Guruvadoo L
and Haeussler M: The UCSC genome browser database: 2015 update.
Nucleic Acids Res. 43:D670–D681. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu C, Bai B, Skogerbø G, Cai L, Deng W,
Zhang, Bu D, Zhao Y and Chen R: NONCODE: An integrated knowledge
database of non-coding RNAs. Nucleic Acids Res. 33:D112–D115. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: Limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Reczko M, Maragkakis M, Alexiou P, Grosse
I and Hatzigeorgiou AG: Functional microRNA targets in protein
coding sequences. Bioinformatics. 28:771–776. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Betel D, Koppal A, Agius P, Sander C and
Leslie C: Comprehensive modeling of microRNA targets predicts
functional non-conserved and non-canonical sites. Genome Biol.
11:R902010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang X: miRDB: A microRNA target
prediction and functional annotation database with a wiki
interface. RNA. 14:1012–1017. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kertesz M, Iovino N, Unnerstall U, Gaul U
and Segal E: The role of site accessibility in microRNA target
recognition. Nat Genet. 39:1278–1284. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dweep H, Sticht C, Pandey P and Gretz N:
miRWalk-database: Prediction of possible miRNA binding sites by
‘walking’ the genes of three genomes. J Biomed Inform. 44:839–847.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xiao F, Zuo Z, Cai G, Kang S, Gao X and Li
T: miRecords: An integrated resource for microRNA-target
interactions. Nucleic Acids Res. 37:D105–D110. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cao C, Sun J, Zhang D, Guo X, Xie L, Li X,
Wu D and Liu L: The long intergenic noncoding RNA UFC1, a target of
microRNA 34a, interacts with the mRNA stabilizing protein HuR to
increase levels of β-catenin in HCC cells. Gastroenterology.
148(415–426): e4182015. View Article : Google Scholar
|
|
28
|
Chen J, Fu Z, Ji C, Gu P, Xu P, Yu N, Kan
Y, Wu X, Shen R and Shen Y: Systematic gene microarray analysis of
the lncRNA expression profiles in human uterine cervix carcinoma.
Biomed Pharmacother. 72:83–90. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang F, Zhang L, Huo XS, Yuan JH, Xu D,
Yuan SX, Zhu N, Zhou WP, Yang GS, Wang YZ, et al: Long noncoding
RNA high expression in hepatocellular carcinoma facilitates tumor
growth through enhancer of zeste homolog 2 in humans. Hepatology.
54:1679–1689. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kohl M, Wiese S and Warscheid B:
Cytoscape: Software for visualization and analysis of biological
networks. Methods Mol Biol. 696:291–303. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wong QW, Ching AK, Chan AW, Choy KW, To
KF, Lai PB and Wong N: MiR-222 overexpression confers cell
migratory advantages in hepatocellular carcinoma through enhancing
AKT signaling. Clin Cancer Res. 16:867–875. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Goto Y, Kojima S, Nishikawa R, Kurozumi A,
Kato M, Enokida H, Matsushita R, Yamazaki K, Ishida Y, Nakagawa M,
et al: MicroRNA expression signature of castration-resistant
prostate cancer: The microRNA-221/222 cluster functions as a tumour
suppressor and disease progression marker. Br J Cancer.
113:1055–1065. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang Y, Takahashi S, Tasaka A, Yoshima T,
Ochi H and Chayama K: Involvement of microRNA-224 in cell
proliferation, migration, invasion and anti-apoptosis in
hepatocellular carcinoma. J Gastroenterol Hepatol. 28:565–575.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bi Q, Tang S, Xia L, Du R, Fan R, Gao L,
Jin J, Liang S, Chen Z, Xu G, et al: Ectopic expression of MiR-125a
inhibits the proliferation and metastasis of hepatocellular
carcinoma by targeting MMP11 and VEGF. PLoS One. 7:e401692012.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ding J, Huang S, Wang Y, Tian Q, Zha R,
Shi H, Wang Q, Ge C, Chen T, Zhao Y, et al: Genome-wide screening
reveals that miR-195 targets the TNF-α/NF-κB pathway by
down-regulating IκB kinase alpha and TAB3 in hepatocellular
carcinoma. Hepatology. 58:654–666. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yang X, Yu J, Yin J, Xiang Q, Tang H and
Lei X: MiR-195 regulates cell apoptosis of human hepatocellular
carcinoma cells by targeting LATS2. Pharmazie. 67:645–651.
2012.PubMed/NCBI
|
|
38
|
Um SH, Mulhall C, Alisa A, Ives AR, Karani
J, Williams R, Bertoletti A and Behboudi S: Alpha-fetoprotein
impairs APC function and induces their apoptosis. J Immunol.
173:1772–1778. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Duvoux C, Roudotâ-'Thoraval F, Decaens T,
Pessione F, Badran H, Piardi T, Francoz C, Compagnon P, Vanlemmens
C, Dumortier J, et al: Liver transplantation for hepatocellular
carcinoma: A model including α-fetoprotein improves the performance
of Milan criteria. Gastroenterology. 143(986–994): e32012.
View Article : Google Scholar
|
|
40
|
Xue Y, Bowen B, Orr A, Koral K, Haynes M,
Bell A, Paranjpe S, Mars W and Michalopoulos G: GPC3-CD81 axis in
the HCV mediated liver carcinogenesis. FASEB J. 29:611–619.
2015.PubMed/NCBI
|
|
41
|
Lei CJ, Yao C, Pan QY, Long HC, Li L,
Zheng SP, Zeng C and Huang JB: Lentivirus vectors construction of
SiRNA targeting interference GPC3 gene and its biological effects
on liver cancer cell lines Huh-7. Asian Pac J Trop Med. 7:780–786.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Koo CY, Muir KW and Lam EW: FOXM1: From
cancer initiation to progression and treatment. Biochim Biophys
Acta. 1819:28–37. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhao J, Liu Y, Zhang W, Zhou Z, Wu J, Cui
P, Zhang Y and Huang G: Long non-coding RNA Linc00152 is involved
in cell cycle arrest, apoptosis, epithelial to mesenchymal
transition, cell migration and invasion in gastric cancer. Cell
Cycle. 14:3112–3123. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Pickard M, Mourtada-Maarabouni M and
Williams G: Long non-coding RNA GAS5 regulates apoptosis in
prostate cancer cell lines. Biochim Biophys Acta. 1832:1613–1623.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Xu C, Shao Y, Xia T, Yang Y, Dai J, Luo L,
Zhang X, Sun W, Song H, Xiao B and Guo J: lncRNA-AC130710 targeting
by miR-129-5p is upregulated in gastric cancer and associates with
poor prognosis. Tumour Biol. 35:9701–9706. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Braconi C, Kogure T, Valeri N, Huang N,
Nuovo G, Costinean S, Negrini M, Miotto E, Croce CM and Patel T:
microRNA-29 can regulate expression of the long non-coding RNA gene
MEG3 in hepatocellular cancer. Oncogene. 30:4750–4756. 2011.
View Article : Google Scholar : PubMed/NCBI
|