Introduction
Glucagon-like peptide-1 (GLP-1) is an incretin
hormone encoded by the glucagon GCG gene (1). GLP-1 is secreted from the
gastrointestinal tract into the circulation in response to nutrient
ingestion (1,2). GLP-1 stimulates insulin secretion
from β-cells and inhibits glucagon secretion from α-cells (3) and has previously been used for
development of novel strategies to treat type 2 diabetes. In a
clinical study involving patients with type 2 diabetes,
administering a subcutaneous infusion of GLP-1 for 6 weeks resulted
in improved insulin sensitivity and β-cell function (4). Ding et al (5) reported that GLP-1 decreased the
levels of serum glucose and improved insulin resistance. Exendin-4,
a GLP-1 receptor agonist, has been reported to inhibit the
expression of glucose-6-phosphatase (G6Pase) and
phosphoenolpyruvate carboxykinase (PEPCK) in the hepatocytes of
young adult mice via an insulin-independent mechanism (6). Several studies have demonstrated that
the GLP-1 receptor is a member of the 7 transmembrane family of G
protein coupled receptors and is expressed in the human hepatocyte
cell lines HepG2 and Huh7 as well as C57BL/6J mice with
high-fat-diet-induced obesity (6–8).
However, the exact mechanism underlying GLP-1-mediated improvement
of liver insulin resistance remains to be elucidated.
Evolutionarily conserved proto-oncogene Wnt (Wnt)
signaling pathways are present in multicellular eukaryotes and
serve roles in embryonic development, morphogenesis, stem cell
regulation, liver zonation and metabolism (9). Wnt genes encode 19 Wnt proteins and
are evolutionarily conversed (9).
To date, two different Wnt signaling pathways have been identified,
including the canonical and non-canonical pathway (10). The canonical Wnt signaling pathway
components include the frizzled family of cell surface receptors,
T-cell factor (TCF) family transcription factors, co-receptors and
the downstream degradation complex containing axin/glycogen
synthase kinase (GSK)-3β/adenomatosis polyposis coli (APC) and
β-catenin (11–13). Interactions between Wnt and
frizzled receptors and/or co-receptors, including low-density
lipoprotein receptor-related protein (LRP) 5 and 6, leads to the
activation of the intracellular protein disheveled (Dsh). These
events trigger an intracellular signaling cascade, which eventually
results in phosphorylation and inactivation of glycogen synthase
kinase-3β (GSK3β), and decreased GSK3β-induced phosphorylation of
β-catenin (14). Accumulation and
nuclear translocation of β-catenin, and increased levels of nuclear
β-catenin lead to the activation of over 60 Wnt-responsive genes
via interactions with transcription factors, including TCF, TCF7,
TCF7 like 1 (TCF7L1), TCF7L2 and lymphoid enhancer factor (LEF)
(14).
Genetic associations between TCF7L2 polymorphisms
and type 2 diabetes has previously been reported in multiple ethnic
populations (15). Several studies
have investigated the role of the Wnt signaling pathway in the
pathogenesis of diabetes (16–19).
In TCF7L2 gene knockout mice, the number of pancreatic β-cells was
significantly reduced along with the reduction of the deficiencies
in insulin secretion and glucose tolerance (16). Liver-specific TCF7L2 knockout mice
demonstrated increased hepatic glucose production and
overexpression of liver-specific TCF4, leading to reduced hepatic
glucose levels (17). TCF7
knockdown in HepG2 cells resulted in increased gluconeogenic gene
expression, whereas restoration of hepatic TCF7 levels decreased
the expression of gluconeogenic genes and reduced hepatic glucose
output (18). Using the
liver-specific dominant-negative TCF7L2 transgenic mouse model
LTCFDN, Ip et al (19)
demonstrated that the Wnt signaling pathway and its effector
β-catenin/TCF serve a beneficial role in suppressing hepatic
gluconeogenesis.
GLP-1 has been demonstrated to directly activate the
Wnt signaling pathway (20). Liu
and Habener (21) reported that
GLP-1 and exendin-4 induced Wnt signaling in pancreatic β-cells and
INS-1 cells independent of GSK3β, as well as increasing nuclear
levels of β-catenin. These interactions led to transcriptional
activation of genes associated with β-cell proliferation via
interacting with TCF7L2 (21).
Furthermore, it has been demonstrated that lithium, a GSK3β
inhibitor, stimulates the synthesis of GLP-1 and activates the
transcription of proglucagon in intestinal endocrine cell lines
(14). These observations suggest
that Wnt signaling effectors β-catenin and TCF are able to control
the production and function of GLP-1 in islets and
insulin-secreting INS-1 cells (21). However, it has not yet been
investigated whether GLP-1 improves liver insulin resistance via
the Wnt signaling pathway.
The aim of the present study was to investigate
whether the Wnt signaling pathway mediates the beneficial effects
of liraglutide on insulin resistance and hepatic glucose metabolism
using in vitro and in vivo diabetic models. The
results of the present study demonstrate that the interaction
between forkhead box (Fox) O1, the main target of insulin
signaling, and β-catenin regulates hepatic gluconeogenesis in
response to liraglutide.
Materials and methods
Animal studies
Male db/db mice (n=12, 40–44 g) and C57BL/6 mice
(n=6, 39–42 g) were purchased from the Institute for Animal
Reproduction of Experimental Animal Center in Nanjing Medical
College (Nanjing, China). C57BL/6 mice were used as the control
group. The mice were bred under standard laboratory conditions and
used for experimentation when 6 weeks old. All animals were
maintained in a temperature-controlled environment (22–24°C; 50–60%
humidity) with a 12-h light/dark cycle and free access to drinking
water and normal chow (60, 15 and 25% calories from carbohydrates,
fats and proteins, respectively). All animal care and experimental
procedures were approved by the Institutional Animal Care and Use
Committee of Tongji Medical College, Huazhong University of Science
and Technology (Wuhan, China). Male db/db mice were divided into 2
groups: Saline and liraglutide (n=6/group) and received
intraperitoneal injections of saline or liraglutide (Novo Nordisk
Pharmaceutical Co., Ltd., Tianjin, China) once daily at a
concentration of 200 µg/kg body weight for 8 weeks. Animals were
monitored each 2 weeks for alterations in body weight, plasma
glucose and insulin. Every 2 weeks, the tail vein blood was
harvested from the tail vein and plasma was stored at −80°C for
further analysis. A Glucose Oxidase and Peroxidase Assay (GOD-POD)
kit (Shanghai Mingdian Bioengineering, Co., Ltd., Shanghai, China)
and an Insulin ELISA kit (cat. no. EZHIASF-14K; EMD Millipore,
Billerica, MA, USA) were used to quantify levels of glucose and
insulin, respectively. The homeostasis model assessment for insulin
resistance (HOMA-IR) was calculated using the following equation:
Fasting insulinxfasting glucose/22.5 (22). Animals were fasted overnight in the
8th week of the experiment and subsequently sacrificed by cervical
dislocation. Liver tissues were harvested, frozen in liquid
nitrogen and stored at −80°C until RNA and protein extraction.
Hematoxylin and eosin (H&E)
staining
Liver tissues were fixed in 10% formalin for 24 h at
room temperature and embedded in paraffin. Paraffin blocks were cut
using a microtome into 5-µm sections. Following deparaffinization
and dehydration, the sections were stained with 0.5% (w/v)
hematoxylin (5–10 min) and eosin (1–2 min; H&E staining) at
room temperature. Histological slides were examined by light
microscopy (magnification, ×200). Histological features, including
steatosis, inflammation and hepatocellular injury were
histologically graded by two pathologists. The nonalcoholic fatty
liver disease (NAFLD) activity score (NAS) was quantified by
summing the scores of steatosis (0–3), lobular inflammation (0–2)
and hepatocellular ballooning (0–2), as previously described
(23).
Cell culture
The HepG2 human hepatoma cell line was purchased
from American Type Culture Collection (ATCC; Manassas, VA, USA).
Cells were grown and cultured in Dulbecco's modified Eagle's medium
(DMEM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
containing 10% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc.), 10 mmol/l
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany), 100 U/ml penicillin, and 100 mg/ml
streptomycin. Cells were maintained at 37°C in a humidified
incubator, with 5% CO2.
Insulin resistance cell model
At 70–80% confluence, cells were treated with 250
µmol/l palmitate (Sigma-Aldrich; Merck KGaA), one of the most
common free fatty acids, and high glucose (25 mmol/l) DMEM culture
medium for 24 h to establish the in vitro insulin resistant
model. Glucose consumption studies were performed to evaluate the
insulin resistant model. Cells were washed three times with PBS and
the culture medium was replaced with low glucose DMEM (5.5 mmol/l
glucose). Insulin was added to the culture medium at a
concentration of 10−7 mol/l for 2 h. Following
incubation, culture medium was collected and tested for glucose
consumption was investigated as previously described (24), using the GOD-POD kit according to
the manufacturer's instructions.
Small interfering RNA (siRNA)
transfection
Following successful establishment of the in
vitro insulin resistant model, β-catenin siRNA (50 nM;
Guangzhou RiboBio Co., Ltd., Guangzhou, China) was transfected into
the cells via incubation with MegaTran 1.0 (OriGene Technologies,
Inc., Rockville, MD, USA) for 48 h at 37°C. β-catenin siRNA was
diluted in opti-MEM (Gibco; Thermo Fisher Scientific, Inc.) to
obtain a final concentration of 100 nmol/l. At 24 h following
transfection, 100 nM liraglutide was added to cell cultures for 24
h. HepG2 cells were harvested and used for western blot analysis.
Following incubation, culture medium was collected and glucose
consumption was investigated as previously described (24), using the GOD-POD kit according to
the manufacturer's instructions.
Glucose production assay
HepG2 cells were plated in 24-well cell culture
plates at a density of 2×105 cells/well and allowed to
grow for 24 h at 37°C. HepG2 cells were transfected with β-catenin
siRNA as above. Subsequently, cells were washed three times with
PBS to remove glucose and treated for 24 h with glucose production
medium (glucose- and phenol red-free DMEM containing
gluconeogenesis substrates: 10 mM sodium lactate and 1 mM sodium
pyruvate), 0.1 mM adenosine 3′,5′-cyclic monophosphate (cAMP) and
100 nM dexamethasone (Dex) to induce gluconeogenesis in the absence
or presence of 100 nM liraglutide. Glucose production was measured
using a Glucose Assay kit (Shanghai Mingdian Bioengineering, Co.,
Ltd.) according to the manufacturer's instrucctions.
Western blot analysis
HepG2 cells were lysed in radioimmunoprecipitation
assay buffer (RIPA; Beyotime Institute of Biotechnology, Beijing,
China) containing 1% Triton X-100 and a mixture of protease and
phosphatase inhibitors. Liver tissue (30 mg wet weight) was
homogenized at 4°C in 400 µl RIPA buffer containing 1% Triton X-100
and a mixture of protease and phosphatase inhibitors. Following a
30 min-incubation on ice, total protein was obtained by
centrifugation at 4,025 × g for 20 min at 4°C. Protein content was
measured using the bicinchoninic acid protein assay and 30 µg
protein was separated by 10% SDS-PAGE. Separated proteins were
electrically transferred onto polyvinylidene difluoride membranes.
Following transfer, membranes were blocked with 5% bovine serum
albumin (MP Biomedicals, LLC, Santa Ana, CA, USA) for 1 h at room
temperature, washed in Tris-buffered saline with 0.08% Tween and
then incubated overnight at 4°C with the following primary
antibodies: β-catenin (1:1,000; cat. no. sc-7963), TCF7L2 (1:1,000;
cat. no. sc-166699), PEPCK (1:1.000; cat. no. sc-32879), G6Pase
(1:1,000; cat. no. sc-25840), GSK3β (1:1,000; cat. no. sc-81462),
phospho-Ser473-Akt (1:1,000; cat. no. sc-33437), total Akt
(1:1,000; cat. no. sc-8312), β-actin (1:2,000; cat. no. sc-47778;
all from Santa Cruz Biotechnology, Inc., Dallas, TX, USA),
p-Ser9-GSK3β (1:1,000; cat. no. 9323), p-Ser256-FoxO1 (1:1,000;
cat. no. 9461) and total FoxO1 (1:1,000; cat. no. 2880; all from
Cell Signaling Technology, Danvers, MA, USA). The membranes were
washed, and incubated at room temperature for 1 h with the
following secondary antibodies: Horseradish peroxidase
(HRP)-conjugated sheep anti-rabbit immunoglobulin (Ig)G (1:2,000;
cat. no. SA00001-2; ProteinTech Group, Inc., Chicago, IL, USA) or
HRP-conjugated goat anti-mouse IgG (1:2,000; cat. no. SA00001-1;
ProteinTech Group, Inc.). Proteins were visualized using enhanced
chemiluminescent detection reagent (Beyotime Institute of
Biotechnology), and quantified using Quantity One software (version
4.6.2; Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from liver tissue using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. cDNA was synthesized
using an RT-qPCR kit (cat. no. 10928042; Invitrogen; Thermo Fisher
Scientific, Inc.). qPCR was performed to detect mouse β-catenin,
TCF7L2, PEPCK, G6Pase, FoxO1 and GSK3β using QuantiTect SYBR Green
PCR kit (Applied Biosystems; Thermo Fisher Scientific, Inc.) and a
real-time PCR detection system (Bio-Rad Laboratories, Inc.). The
following primer pairs were used for the qPCR: β-catenin forward,
5′-GTTCTACGCCATCACGACAC-3′ and reverse, 5′-GACAGCACCTTCAGCACTCT-3′;
TCF7L2 forward, 5′-AATCCTTGCCTTTCGCTTCC-3′ and reverse,
5′-TCTGTGACTTGGCGTCTTGG-3′; pck1 forward,
5′-ACAGTCATCATCACCCAAGAGC-3′ and reverse,
5′-GGGCGAGTCTGTCAGTTCAATA-3′; G6Pase forward,
5′-CATCAATCTCCTCTGGGTGGC-3′ and reverse,
5′-GCTGTTGCTGTAGTAGTCGGTGTC-3′; FoxO1 forward,
5′-TGCTTTTATTACCCTGTGAGTTGTG-3′ and reverse,
5′-ATCGTGACAAAAGCCAACAGC-3′; Gsk3β forward,
5′-GGTCAGTTTCACAGGGTTATGC-3′ and reverse,
5′-AGATGGAAGTGGTCACGCTAAT-3′; β-actin forward,
5′-AGAGGGAAATCGTGCGTGAC-3′ and reverse,
5′-CAATAGTGATGACCTGGCCGT-3′. The following thermocycling conditions
were used for the PCR: Initial denaturation at 95°C for 5 min
followed by 40 cycles of 95°C for 15 sec, 60°C for 15 sec and 72°C
for 20 sec. Relative quantification was performed using the
2−ΔΔCq method (25).
Transcript levels were normalized to the internal reference gene
β-actin.
Statistical analysis
Data are presented as the mean ± standard error of
the mean. Statistical comparisons were performed using the unpaired
two-tailed t-test or one-way analysis of variance followed by
Newman-Keuls post hoc test for multiple group analyses. Analysis
was performed using SPSS 17.0 software (SPSS, Inc., Chicago, IL,
USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
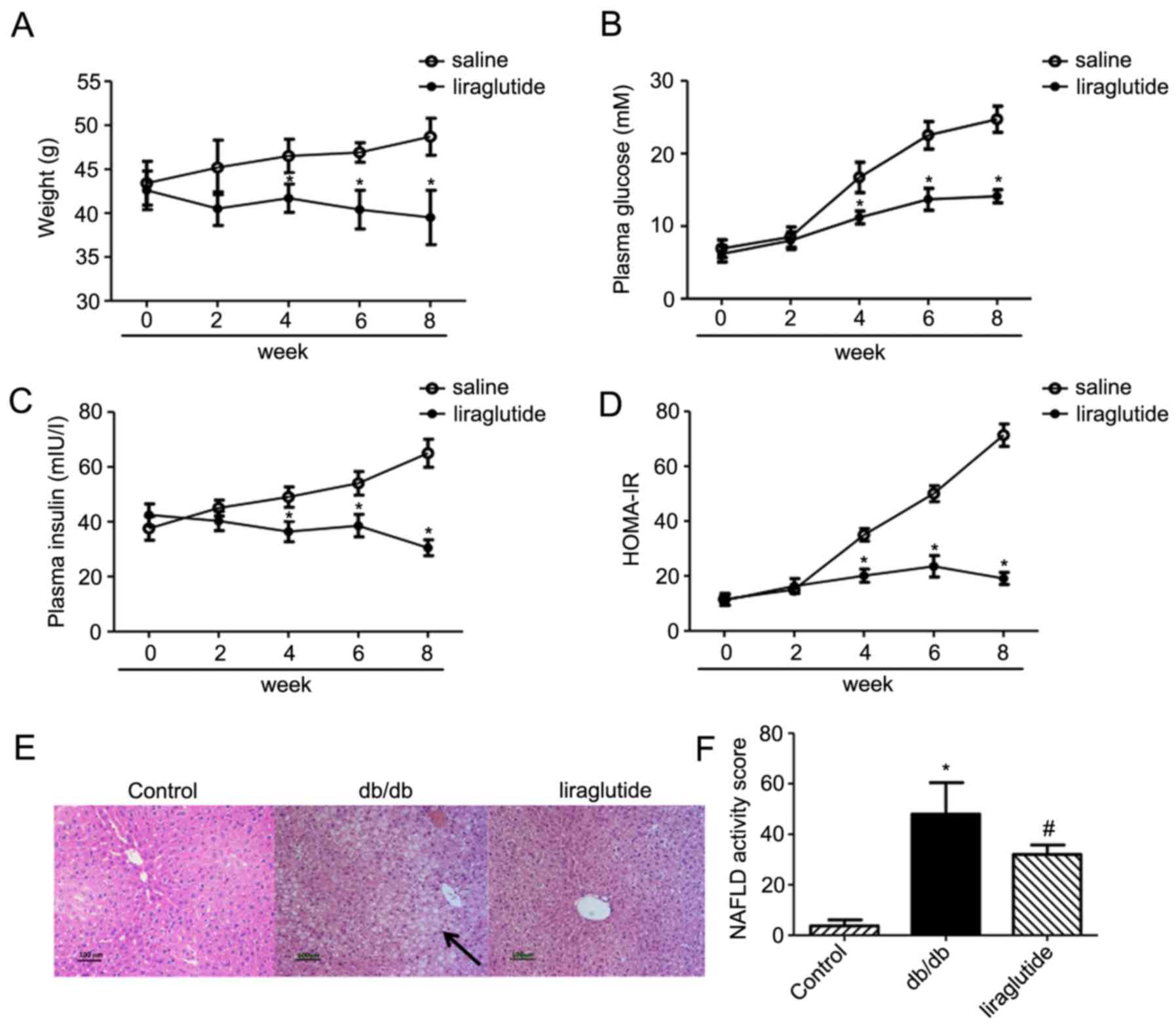
Liraglutide significantly decreases
body weight, plasma glucose concentration, insulin levels and
HOMA-IR
The body weight of db/db mice treated with
liraglutide decreased from week 4 onwards compared with the
saline-treated control group (all P<0.05; Fig. 1A). The final body weight of the
control group at week 8 was significantly increased compared with
the liraglutide treated group (39.5±3.1 g in the liraglutide
treated group vs. 48.7±2.1 g in the control group; P<0.05).
Quantification of plasma glucose levels revealed
significantly reduced glucose in the liraglutide treated group
(14.1±0.9 mM), compared with the control group (24.7±1.8 mM) at
week 8 (P<0.05; Fig. 1B).
Similarly, at week 8 the plasma insulin concentration in the
liraglutide treated group (30.5±2.9 mIU/l) was significantly lower
compared with the saline treated group (65.0±5.4 mIU/l; P<0.05;
Fig. 1C). Treatment with
liraglutide significantly improved HOMA-IR insulin resistance
(19.1±2.2), compared with the control group (71.4±4.1; P<0.05;
Fig. 1D).
Liraglutide alters liver
morphology
Hepatic accumulation of lipids increased in the
db/db group compared with control group and decreased in mice
treated with liraglutide (Fig.
1E). Additionally, NAFLD activity significantly decreased in
the liraglutide group compared with the saline group (P<0.05;
Fig. 1F).
Liraglutide activates the Wnt
signaling pathway and downregulates liver gluconeogenesis in
vivo
The present study aimed to identify the signaling
pathway and potential mechanisms underlying the effects of
liraglutide on insulin resistance. GLP-1 is an activator of the
canonical Wnt signaling pathway; as such, changes in the expression
of β-catenin, GSK3β and TCF7L2 in the liver samples were analyzed
using western blotting and RT-qPCR. Levels of β-catenin protein,
but not mRNA, increased significantly in the db/db group treated
with liraglutide compared with the control group treated with
saline (P<0.05; Fig. 2A-C).
Furthermore, levels of TCF7L2 mRNA and protein, levels of GSK3β
mRNA, and levels of p-GSK3β protein were significantly increased in
liraglutide-treated mice compared with the saline group (P<0.05;
Fig. 2A-C). Furthermore, treatment
with liraglutide significantly inhibited kinase enzymes G6Pase and
PEPCK at the mRNA and protein level (all P<0.05; Fig. 2A-C). Upstream of PEPCK and G6Pase,
FoxO1 is a transcription factor that promotes gluconeogenesis in
the liver, whereas p-FoxO1 suppresses gluconeogenesis (26). The levels of total and p-FoxO1 were
assessed in the present study (Fig.
2A-C). The results demonstrated that the levels of p-FoxO1
protein and therefore the ratio of p-FoxO1/FoxO1 were significantly
elevated in liraglutide treated db/db mice compared with
saline-treated mice (P<0.05). These results suggest that
liraglutide inactivates FoxO1 by phosphorylation and therefore
inhibits the downstream activity of G6Pase and PEPCK. The above
data indicate that liraglutide improves insulin resistance and
reduces liver gluconeogenesis; it may be hypothesized that these
effects are mediated by the β-catenin/Wnt signaling pathway.
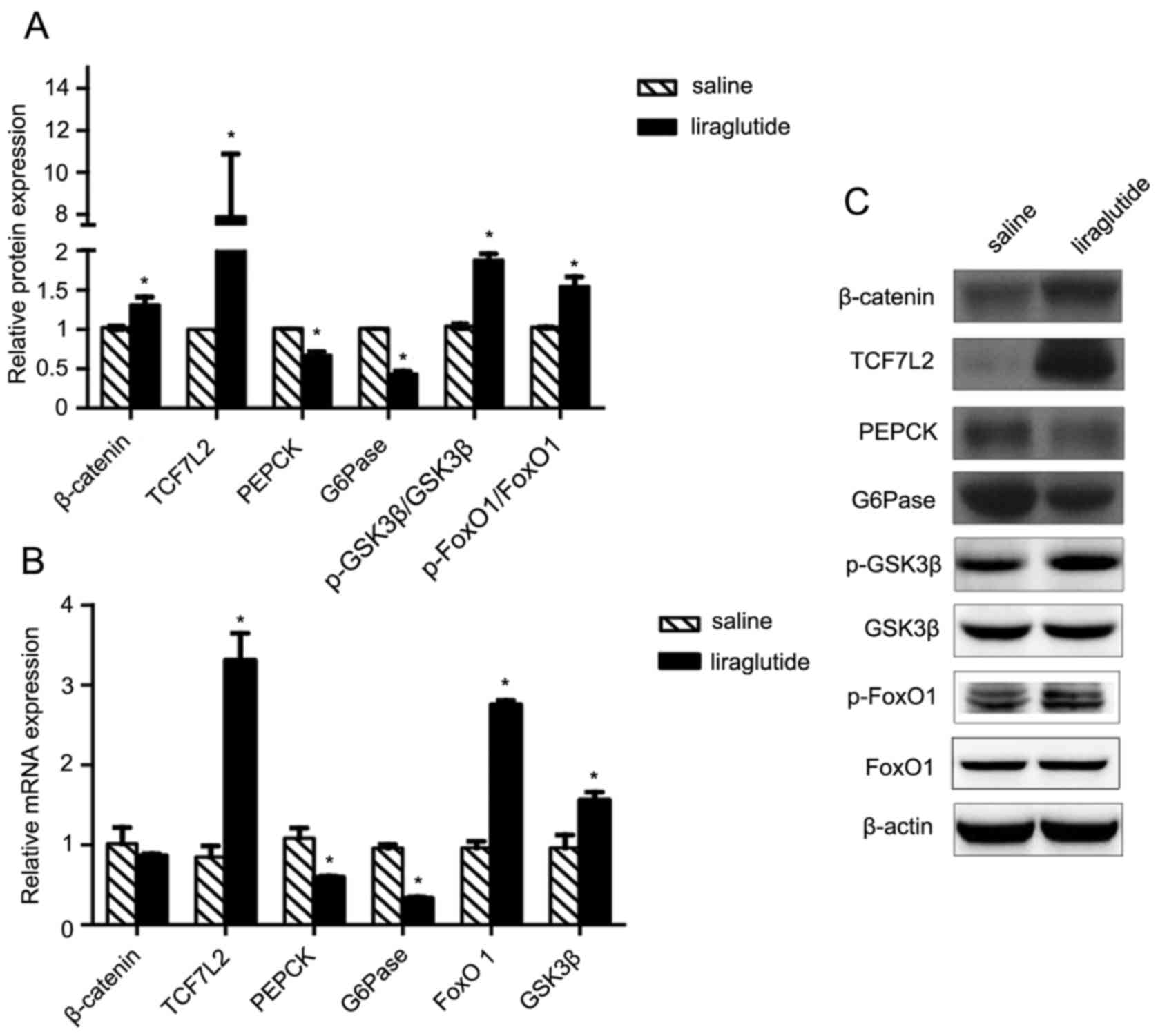 | Figure 2.Liraglutide activates the Wnt
signaling pathway and decreases liver gluconeogenesis in
vivo. (A) Protein and (B) mRNA levels of Wnt signaling pathway
molecules, including β-catenin, TCF7L2 and p-GSK3β, and molecules
associated with liver gluconeogenesis, including PEPCK, G6Pase and
p-FoxO1. β-actin was used as a loading control. (C) Representative
western blot images of the Wnt signaling pathway molecules and
molecules associated with liver gluconeogenesis. All values are
expressed as the mean ± standard error of the mean. *P<0.05 vs.
control. TCF7L2, transcription factor 7 like 1; p, phosphorylated;
GSK3β, glycogen synthase kinase-3β; PEPCK, phosphoenolpyruvate
carboxykinase; G6Pase, glucose-6-phosphatase; FoxO1, forkhead box
O1; Wnt, proto-oncogene Wnt. |
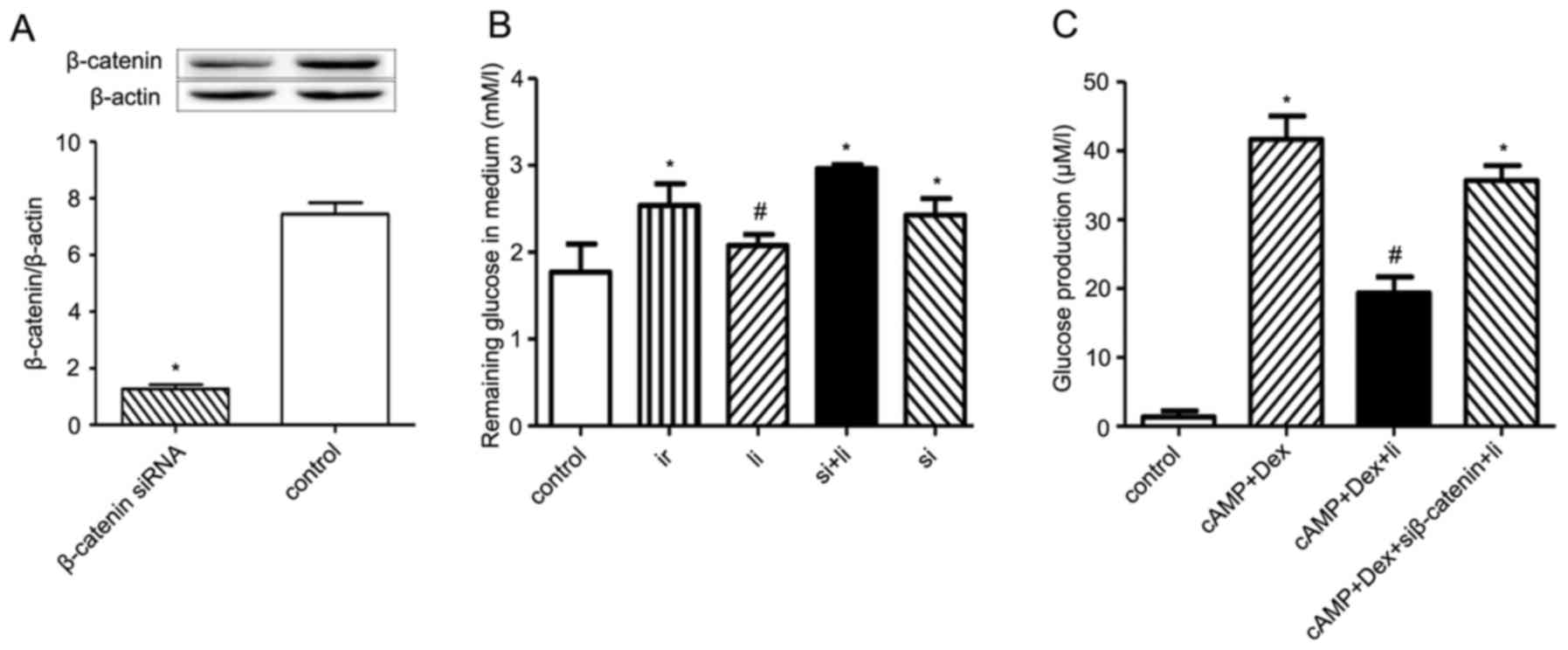
Liraglutide increases glucose uptake
and decreases glucose production in hepatocytes, but this effect is
reversed in hepatocytes transfected with β-catenin siRNA
To investigate the effect of β-catenin on
liraglutide-induced decrease in insulin resistance in vitro,
HepG2 human hepatocytes were transfected with β-catenin siRNA for
48 h. The transfection efficiency was subsequently determined by
verifying β-catenin protein levels using western blot analysis
(Fig. 3A). The results demonstrate
that the expression of β-catenin was significantly reduced in
transfected cells (1.27±0.15) compared with the control cells
(7.44±0.40; P<0.05).
The effects of liraglutide on glucose uptake were
investigated by establishing an in vitro insulin resistance
model in palmitate- and high glucose-stimulated human hepatocytes.
The amount of glucose in cell culture supernatants was quantified
by the GOD-POD assay. Insulin resistant cells had a lower glucose
uptake rate compared with the control cells (P<0.05, Fig. 3A). Following treatment with
liraglutide, the rate of glucose uptake increased significantly
compared with the insulin resistant cells (P<0.05). Furthermore,
silencing β-catenin resulted in a decreased glucose uptake rate
(P<0.05); however, treatment with liraglutide in β-catenin
silenced cells had no effect on glucose uptake (Fig. 3B). In the glucose production
experiment, liraglutide significantly suppressed cAMP+Dex-induced
glucose production (P<0.05; Fig.
3C). Silencing β-catenin resulted in increased glucose
production in the liraglutide treated group compared with the
control (P<0.05). These results indicate that liraglutide
increased glucose uptake and suppressed glucose production in
hepatocytes via a β-catenin dependent mechanism.
Liraglutide activates the Wnt
signaling pathway and decreases liver gluconeogenesis in vitro
To investigate whether the effects of liraglutide
are mediated by the Wnt signaling pathway in vitro, HepG2
cells were transfected with β-catenin siRNA and treated with or
without 100 nM liraglutide. Western blot analysis revealed that
liraglutide increased the expression of β-catenin protein and that
this effect was reversed following β-catenin silencing (P<0.05;
Fig. 4A and B). Furthermore, as
demonstrated in Fig. 4A and C,
liraglutide significantly increased the p-GSK3β/GSK3β ratio,
(P<0.05). The effect of liraglutide on GSK3β was partially
inhibited in cells transfected with β-catenin siRNA. Treatment with
liraglutide resulted in significantly decreased PEPCK protein
expression compared with the control group; however, PEPCK
expression was increased following transfection with β-catenin
siRNA (both P<0.05; Fig. 4A and
D). Furthermore, the p-FoxO1/FoxO1 ratio was significantly
increased in liraglutide-treated hepatocytes compared with both
control and β-catenin siRNA transfected cells (both P<0.05;
Fig. 4A and E). Treatment with
liraglutide in β-catenin-silenced cells decreased the ratio of
pFoxO1/FoxO1, suggesting that liraglutide exerted its effects on
FoxO1 and PEPCK via the canonical Wnt signaling pathway.
Additionally, the levels of pAKT and total AKT, which are
associated with the insulin signaling pathway, were analyzed
(Fig. 4A and F). Western blot
analysis indicated that liraglutide significantly increased the
pAKT/AKT ratio compared with the control group (P<0.05).
Transfection of hepatocytes with β-catenin siRNA had no significant
effect on the ratio of pAKT/AKT, which remained similar to that
identified in liraglutide-only treated cells. These results suggest
that liraglutide acts mainly via the canonical β-catenin Wnt
signaling pathway in hepatocytes.
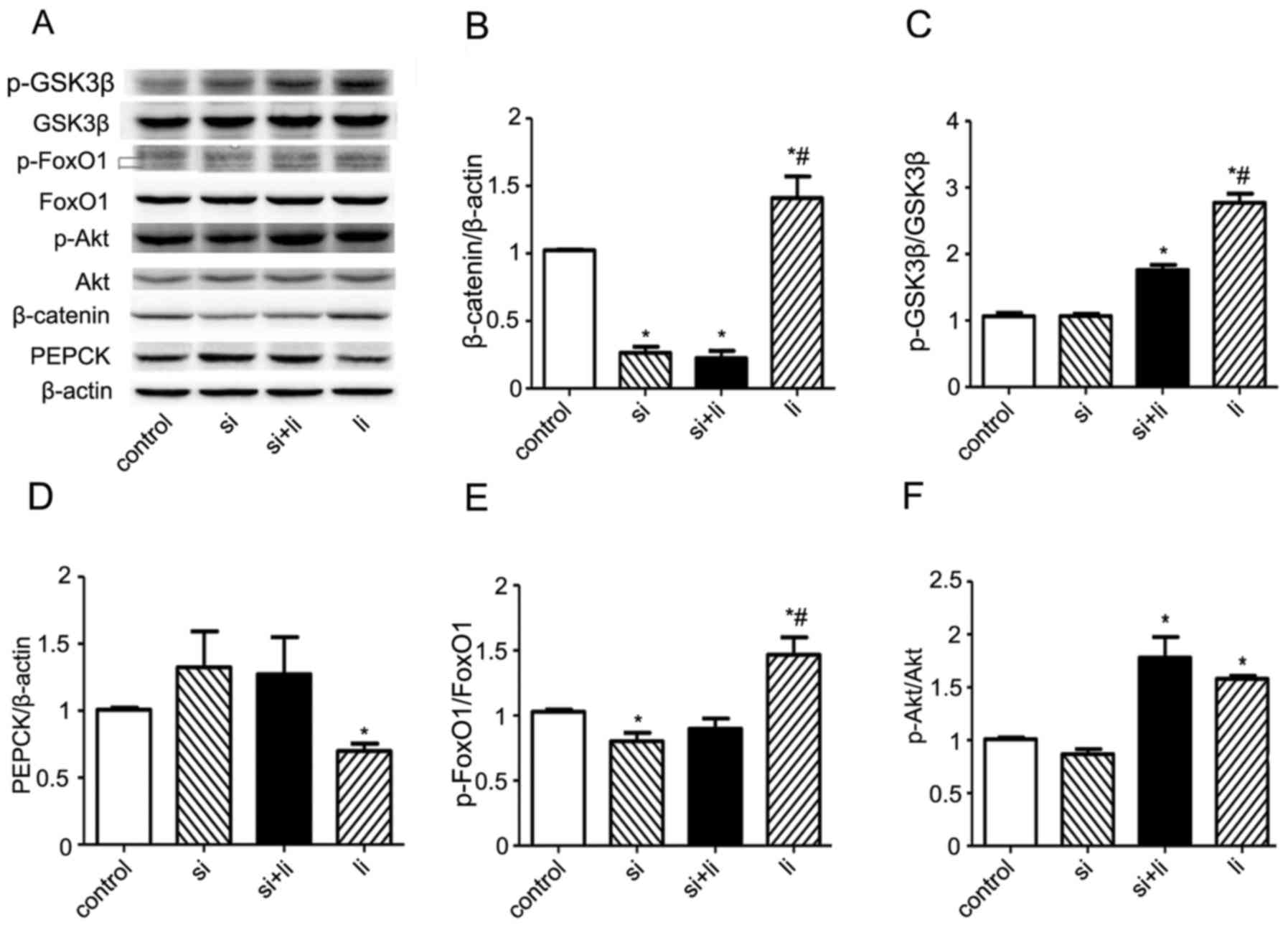 | Figure 4.Liraglutide activates the Wnt
signaling pathway and decreases liver gluconeogenesis in
vitro. (A) Representative western blot images of the Wnt
signaling pathway molecules, insulin signaling pathway molecules
and molecules associated with liver gluconeogenesis. Protein levels
of Wnt signaling pathway molecules, including (B) β-catenin and (C)
total/p-GSK3β. Expression levels of liver gluconeogenesis
molecules, including (D) PEPCK and (E) total/pFoxO1. Expression
levels of insulin signaling pathway molecules, including (F)
p-AKT/Akt. The blots are representative of three independent
experiments with similar results. β-actin was used as a loading
control. All values are expressed as the mean ± standard error of
the mean. *P<0.05 vs. con and #P<0.05 vs. si. Ir, insulin
resistance; li, liraglutide; si, β-catenin small interfering RNA;
Wnt, proto-oncogene Wnt; TCF7L2, transcription factor 7 like 1; p,
phosphorylated; GSK3β, glycogen synthase kinase-3β; PEPCK,
phosphoenolpyruvate carboxykinase; G6Pase, glucose-6-phosphatase;
FoxO1, forkhead box O1; Akt, RAC-alpha serine/threonine-protein
kinase. |
Discussion
Liraglutide, a GLP-1 receptor agonist, is used for
the treatment of diabetes. In addition to stimulating insulin
secretion, liraglutide confers cardio-protective effects,
suppresses appetite and delays gastric emptying (27,28).
In patients with type 2 diabetes, liraglutide decreases the
concentration of plasma glucose, hemoglobin A1c, fructosamine and
free fatty acids, as well as improving insulin sensitivity and
β-cell function (4). Furthermore,
liraglutide inhibits glucagon release, improves insulin resistance
and protects other organs, including the brain, cardiac tissue and
muscles, from high glucose in an insulin-independent manner
(3,29,30).
The results of the present study demonstrate that liraglutide
markedly decreases plasma glucose, insulin, body weight and
HOMA-IR. HOMA-IR is a method used to assess insulin resistance,
which is inversely associated with insulin sensitivity as measured
using an euglycemic clamp (22).
Su et al (31) demonstrated
that GLP-1 significantly decreased HOMA-IR in db/db mice. Lee et
al (8) reported that exendin-4
improves lobular inflammation and markedly reduces the accumulation
of lipids, including free fatty acids and triglycerides in the
fatty liver of high fat diet-induced obese C57BL/6J mice, thereby
improving hepatic steatosis. In the present study, H&E staining
of liver specimens from db/db mice treated with liraglutide
revealed improved lobular structure, with reduced lobular
inflammation and fewer lipid droplets within cells compared with
the saline treated control group.
The liver regulates glucose metabolism and expresses
the GLP-1 receptor (6). Numerous
studies have demonstrated that GLP-1 reduces glucose production,
gluconeogenesis, fatty acid synthesis and oxidative stress, as well
as improving insulin resistance (5,32).
However, the mechanism underlying GLP-1-mediated improvements in
insulin resistance remains to be elucidated. The therapeutic effect
of GLP-1 against insulin resistance may be secondary to the effects
of weight loss and attenuation of hyperglycemia (32). Previous studies have also
demonstrated that GLP-1 may affect insulin resistance directly. Lee
et al (8) reported that
GLP-1 improved liver insulin resistance via activation of the
sirtuin 1/adenosine monophosphate-activated protein kinase
signaling pathway in C57BL/6J mice and HepG2 and Huh7 cell lines,
whilst also increasing the expression of phosphorylated FoxO1 and
GLUT2. GLP-1 promotes glycogen synthesis via activation of the
phosphatidylinositol 4,5-bisphosphate 3-kinase/AKT signaling
pathway in primary rat hepatocytes and this effect of GLP-1 is
similar to that induced by insulin in liver (33). In HepG2 cells, silencing FoxO1
resulted in reduced expression of PEPCK and G6Pase (34). GLP-1 inhibited the expression of
G6Pase and PEPCK in the liver and increased the phosphorylation of
AKT and FoxO1 in young mice (6).
In the present study, treatment with liraglutide resulted in
decreased expression of PEPCK and G6Pase and increased p-FoxO1
levels compared with the control group. PEPCK and G6Pase serve
roles in liver gluconeogenesis and decreased levels of these
enzymes reduce the production of glucose, suggesting that
liraglutide may inhibit the gluconeogenesis pathway. FoxO1
activates the expression of both PEPCK and G6Pase, whereas p-FoxO1
is targeted for proteasomal degradation (26). Furthermore, liraglutide-induced
p-FoxO1 may also contribute to the suppression of gluconeogenesis.
These observations support the results obtained in a previous
study, which reported that GLP-1 improved insulin resistance via
inhibiting PEPCK and G6Pase (6).
The Wnt signaling pathway in the liver is associated
with diabetes, glucose and lipid metabolism (35,36).
GLP-1 binds the GLP-1 receptor on cell membranes and activates
cAMP-dependent protein kinase A (PKA) via the secondary messenger
cAMP. GLP-1-mediated activation of PKA transmits the signal
intracellularly and phosphorylates β-catenin to activate the
canonical Wnt pathway (14).
Activation of the Wnt signaling pathway induces the transcription
of genes, including a diabetes risk gene TCF7L2 that stimulates the
proliferation of pancreatic β cells (14,20,21,37).
TCF7L2 silencing results in significantly increased expression of
PEPCK and G6Pase (35,38). FoxO1 and TCF7L2 competitively bind
β-catenin to regulate the hepatic metabolism of glucose. Under
starvation, FoxO1 competes with TCF7L2 and binds β-catenin, which
leads to increased gluconeogenesis (19). Insulin inhibits FoxO1 by
stimulating its phosphorylation, thereby allowing β-catenin to bind
TCF7L2 and inhibit liver gluconeogenesis (39,40).
In the present study, markedly increased levels of Wnt signaling
pathway molecules TCF7L2, β-catenin and phosphorylated GSK3β were
identified in mice treated with liraglutide, indicating a potential
association with liver glucose metabolism under diabetic
conditions. Db/db mice demonstrate >10-fold levels of plasma
leptin compared with wild-type mice, which may also contribute to
insulin resistance. GLP-1 markedly decreases levels of plasma
leptin in db/db mice (31,41). It is therefore possible that the
effect of liraglutide on liver gluconeogenesis in the present study
may result from decreased leptin levels. To study the association
between liraglutide and the Wnt signaling pathway, an in
vitro insulin resistance cell model transfected with β-catenin
siRNA was also used in the present study. The results demonstrate
that treatment with liraglutide increases p-FoxO1 expression whilst
downregulating PEPCK and the glucose uptake rate; β-catenin
silencing effectively reversed these effects. The results of the
present study suggest that liraglutide improves insulin resistance
by increasing glucose uptake via β-catenin/Wnt signaling.
Levels of pAKT/AKT, which serve a role in the
insulin signaling pathway, were investigated. The results of the
present study demonstrate that liraglutide markedly increases the
ratio of pAKT/AKT compared with control hepatocytes. Silencing
β-catenin had no effect on the pAKT/AKT ratio, suggesting that the
effect of liraglutide on AKT phosphorylation is not mediated by
β-catenin/Wnt signaling. AKT acts upstream of GSK3β and β-catenin.
Therefore, liraglutide may participate in the insulin pathway by
activating the canonical Wnt signaling pathway via increased
phosphorylation of GSK3β and not AKT (42,43).
In conclusion, the present study demonstrated that
liraglutide decreases plasma glucose levels, body weight and
HOMA-IR in mice with diabetes. Liraglutide reduced lobular
inflammation and accumulation of lipid droplets, increased
β-catenin, TCF7L2 and pGSK3β expression levels and led to the
inactivation of FoxO1 and its downstream target genes, PEPCK and
G6Pase. In HepG2 cells, liraglutide increased glucose uptake and
reduced hepatic gluconeogenesis; these effects were reversed in
cells transfected with β-catenin siRNA. Therefore, the present
study suggests that treatment with liraglutide may increase the
rate of glucose uptake and improve liver insulin resistance by
activating the canonical Wnt signaling pathway. The results of the
present study provide a novel mechanism by which liraglutide may
improve insulin resistance in the liver. Furthermore, targeting the
canonical Wnt signaling may have therapeutic potential for the
treatment of altered hepatic physiology in insulin resistance and
type 2 diabetes.
Acknowledgements
The present study was supported by a grant from the
National Natural Science Foundation of China (grant no. 81370941,
YG.).
References
|
1
|
Baggio LL and Drucker DJ: Biology of
incretins: GLP-1 and GIP. Gastroenterology. 132:2131–2157. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chiang YT, Ip W and Jin T: The role of the
Wnt signaling pathway in incretin hormone production and function.
Front Physiol. 3:2732012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Salehi M, Aulinger B, Prigeon RL and
D'Alessio DA: Effect of endogenous GLP-1 on insulin secretion in
type 2 diabetes. Diabetes. 59:1330–1337. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zander M, Madsbad S, Madsen JL and Holst
JJ: Effect of 6-week course of glucagon-like peptide 1 on glycaemic
control, insulin sensitivity, and beta-cell function in type 2
diabetes: A parallel-group study. Lancet. 359:824–830. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ding X, Saxena NK, Lin S, Gupta NA and
Anania FA: Exendin-4, a glucagon-like protein-1 (GLP-1) receptor
agonist, reverses hepatic steatosis in ob/ob mice. Hepatology.
43:173–181. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fan R, Kang Z, He L, Chan J and Xu G:
Exendin-4 improves blood glucose control in both young and aging
normal non-diabetic mice, possible contribution of beta cell
independent effects. PLoS One. 6:e204432011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gupta NA, Mells J, Dunham RM, Grakoui A,
Handy J, Saxena NK and Anania FA: Glucagon-like peptide-1 receptor
is present on human hepatocytes and has a direct role in decreasing
hepatic steatosis in vitro by modulating elements of the
insulin signaling pathway. Hepatology. 51:1584–1592. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee J, Hong SW, Chae SW, Kim DH, Choi JH,
Bae JC, Park SE, Rhee EJ, Park CY, Oh KW, et al: Exendin-4 improves
steatohepatitis by increasing Sirt1 expression in high-fat
diet-induced obese C57BL/6J mice. PLoS One. 7:e313942012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Thompson MD and Monga SP: WNT/beta-catenin
signaling in liver health and disease. Hepatology. 45:1298–1305.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ackers I and Malgor R: Interrelationship
of canonical and non-canonical Wnt signalling pathways in chronic
metabolic diseases. Diab Vasc Dis Res. Nov 1–2017.(Epub ahead of
print). PubMed/NCBI
|
|
11
|
Shao W, Wang D, Chiang YT, Ip W, Zhu L, Xu
F, Columbus J, Belsham DD, Irwin DM, Zhang H, et al: The Wnt
signaling pathway effector TCF7L2 controls gut and brain
proglucagon gene expression and glucose homeostasis. Diabetes.
62:789–800. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jin T and Liu L: The Wnt signaling pathway
effector TCF7L2 and type 2 diabetes mellitus. Mol Endocrinol.
22:2383–2392. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Akiyama T: Wnt/beta-catenin signaling.
Cytokine Growth Factor Rev. 11:273–282. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xiong X, Shao W and Jin T: New insight
into the mechanisms underlying the function of the incretin hormone
glucagon-like peptide-1 in pancreatic β-cells: The involvement of
the Wnt signaling pathway effector β-catenin. Islets. 4:359–365.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Grant SF, Thorleifsson G, Reynisdottir I,
Benediktsson R, Manolescu A, Sainz J, Helgason A, Stefansson H,
Emilsson V, Helgadottir A, et al: Variant of transcription factor
7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet.
38:320–323. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
da Silva Xavier G, Mondragon A, Sun G,
Chen L, McGinty JA, French PM and Rutter GA: Abnormal glucose
tolerance and insulin secretion in pancreas-specific Tcf7l2-null
mice. Diabetologia. 55:2667–2676. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Boj SF, van Es JH, Huch M, Li VS, José A,
Hatzis P, Mokry M, Haegebarth A, van den Born M, Chambon P, et al:
Diabetes risk gene and Wnt effector Tcf7l2/TCF4 controls hepatic
response to perinatal and adult metabolic demand. Cell.
151:1595–1607. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kaur K, Vig S, Srivastava R, Mishra A,
Singh VP, Srivastava AK and Datta M: Elevated hepatic miR-22-3p
expression impairs gluconeogenesis by silencing the Wnt-responsive
transcription factor Tcf7. Diabetes. 64:3659–3669. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ip W, Shao W, Song Z, Chen Z, Wheeler MB
and Jin T: Liver-specific expression of dominant-negative
transcription factor 7-like 2 causes progressive impairment in
glucose homeostasis. Diabetes. 64:1923–1932. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gustafson B and Smith U: WNT signalling is
both an inducer and effector of glucagon-like peptide-1.
Diabetologia. 51:1768–1770. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu Z and Habener JF: Glucagon-like
peptide-1 activation of TCF7L2-dependent Wnt signaling enhances
pancreatic beta cell proliferation. J Biol Chem. 283:8723–8735.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Matthews DR, Hosker JP, Rudenski AS,
Naylor BA, Treacher DF and Turner RC: Homeostasis model assessment:
Insulin resistance and beta-cell function from fasting plasma
glucose and insulin concentrations in man. Diabetologia.
28:412–419. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang Z, Li B, Meng X, Yao S, Jin L, Yang
J, Wang J, Zhang H, Zhang Z, Cai D, et al: Berberine prevents
progression from hepatic steatosis to steatohepatitis and fibrosis
by reducing endoplasmic reticulum stress. Sci Rep. 6:208482016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yin J, Hu R, Chen M, Tang J, Li F, Yang Y
and Chen J: Effects of berberine on glucose metabolism in
vitro. Metabolism. 51:1439–1443. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tikhanovich I, Cox J and Weinman SA:
Forkhead box class O transcription factors in liver function and
disease. J Gastroenterol Hepatol. 28 Suppl 1:S125–S131. 2013.
View Article : Google Scholar
|
|
27
|
Desouza CV, Gupta N and Patel A:
Cardiometabolic effects of a new class of antidiabetic agents. Clin
Ther. 37:1178–1194. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
van Can J, Sloth B, Jensen CB, Flint A,
Blaak EE and Saris WH: Effects of the once-daily GLP-1 analog
liraglutide on gastric emptying, glycemic parameters, appetite and
energy metabolism in obese, non-diabetic adults. Int J Obes (Lond).
38:784–793. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nyström T, Gutniak MK, Zhang Q, Zhang F,
Holst JJ, Ahrén B and Sjöholm A: Effects of glucagon-like peptide-1
on endothelial function in type 2 diabetes patients with stable
coronary artery disease. Am J Physiol Endocrinol Metab.
287:E1209–E1215. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Alcántara AI, Morales M, Delgado E,
López-Delgado MI, Clemente F, Luque MA, Malaisse WJ, Valverde I and
Villanueva-Peñacarrillo ML: Exendin-4 agonist and
exendin(9–39)amide antagonist of the GLP-1(7–36)amide effects in
liver and muscle. Arch Biochem Biophys. 341:1–7. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Su H, He M, Li H, Liu Q, Wang J, Wang Y,
Gao W, Zhou L, Liao J, Young AA and Wang MW: Boc5, a non-peptidic
glucagon-like Peptide-1 receptor agonist, invokes sustained
glycemic control and weight loss in diabetic mice. PLoS One.
3:e28922008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cho YM, Fujita Y and Kieffer TJ:
Glucagon-like peptide-1: Glucose homeostasis and beyond. Annu Rev
Physiol. 76:535–559. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Redondo A, Trigo MV, Acitores A, Valverde
I and Villanueva-Peñacarrillo ML: Cell signalling of the GLP-1
action in rat liver. Mol Cell Endocrinol. 204:43–50. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nerurkar PV, Nishioka A, Eck PO, Johns LM,
Volper E and Nerurkar VR: Regulation of glucose metabolism via
hepatic forkhead transcription factor 1 (FoxO1) by Morinda
citrifolia (noni) in high-fat diet-induced obese mice. Br J Nutr.
108:218–228. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ip W, Shao W, Chiang YT and Jin T: The Wnt
signaling pathway effector TCF7L2 is upregulated by insulin and
represses hepatic gluconeogenesis. Am J Physiol Endocrinol Metab.
303:E1166–E1176. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu H, Fergusson MM, Wu JJ, Rovira II, Liu
J, Gavrilova O, Lu T, Bao J, Han D, Sack MN and Finkel T: Wnt
signaling regulates hepatic metabolism. Sci Signal. 4:ra62011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hino S, Tanji C, Nakayama KI and Kikuchi
A: Phosphorylation of beta-catenin by cyclic AMP-dependent protein
kinase stabilizes beta-catenin through inhibition of its
ubiquitination. Mol Cell Biol. 25:9063–9072. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Norton L, Fourcaudot M, Abdul-Ghani MA,
Winnier D, Mehta FF, Jenkinson CP and Defronzo RA: Chromatin
occupancy of transcription factor 7-like 2 (TCF7L2) and its role in
hepatic glucose metabolism. Diabetologia. 54:3132–3142. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hoogeboom D, Essers MA, Polderman PE,
Voets E, Smits LM and Burgering BM: Interaction of FOXO with
beta-catenin inhibits beta-catenin/T cell factor activity. J Biol
Chem. 283:9224–9230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Arai T, Kano F and Murata M: Translocation
of forkhead box O1 to the nuclear periphery induces histone
modifications that regulate transcriptional repression of PCK1 in
HepG2 cells. Genes Cells. 20:340–357. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Patel V, Joharapurkar A, Gandhi T, Patel
K, Dhanesha N, Kshirsagar S, Dhote V, Detroja J, Bahekar R and Jain
M: Omeprazole improves the anti-obesity and antidiabetic effects of
exendin-4 in db/db mice (−4 db/db)*. J Diabetes. 5:163–171. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yi F, Sun J, Lim GE, Fantus IG, Brubaker
PL and Jin T: Cross talk between the insulin and Wnt signaling
pathways: Evidence from intestinal endocrine L cells.
Endocrinology. 149:2341–2351. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sun J, Wang D and Jin T: Insulin alters
the expression of components of the Wnt signaling pathway including
TCF-4 in the intestinal cells. Biochim Biophys Acta. 1800:344–351.
2010. View Article : Google Scholar : PubMed/NCBI
|