Introduction
While >80% of the incidence occurs in sub-Saharan
Africa and East Asia, cases of hepatocellular carcinoma (HCC) have
been rapidly increasing in Western countries (1). Despite its global importance, HCC is
relatively under-researched compared with other lethal cancer
types, which is possibly due to the high complexity and
heterogeneity of HCC (2). It is
widely accepted that chronic liver diseases, including liver
cirrhosis and hepatitis, are important steps in HCC tumorigenesis.
A clinical investigation demonstrated that >80% of patients with
HCC had suffered from liver cirrhosis, which is caused by hepatitis
B virus (HBV) or hepatitis C virus (HCV) infection, alcoholism,
metabolic disorders or exposure to toxic chemicals (3). Among these factors, HBV infection is
most prevalent in Asia, while HCV infection is more common in
Western countries. The majority of aflatoxin-associated liver
cirrhosis occurs in Southeast Asia (4). Therefore, an improved understanding
of the pathogenesis of HCC is required in order to develop novel
treatment strategies.
A previous study identified a gene termed COMMD7,
which is mapped to 20q11.22, by sequence analysis and homology
comparison. COMMD7 cDNA fragments were observed to be highly
expressed in HCC samples via suppression subtractive hybridization.
The pro-cancer properties of COMMD7 were additionally demonstrated,
since knockdown of this gene by short hairpin RNA reduced HCC cell
proliferation and tumor growth in a mouse model (5). It was additionally observed that
COMMD7 is capable of stimulating C-X-C motif chemokine 10 (CXCL10)
expression and, in turn, may promote HCC cell proliferation and
metastasis in an autocrine manner (Zheng et al., unpublished
data). However, the value of targeting CXCL10 signal transduction
in treating COMMD7-positive tumors, or the molecular mechanisms
underlying COMMD7-mediated CXCL10 expression, has not been
completely addressed.
In the present study, using multiple in vitro
and in vivo models, it was demonstrated that inhibition of
the CXCL10/C-X-C chemokine receptor type 3 (CXCR3) axis attenuated
COMMD7-mediated HCC proliferation. The present mechanistic study
demonstrated that COMMD7 activated CXCL10 by modulating nuclear
factor (NF)-κB and oxidative stress.
Materials and methods
Cell culture
The human HCC cancer cell line Huh7 was purchased
from the American Type Culture Collection (Manassas, VA, USA). All
cells were maintained in Dulbecco's modified Eagle's medium (Thermo
Fisher Scientific, Inc., Waltham, MA, USA) containing 10% fetal
calf serum (HyClone; GE Healthcare Life Sciences, Logan, UT, USA),
penicillin (107 U/l) and streptomycin (10 mg/l) at 37°C
in a humidified chamber containing 5% CO2.
Reagents
Rabbit anti-human Ki67 monoclonal antibody (cat. no.
ab92742), rabbit anti-human total p65 monoclonal antibody (cat. no.
ab32536), rabbit anti-human phosphorylated p65 monoclonal antibody
(cat. no. ab76302) and rabbit anti-human β-actin monoclonal
antibody (cat. no. ab8227) were purchased from Abcam (Cambridge,
MA, USA). The CXCL10 ELISA kit (cat. no. ab83700) was purchased
from Abcam. The NF-κB inhibitor, celastrol, was purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). The NF-κB
inhibitor, Bay 11–7085, was purchased from Cayman Chemical Company
(Ann Arbor, MI, USA). A pan inhibitor of ROS production, N-acetyl
cysteine (NAC), was purchased from Abcam (cat. no. ab143032). As
CXCR3 is the principal receptor for CXCL10, a commercially
available chemical antagonist, NBI-74330 (6) was used in the present study (Thermo
Fisher Scientific, Inc., Waltham, MA, USA).
pGenesil-COMMD7 plasmid
establishment
The COMMD7 cDNA was extracted from the human HCC
cell lines of Huh7 cells. The primer sequences for the COMMD7 was
listed as: Forward, 5′-AGTGGCTTTCTCCTCACTAAGACC-3′ and reverse,
5′-GGAAAGATTTCTGGCTCAGCTC-3′. The amplified COMMD7 was directly
cloned into the plasmid pGenesil-1 vector (Genesil Biotechnology,
Wuhan, China), and then transformed into the DH5α E. coli
(Tiangen, Beijing, China). The transformed colonies were selected
by the kanamycin resistance. The established pGenesil-COMMD7
plasmid was transfected into the Huh7 by using the Lipofectamine™
2000 (Invitrogen; Thermo Fisher Scientific, Inc.) at the condition
of 5% CO2 and at 37°C. Then, the over-expression of the
COMMD7 in Huh7 cells were observed.
Bromodeoxyuridine (BrdU) labeling
assay
BrdU was purchased from Roche Diagnostics
(Indianapolis, IN, USA). BrdU was added to the cell culture at a
working concentration of 10 mM. Following incubation for 18 h, BrdU
signaling was measured using the BrdU Labeling and Detection kit
III (Roche Diagnostics).
In vivo study
The present study was approved by the Laboratory
Animal Welfare and Ethics Committee of Third Military Medical
University (Chongqing, China). A total of 7 Balb/C mice, weighting
25–30 g were humanely housed and treated at room temperature (about
20°C), 40% humidity and a 12-h light/dark cycle. All the mice were
given free access to food and water. A total of 5×107
Huh7 cells were subcutaneously injected into male athymic nude mice
(n=7). Animals were sacrificed 20 days post-injection. The tumor
volumes were evaluated as follows: Tumor volume
(mm3)=(length × width2)/2.
ELISA
The CXCL10 levels were examined by using the ELISA
kit (Abcam) according to the manufacturer's protocols.
Western blotting
Cells were washed and treated with lysis buffer (50
mM Tris base, 1.0 mM EDTA, 150 mM NaCl, 0.1% SDS, 1% Triton X-100,
1% sodium deoxycholate, 1 mM phenylmethylsulfonyl fluoride) for 30
min on ice. The concentrations of protein samples were determined
by using BCA assay (Thermo Fisher Scientific, Inc.) and 2 µg
protein samples were separated by 15% SDS-PAGE, and translocated to
polyvinylidene fluoride membranes. Following incubation with 5%
skimmed milk for 1 h at room temperature, the blots were incubated
with primary antibodies (1:3,000) and rabbit anti-human β-actin
monoclonal (1:2,000) for 2 h and secondary antibody (1:2,000) for 1
h at room temperature. Finally, the blots were visualized by
enhanced chemiluminescence (Amersham; GE Healthcare, Chicago, IL,
USA).
Reactive oxygen species (ROS)
measurement
CM-H2DCFDA (general oxidative stress indicator; DCF)
was purchased from Thermo Fisher Scientific, Inc. An inverted
fluorescence microscope (IX73; Olympus Corporation, Japan;
magnification, ×200) was used to visualize the images. The
glutathione (GSH)/oxidized glutathione (GSSG) assay kit was
purchased from Abcam. Intracellular ROS were examined according to
the manufacturers' protocols (with the cell density of
105 cells/ml).
Statistical analysis
The data in this study were analyzed by using SPSS
statistical analysis software version 18.0 (SPSS, Inc., Chicago,
IL, USA). Statistical analysis of data was performed using one-way
analysis of variance followed by the Tukey test. Data are presented
as the mean ± standard error of the mean. All of the experiments
were repeated at least 3 times. P<0.05 was considered to
indicate a statistically significant difference.
Results
Disruption of the CXCL10/CXCR3 axis
reduces COMMD7-mediated HCC cell proliferation
To determine whether COMMD7-mediated HCC cell
proliferation depended on CXCL10, the present study aimed to
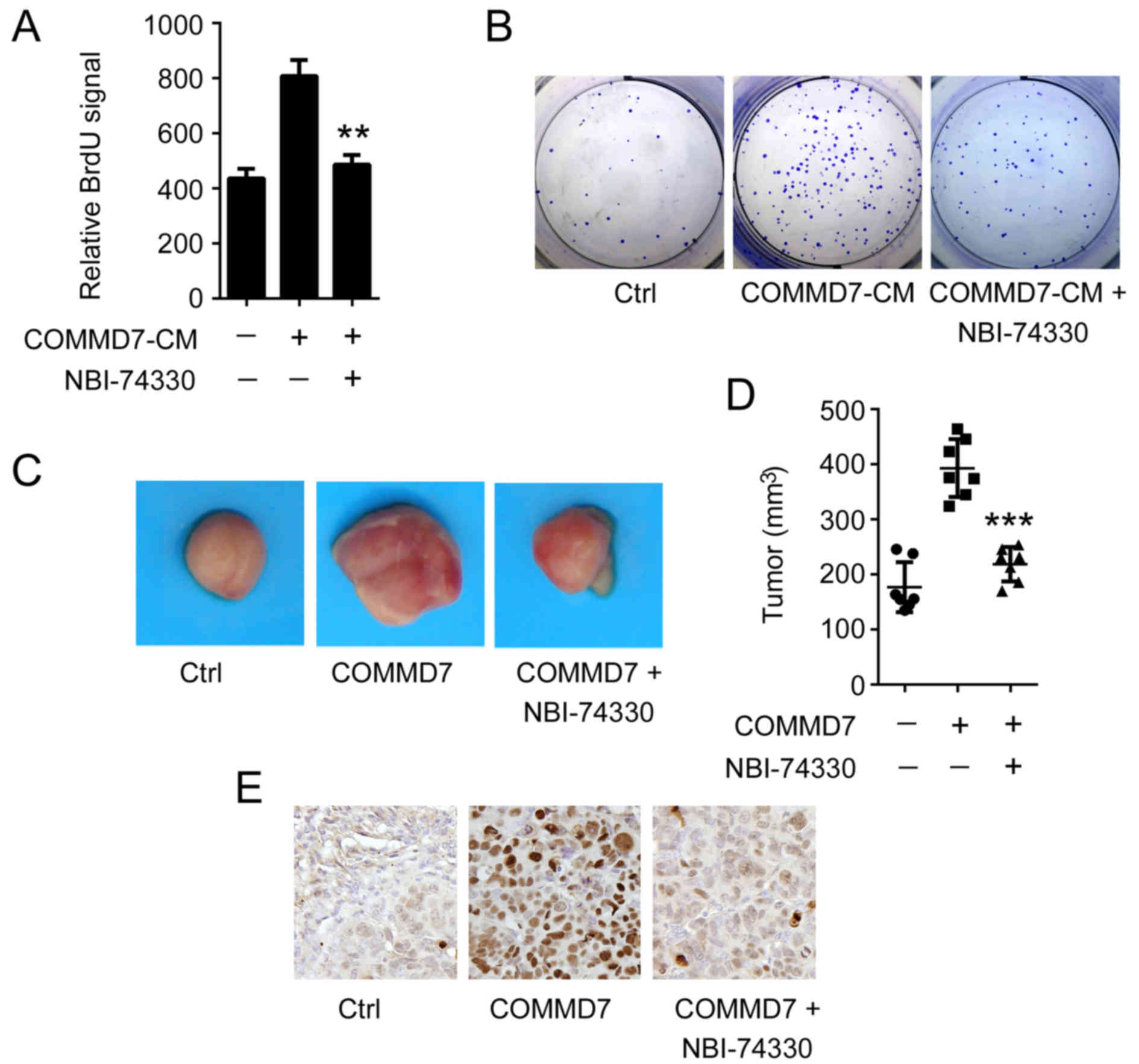
inhibit CXCL10 signal transduction. As presented in Fig. 1A, for the BrdU assay, the
conditioned medium (CM) derived from the COMMD7-over-expressing
Huh7 cells markedly induced the proliferation of naïve Huh7 cells.
However, the CM-mediated proliferation was significantly reduced in
NBI-74330-treated cells. This observation was further supported by
the clonogenic formation assay. As in Fig. 1B, COMMD7-induced cell clone
formation was eliminated when cells were treated with
NBI-74330.
To further support the in vitro observations,
a mouse xenograft model was employed to determine the role of
CXCL10 in the COMMD7-mediated proliferation of HCC tumor growth. To
this end, Huh7 sub-clones that stably expressed COMMD7 or mock
vector were established. These two sub-clones were subcutaneously
transplanted into nude mice, respectively, and the mice were
treated with or without NBI-74330. As presented in Fig. 1C, as expected, the tumor growth was
enhanced in the COMMD7-expressing group compared with the mock
vector-expressing group. Notably, inhibition of CXCL10 signal
transduction by NBI-74330 substantially reduced tumor growth
(Fig. 1C). The tumor volume in the
NBI-74330-treated group decreased by 41.17% (P<0.001), compared
with the vehicle-treated group (Fig.
1D). To further examine the cell proliferation status in tumor
xenografts, immunostaining of Ki67 was performed. As shown in
Fig. 1E, overexpression of COMMD7
induced more Ki67-positive cells, while treatment with NBI-74330
markedly reduced the number of Ki67-positive cells. These results
suggested that the CXCL10/CXCR3 axis may be required for
COMMD7-mediated HCC cell proliferation.
COMMD7 activates CXCL10 by modulating
NF-κB
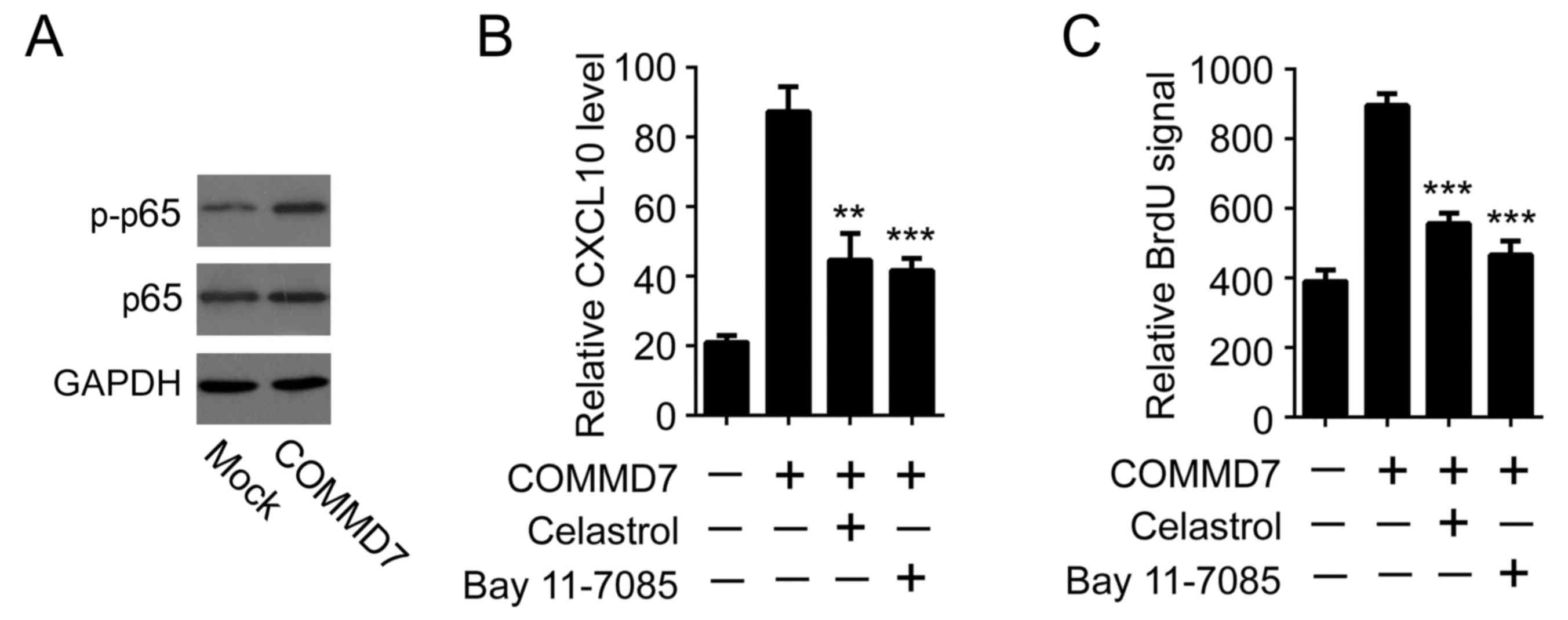
The present study aimed to determine the mechanisms
responsible for COMMD7-induced CXCL10 expression. It was previously
reported that COMMD7 is required for tumor necrosis factor
(TNF)-α-induced NF-κB activation (5). As a pilot result, it was observed
that overexpression of COMMD7 augmented p65 phosphorylation,
suggesting that overexpression of COMMD7 alone may be sufficient to
induce NF-κB activation (Fig. 2A).
Considering that NF-κB was able to directly bind to the CXCL10
promoter and initiate CXCL10 expression, it was thus examined
whether NF-κB was involved in COMMD7-induced CXCL10 expression. To
this end, two different NF-κB inhibitors, celastrol and Bay
11–7085, were used (7,8). In line with previous data, the
overexpression of COMMD7 induced a significant upregulation of
CXCL10 expression in Huh7 cells. Notably, inhibition of NF-κB by
either celastrol or Bay 11–7085 impeded COMMD7-mediated CXCL10
expression (Fig. 2B). Furthermore,
the pro-proliferative effects of CM derived from
COMMD7-overexpressing cells were abolished when cells were
pre-incubated with either celastrol or Bay 11–7085 (Fig. 2C). These results suggested that
COMMD7 may activate CXCL10 by modulating NF-κB.
COMMD7 activates NF-κB by modulating
intracellular ROS
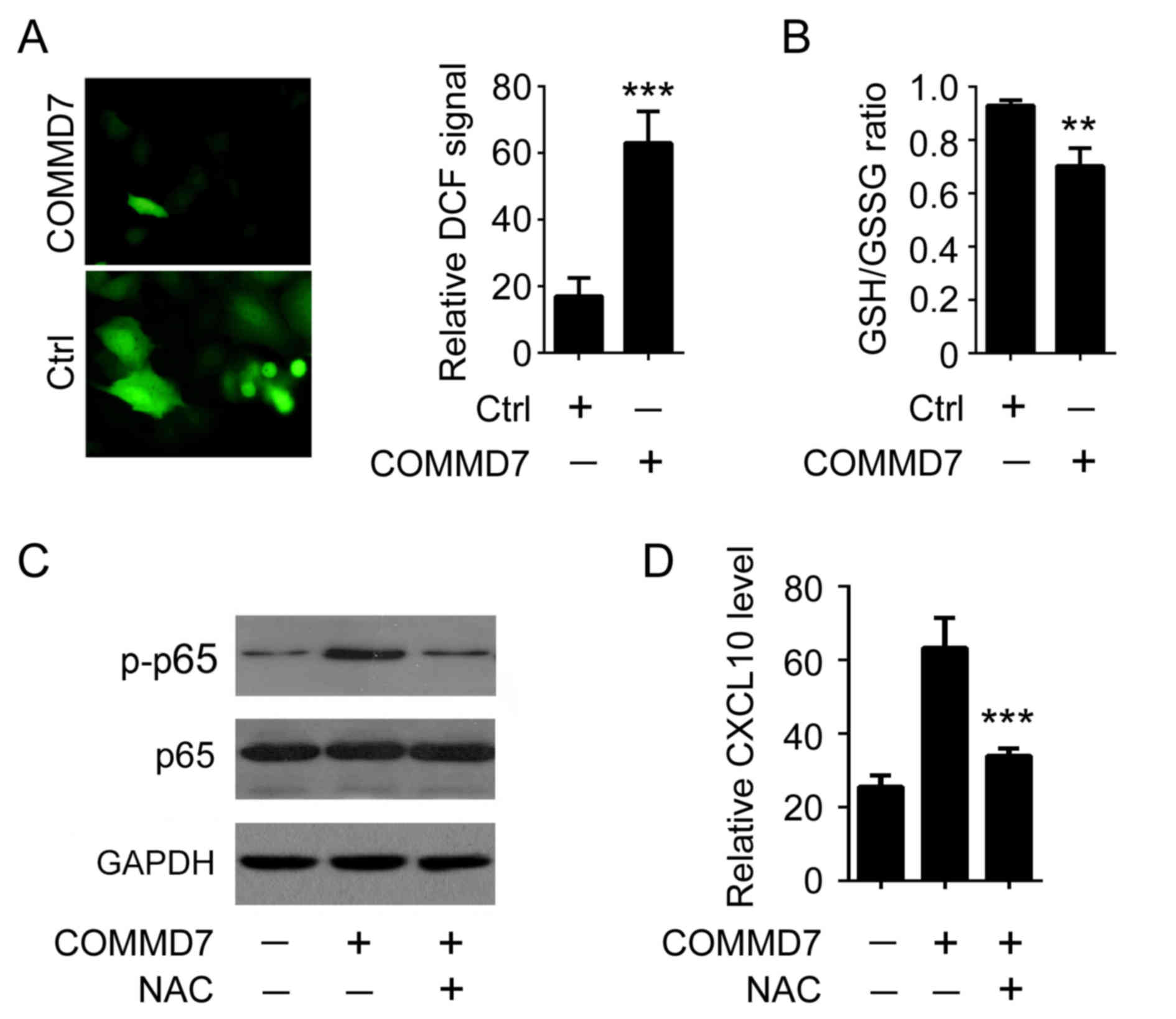
It has been reported that oxidative stress is
involved in interferon- or TNF-induced NF-κB expression (9). To examine the potential signaling
cascades underlying COMMD7-mediated NF-κB activation, of particular
interest, the impact of COMMD7 expression on the level of
intracellular ROS was examined. It was demonstrated that
overexpression of COMMD7 in Huh7 cells augmented intracellular ROS
level by >3-fold, as demonstrated by the use of the specific ROS
probe DCF (Fig. 3A). Similarly,
overexpression of COMMD7 reduced the GSH/GSSG ratio, an additional
hallmark of oxidative stress (10)
(Fig. 3B). To determine whether
ROS are involved in COMMD7-mediated NF-κB activation, NAC, a pan
inhibitor of ROS production, was used (11). As presented in Fig. 3C, COMMD7-mediated p65
phosphorylation was attenuated when cells were treated with NAC,
suggesting that oxidative stress was required for COMMD7-mediated
NF-κB activation. Accordingly, the elimination of ROS by NAC
reduced COMMD7-mediated CXCL10 expression (Fig. 3D). These results indicated that
COMMD7 activates NF-κB by modulating intracellular ROS
production.
Discussion
HCC is considered to be one of the most important
life-threatening tumors worldwide, and remains a notable problem
for public health (12). Multiple
genetic and environmental factors, including mutations of oncogenes
and tumor suppressor genes, infection with oncogenic microbes and
metabolic imbalances, are associated with the development of HCC
(13). Therefore, the
identification of novel proteins with aberrant expression or
function in HCC is required for early diagnosis and discovery of
novel drug targets.
Chemokines refer a family of structurally similar
extracellular proteins which govern leukocyte trafficking. It is
well accepted that chemokines are involved in tumorigenesis by
regulating immune cell and tumor cells (14). CXCL10 initiates its downstream
signaling cascade by interacting with and activating CXCR3
(15). Aberrant regulation of
CXCL10 was demonstrated to be associated with metastasis of
colorectal cancer (16).
Previously, it was reported that CXCL10 expression was positively
correlated with COMMD7 expression in a multitude of HCC cell lines.
In addition, previous studies (17,18)
also reported that CXCR3 was predominately expressed and
upregulated significantly in HCC cancer tissues compared with
adjacent non-cancerous tissues. Meanwhile, the expression of CXCR3
was significantly increased in certain hepatic cell lines,
including HepG2 (19), Huh-7
(20) and SMMC-7721 (21). Therefore, it was confirmed that
CXCR3 was highly expressed in HCC cell lines and HCC human samples
(17,18). Overexpression of COMMD7 induced
CXCL10 production in the culture medium. Furthermore, inhibiting
CXCL10 signaling via treatment with a neutralizing antibody
markedly inhibited CM-mediated HCC cell proliferation (17). These results suggested that CXCL10
served an essential role in COMMD7-mediated HCC cell proliferation.
The present study further tested whether CXCL10 signaling was a
potential target for treating COMMD7-positive tumors. It was
demonstrated that treatment with the CXCR3 inhibitor NBI-74330,
markedly reduced HCC cell proliferation in vitro and in
vivo model. Moreover, it was additionally demonstrated that
NF-κB is a prerequisite for CXCL10 expression.
Oxidative stress is characterized as an imbalance
between producing and scavenging free radicals and reactive
metabolites, termed ROS. ROS were historically considered to be
toxic byproducts from metabolism, which lead to damage to important
macromolecules in cells (22). A
recent study, however, indicated that ROS may function as essential
physiological regulators of diverse biological processes, including
gene expression (23). In the
present study, it was demonstrated that ROS served an important
role in COMMD7-mediated CXCL10 expression. Overexpression of COMMD7
induced severe oxidative stress in HCC cells, illustrated by the
increased DCF signal and reduced GSH/GSSG ratio. In addition,
phosphorylation of p65 and expression of CXCL10 were markedly
reduced in COMMD7-expressing HCC cells following the inhibition of
ROS. Further work is required to examine the primary source of
COMMD7-induced ROS production.
In conclusion, the present data suggest a
COMMD7-initiated pro-tumor pathway. COMMD7 is upregulated in HCC
and triggers ROS production. As a consequence of accumulated
cellular ROS, NF-κB is activated and, in turn, activates CXCL10
expression. CXCL10 finally promotes HCC cell proliferation in an
autocrine manner. The present study highlighted the role of COMMD7
in the development of HCC, and provides new options for anticancer
drug design.
Acknowledgements
The present study was supported by the National
Natural Science Fund (grant no. NSFC 81372561).
References
|
1
|
El-Serag HB: Epidemiology of viral
hepatitis and hepatocellular carcinoma. Gastroenterology.
142:1264–1273.e1. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bruix J, Gores GJ and Mazzaferro V:
Hepatocellular carcinoma: Clinical frontiers and perspectives. Gut.
63:844–855. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Arzumanyan A, Reis HM and Feitelson MA:
Pathogenic mechanisms in HBV- and HCV-associated hepatocellular
carcinoma. Nat Rev Cancer. 13:123–135. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Morgan RL, Baack B, Smith BD, Yartel A,
Pitasi M and Falck-Ytter Y: Eradication of hepatitis C virus
infection and the development of hepatocellular carcinoma: A
meta-analysis of observational studies. Ann Intern Med.
158:329–337. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zheng L, Liang P, Li J, Huang XB, Liu SC,
Zhao HZ, Han KQ and Wang Z: ShRNA-targeted COMMD7 suppresses
hepatocellular carcinoma growth. PLoS One. 7:e454122012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
van Wanrooij EJ, de Jager SC, van Es T, de
Vos P, Birch HL, Owen DA, Watson RJ, Biessen EA, Chapman GA, van
Berkel TJ and Kuiper J: CXCR3 antagonist NBI-74330 attenuates
atherosclerotic plaque formation in LDL receptor-deficient mice.
Arterioscler Thromb Vasc Biol. 28:251–257. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ni H, Zhao W, Kong X, Li H and Ouyang J:
NF-kappa B modulation is involved in celastrol induced human
multiple myeloma cell apoptosis. PLoS One. 9:e958462014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Matteucci C, Minutolo A, Marino-Merlo F,
Grelli S, Frezza C, Mastino A and Macchi B: Characterization of the
enhanced apoptotic response to azidothymidine by pharmacological
inhibition of NF-kB. Life Sci. 127:90–97. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gilmore TD: Introduction to NF-kappaB:
Players, pathways, perspectives. Oncogene. 25:6680–6684. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Meimaridou E, Kowalczyk J, Guasti L,
Hughes CR, Wagner F, Frommolt P, Nürnberg P, Mann NP, Banerjee R,
Saka HN, et al: Mutations in NNT encoding nicotinamide nucleotide
transhydrogenase cause familial glucocorticoid deficiency. Nat
Genet. 44:740–742. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chang JH, Kim YJ, Han SH and Kang CY:
IFN-gamma-STAT1 signal regulates the differentiation of inducible
Treg: Potential role for ROS-mediated apoptosis. Eur J Immunol.
39:1241–1251. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Altekruse SF, McGlynn KA and Reichman ME:
Hepatocellular carcinoma incidence, mortality, and survival trends
in the United States from 1975 to 2005. J Clin Oncol. 27:1485–1491.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Farazi PA and DePinho RA: Hepatocellular
carcinoma pathogenesis: From genes to environment. Nat Rev Cancer.
6:674–687. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lazennec G and Richmond A: Chemokines and
chemokine receptors: New insights into cancer-related inflammation.
Trends Mol Med. 16:133–144. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Datta D, Flaxenburg JA, Laxmanan S, Geehan
C, Grimm M, Waaga-Gasser AM, Briscoe DM and Pal S: Ras-induced
modulation of CXCL10 and its receptor splice variant CXCR3-B in
MDA-MB-435 and MCF-7 cells: Relevance for the development of human
breast cancer. Cancer Res. 66:9509–9518. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kawada K, Hosogi H, Sonoshita M, Sakashita
H, Manabe T, Shimahara Y, Sakai Y, Takabayashi A, Oshima M and
Taketo MM: Chemokine receptor CXCR3 promotes colon cancer
metastasis to lymph nodes. Oncogene. 26:4679–4688. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ding Q, Xia Y, Ding S, Lu P, Sun L and Liu
M: An alternatively spliced variant of CXCR3 mediates the
metastasis of CD133+ liver cancer cells induced by CXCL9.
Oncotarget. 7:14405–14414. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ling CC, Ng KT, Shao Y, Geng W, Xiao JW,
Liu H, Li CX, Liu XB, Ma YY, Yeung WH, et al: Post-transplant
endothelial progenitor cell mobilization via CXCL10/CXCR3 signaling
promotes liver tumor growth. J Hepatol. 60:103–109. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhu Y, Wang Z, Zhu Y, Wang J, Cai L, Shen
H, Kong Y and Qiu Y: CXCR3 monoclonal antibody inhibits the
proliferation and migration of MCF-7 cells and HepG2 cells in
vitro. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 31:1544–1548. 2015.(In
Chinese). PubMed/NCBI
|
|
20
|
Helbig KJ, Ruszkiewicz A, Semendric L,
Harley HA, McColl SR and Beard MR: Expression of the CXCR3 ligand
I-TAC by hepatocytes in chronic hepatitis C and its correlation
with hepatic inflammation. Hepatology. 39:1220–1229. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guo JQ, Chen L, Ai HW, Jing JN, Zhou JY,
Zhang CY and You SY: A novel fusion protein of IP10-scFv retains
antibody specificity and chemokine function. Biochem Biophys Res
Commun. 320:506–513. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang YT, Chen YY, Lai YH, Cheng CC, Lin
TC, Su YS, Liu CH and Lai PC: Resveratrol alleviates the
cytotoxicity induced by the radiocontrast agent, ioxitalamate, by
reducing the production of reactive oxygen species in HK-2 human
renal proximal tubule epithelial cells in vitro. Int J Mol Med.
37:83–91. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Karthik S, Sankar R, Varunkumar K, Anusha
C and Ravikumar V: Blocking NF-κB sensitizes non-small cell lung
cancer cells to histone deacetylase inhibitor induced extrinsic
apoptosis through generation of reactive oxygen species. Biomed
Pharmacother. 69:337–344. 2015. View Article : Google Scholar : PubMed/NCBI
|