Introduction
Primary lung cancer is the leading cause of global
cancer morbidity and cancer-associated mortality, accounting for
~1.59 million cases of mortality annually (1,2).
Among lung cancer types, >85% of cases are presently identified
as non-small cell lung cancer (NSCLC) (3). The treatment of patients diagnosed
with lung cancer has significantly improved due to medical
advances; however, the prognosis for NSCLC is remains poor and the
5-year overall survival rate is only 15% (4). The poor prognosis of patients with
NSCLC is closely associated with distant metastasis. Consequently,
further investigation is required to understand the potential
mechanisms of metastasis in NSCLC.
Krüppel-like factors (KLFs) belong to the
evolutionary conserved family of zinc finger transcription factors
in mammals (5). KLF4, one of the
earliest identified genes in the KLF family, is widely detected in
a variety of human tissues and is reported to be crucial for
diverse physiological processes, including development,
differentiation and apoptosis, and stemness in embryonic stem cells
(6). KLF4 is a bifunctional
molecule displaying dual functions in different human cancers
depending on the intracellular environment or the cell type. KLF4
can act as an oncogene, and it was previously reported that the
expression levels of KLF4 were elevated in breast cancer and
squamous cell carcinoma (7,8);
whereas, in esophageal, human colon and gastric cancer, KLF4 served
as a tumor suppressor (9–11). Research focusing on the effect of
KLF4 in tumorigenesis has increased in recent years; however, the
underlying mechanism of KLF4 in tumorigenesis is not clear.
MicroRNAs (miRNAs) are single-stranded, non-coding RNA molecules of
18–22 nucleotides that can cause translational inhibition and/or
mRNA degradation by binding to the 3′ untranslated region (UTR) of
target mRNAs (12). It has been
estimated that miRNAs regulate the expression of a third of human
genes (13). miRNAs are involved
in the regulation of diverse biological processes, including
invasion, migration, proliferation and cell apoptosis (14–16).
Abnormal expression of miRNAs, including the upregulation of
miR-429 (17), miR-19a (18) and miR-146a (19), and downregulation of miR-143
(20), miR-3666 (21) and miR-29b (22) have been detected in lung cancer.
Thus, miRNAs have potential as biomarker for diagnosis, targeted
therapy and prognosis.
miR-25 is one of the miR-106b-25 cluster (miR-93,
miR-25 and miR-106b) (23,24). Evidence has highlighted the key
regulatory roles of miR-25 in tumor progression. For instance, Su
et al (25) reported that
upregulated miR-25 in liver cancer tissues led to a shorter
survival time. Li et al (26) also reported that overexpressed
miR-25 increased the proliferation, invasion and migration of
gastric cancer cells by inducing the degradation of transducer of
ERBB2 1 and also demonstrated that serum concentrations of miR-25
were positively associated with poor prognosis in patients with
gastric cancer. However, in certain diseases, miR-25 was
downregulated. For instance, diminished expression of miR-25 in
colorectal cancer induced an increase in expression of angiopoietin
like 2 and resulted in reduced cell clones, inhibited invasion and
migration (27). Furthermore, Wu
et al (28) studied miR-25,
which was reported to promote cell growth and inhibit apoptosis in
NSCLC by reducing modulator of apoptosis 1 expression. Xiang et
al (29) revealed that miR-25
was overexpressed in NSCLC cells and tissues, and promoted the
motility and proliferation of NSCLC cells, in part by diminishing
F-box and WD repeat domain containing 7 expression levels. The
majority of studies have investigated the biological function of
miR-25 in lung cancer, however the underlying mechanism is remains
unknown. The aim of the present study was to research the function
of miR-25 in NSCLC and to improve the understanding of the
underlying mechanism of miR-25 in NSCLC.
In the present study, the expression levels of
miR-25 were examined in NSCLC tissues via reverse
transcription-quantitative polymerase chain reaction (RT-qPCR).
Subsequently, in vitro assays, including cell migration and
invasion assays, were conducted to understand the biological
functions of miR-25 in NSCLC. Additionally, KLF4 was demonstrated
to be a novel target gene of miR-25, and to be involved in the
invasion and migration in NSCLC cells. Finally, the phosphorylation
of extracellular signal-regulated kinase (ERK1/2; p44/42
mitogen-activated protein kinase) demonstrated that the ERK
signaling pathway was downstream of miR-25. Collectively, the
results of the present study revealed that miR-25 promoted the
migration and invasion of NSCLC cells via regulation of the ERK1/2
signaling pathway by targeting KLF4.
Materials and methods
Tissue samples and cell lines
Pairs of normal and NSCLC tissue specimens (n=31; 21
male and 10 female; age, 41–77) were obtained from patients that
received surgery at the Cardiovascular Surgery of First Affiliated
Hospital of Gannan Medical University (Ganzhou, China) between
January 2014 and March 2016. Specimens were placed immediately into
liquid nitrogen following collection and were then stored at −80°C
until use. Written informed consent was obtained from the patients
involved in the present study, which was approved by the Ethics
Committee of Gannan Medical University. Human A549 and Calu1 NSCLC
cell lines, GES-1 and 293T cells used in the present study were
obtained from the Institute of Biochemistry and Cell Biology of the
Chinese Academy of Sciences (Shanghai, China), A549, GES-1 and 293T
cells were cultured in Dulbecco's modified Eagle's medium (DMEM),
Calu1 cells were cultured in McCOYs 5A (HyClone; GE Healthcare Life
Sciences, Logan, UT, USA). The culture medium was supplemented with
1% penicillin and streptomycin (HyClone; GE Healthcare Life
Sciences), and 10% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). The cells were cultivated in a
humidified environment at 37°C with 5% CO2.
RT-qPCR
Total RNA was extracted from the cultured cells and
NSCLC tissues with TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocols. RNA
was dissolved in diethylpyrocarbonate water. RT of mRNA and miRNA
was conducted using ReverTra Ace® qPCR RT kit (Toyobo
Life Science, Osaka, Japan) and miRNA reverse transcription kit
(Promega Corporation, Madison, WI, USA). cDNA was subsequently
harvested and qPCR was conducted with SYBR® Fast qPCR
Mix (Takara Biotechnology Co., Ltd., Dalian, China) using a 7500
Fast Real-Time PCR detection system (Thermo Fisher Scientific,
Inc.). qPCR reaction conditions were as follows: 95°C
pre-denaturation for 3 min, followed by 40 cycles of denaturing at
95°C for 3 sec, annealing and synthesis at 60°C for 30 sec; the Cq
value was then determined. The primers for RT-qPCR was as follows:
miR-25 forward, 5′-GCAGCATTGCACTTGTCTCG-3′ and reverse,
5′-AGTGCAGGGTCCGAGGTATTC-3′; and KLF4 forward,
5′-AAGCCAAAGAGGGGAAGACG-3′ and reverse,
5′-GTGCCTGGTCAGTTCATCTGAG-3′. The 18S and U6 were taken as internal
control for KLF4 and miR-25 respectively. To calculate the
expression levels of miR-25 and KLF4, the 2−ΔΔCq method
was used (30). Overexpression was
defined as a ≥1.5-fold change and underexpression as a ≤0.67-fold
change, based on a previous study (31). All experiments were repeated three
times independently.
Bioinformatics analysis
To identify the targets of miR-25 that may regulate
the migration and invasion of NSCLC, the target prediction tool
TargetScan version 7.1 (http://www.targetscan.org/mmu_71/) was used for target
prediction of miR-25. The Database for Annotation, Visualization
and Integrated Discovery (version 6.7; https://david.ncifcrf.gov) is a functional annotation
tool that provides a comprehensive set of biological annotations to
understand the meaning underlying a large list of target genes. The
Gene Ontology (GO) (version 4.0; http://www.geneontology.org/) and Kyoto Encyclopedia
of Genes and Genomes (KEGG; http://www.genome.jp/kegg/pathway.html) databases were
used for functional clustering analysis and pathway location.
Vector construction
The primers on the flanks of the coding sequences of
KLF4 were: Forward, ATACTCGAGATGAGGCAGCCACCTGGC (Xhol) and reverse,
CCACTAGTTTAAAAATGCCTCTTCATGTGTAAG (SpeI). Primers were designed
using Primer3 (version 4.1.0; http://primer3.ut.ee) according to the reference
sequence in the University of California Santa Cruz Genome Browser
(https://genome.ucsc.edu). The KLF4 cDNA reverse
transcribed from mRNA of GES-1 was used as a template. For the KLF4
vector, KLF4 cDNA was cloned into the pCMV-N-Flag plasmid (Beyotime
Institute of Biotechnology, Shanghai, China) between XhoI and SpeI
restriction sites to construct the KLF4 overexpression plasmid. For
the 3′-UTR of the KLF4 vector, the primers for the 3′-UTR of KLF4
containing the putative binding site of miR-25 was amplified by
PCR. The primers for the 3′-UTR of KLF4 sequence were: Forward,
5′-GGTCTCGAGGGATATGACCCACACTGCC-3′ (Xho1I) and reverse,
3′-TTAAGCGGCCGCCCTTGAGTATGCAAAATACAAACTCC-5′ (NOTI). and cloned
into the psi-CHECK2 vector (Promega Corporation) using the
restriction endonucleases Xho1I and NotI. The insertion regions
were sequenced by Biosune (Platinum Shang Biotechnology, Shanghai,
China) to ensure the accuracy of each base.
Cell transfection
miR-25 mimics/inhibitors and its negative control
(NC) miRNAs (mimic-NC/inhibitor-NC, respectively), siRNA and its
negative control (si-NC) were purchased from Guangzhou RiboBio Co.,
Ltd. (Guangzhou, China). NC miRNAs of 20–30 base pairs served as
the negative control and exhibited minimal effect on cells in the
present study. miR-25 mimics are chemically synthesized,
double-stranded RNAs which mimic mature endogenous miR-25 following
transfection into cells. miR-25 inhibitors are chemically
synthesized, single-stranded modified RNAs that specifically
inhibit endogenous miRNA function following transfection into
cells. Cells were transfected with 50 nM miR-25 mimic (sense,
5′-CAUUGCACUUGUCUCGGUCUGA-3′ and antisense,
3′-GUAACGUGAACAGAGCCAGACU-5′), inhibitor
(5′-UCAGACCGAGACAAGUGCAAUG-3′), mimic-NC (sequence unavailable),
inhibitor-NC (sequence unavailable), small interfering (si)RNA
(hs-KLF4-si forward, 5′-GAGUCAUCUUGUGAGUGGAdTdT-3′ and reverse,
5′-UCCACUCACAAGAUGACUCdTdT-3′) targeting KLF, or si-NC (sequence
unavailable) with 3 µl Lipofectamine® 2000 transfection
reagent (Invitrogen; Thermo Fisher Scientific, Inc.). Plasmid
transfection was conducted using 2 µg KLF4 overexpression plasmid
or KLF4-3′UTR plasmid with 4 µl Lipofectamine® 2000
according to the manufacturer's protocols.
Luciferase reporter assay
293T cells transfected with miR-25/miR-NC mimics and
wild type (WT)/mutated (MUT) psi-CHECK2-KLF4-3′UTR were transferred
into 24-well plates and cultured until 90% confluence was reached.
Luciferase activity was detected using the
Dual-Luciferase® Reporter Assay System kit (cat. no.
E1910; Promega Corporation). After 48 h post-transfection, cells
were collected, centrifuged at 56,669 × g for 1 min at 4°C and
lysed with passive lysis buffer (Promega Corporation). Culture
plates were placed on a rocking platform for 15 min to ensure
complete and even coverage of the cell monolayer with PLB. Cell
lysates were transferred to a tube, and lysate samples were
centrifuged for 30 sec at 56,669 × g at 4°C. Cell lysate (20 µl)
was transferred into the luminometer tube containing Luciferase
Assay Reagent II and was mixed by pipetting two or three times.
Stop & Glo® (100 µl) Reagent was added to the tube
which was vortexed briefly to mix. The relative luciferase activity
was subsequently detected with a microplate reader (Synergy 2;
BioTek China, Beijing, China). The ratio of the Renilla
fluorescence value to the firefly fluorescence value was
calculated.
Wound healing assay
Cells were transfected with miR-25 inhibitor, mimic,
KLF4 siRNA, KLF4 overexpression plasmid, miR-25 inhibitor NC
(in-NC), miR-25 mimic NC (miR-NC), si-NC or empty vector. The
ability of cell migration was evaluated via a wound healing assay.
Calu1 and A549 cells were seeded into a 6-well plate (Corning
Incorporated, Corning, NY, USA) at a density of 1.8×105
cells/ml and cultured, then transferred to 12-well plate 24 h
post-transfection and permitted to grow to a density of 90%. Cells
were scratched with a sterile yellow pipette tip and then washed
several times with PBS to remove non-adherent cells. Leica Image
Analysis (Leica Microsystem, Inc., Buffalo Grove, IL, USA) was used
to capture images; gaps were measured using ImageJ software
(version 1.51t; National Institutes of Health, Bethesda, MD, USA)
to calculate healing percentages.
Transwell assay
Matrigel was pre-cooled and diluted 1:3 in medium
without FBS, 50 µl was added to the upper chamber and placed in the
incubator for 1 h. The cells were suspended with the culture medium
containing 0.1% bovine serum albumin (BSA; Beyotime Institute of
Biotechnology) at a density of 5×105/ml, and 200 µl was
added in the upper chamber. A total of ٥٠٠ µl DMEM or McCOYs 5A
medium containing 15% FBS was added to the lower chamber. The
culture plates were removed from the incubator following incubation
for 24 or 48 h at 5% CO2 and 37°C. The cells from the
upper layer of the chamber's filter membrane were cleared, and the
chamber was washed with PBS, then placed into 95% ethanol to fix
the cells adhering on the undersurface of filter membrane at room
temperature for 10 min, cells were then stained with 0.5% crystal
violet (2 mg/ml; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) at
room temperature for 30 min. The number of cells was counted in
five fields that were randomly selected under a light microscope,
the average number of cells was recorded and analyzed.
Western blotting
Total proteins were harvested from transfected cells
at 48 h after transfection and lysed using a mixture of
radioimmunoprecipitation assay buffer (Beyotime Institute of
Biotechnology), phenylmethane sulfonyl fluoride (Beyotime Institute
of Biotechnology) and protease inhibitor cocktail (Roche Applied
Science, Penzberg, Germany). The protein amount was determined
using a bicinchoninic acid assay kit (Beyotime Institute of
Biotechnology); 30 µg total protein was used for western blotting.
Different sized proteins were separated by the 10% SDS gel, then
electrotransferred to polyvinylidene fluoride membranes (Merck
KGaA). BSA (2–5%) was applied to the membranes, which were
incubated at room temperature for 1 h to remove nonspecific
binding. Subsequently, the membranes were incubated with the
following primary antibodies overnight at 4°C (1:1,000): Matrix
metalloproteinase 11 (MMP11; cat. no. ab201757; Abcam, Cambridge,
UK); E-cadherin (cat. no. ab1416; Abcam); KLF4 (cat. no. 043474;
CST Biological Reagents Co., Ltd., Shanghai, China); phosphorylated
(p)-ERK1/2 (cat. no. 4370S; CST Biological Reagents Co., Ltd.);
ERK1/2 (cat. no. 4695P; CST Biological Reagents Co., Ltd.);
N-cadherin (cat. no. 4061p; CST Biological Reagents Co., Ltd.);
vimentin (cat. no. 12826; Cell Signaling Technology, Inc., Danvers,
MA, USA); anti-β-actin antibody (1:5,000; cat. no. sc-47778; Santa
Cruz Biotechnology, Inc., Dallas, TX, USA) and the membrane was
incubated. The membranes was washed three times with PBS with
Tween-20 to remove the unbound primary antibody, then incubated
with the secondary antibodies of corresponding species for 1 h
[1:5,000, anti-rabbit (cat. no. BS13278; Biogot Technology Co.,
Ltd., Nanjing, China) or anti-mouse (cat. no. BS10043; Biogot
Technology Co., Ltd.)] at 37°C for 1 h. Protein bands were scanned
and visualized under automated chemiluminescence image analysis
system (Tanon 5200; Tanon Science and Technology Co., Ltd.,
Shanghai, China) following incubation with an enhanced
chemiluminescent substrate (Thermo Fisher Scientific, Inc.) for 2
min. β-actin served as the internal control.
Statistical analysis
The data of the present study are presented as the
mean ± standard deviation from three independent experiments and
analyzed with version 21.0 of SPSS (IBM Corp., Armonk, NY, USA).
The difference between two groups was analyzed by Student's t-test.
One-way analysis of variance was to compare multiple groups
followed by Tukey's post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
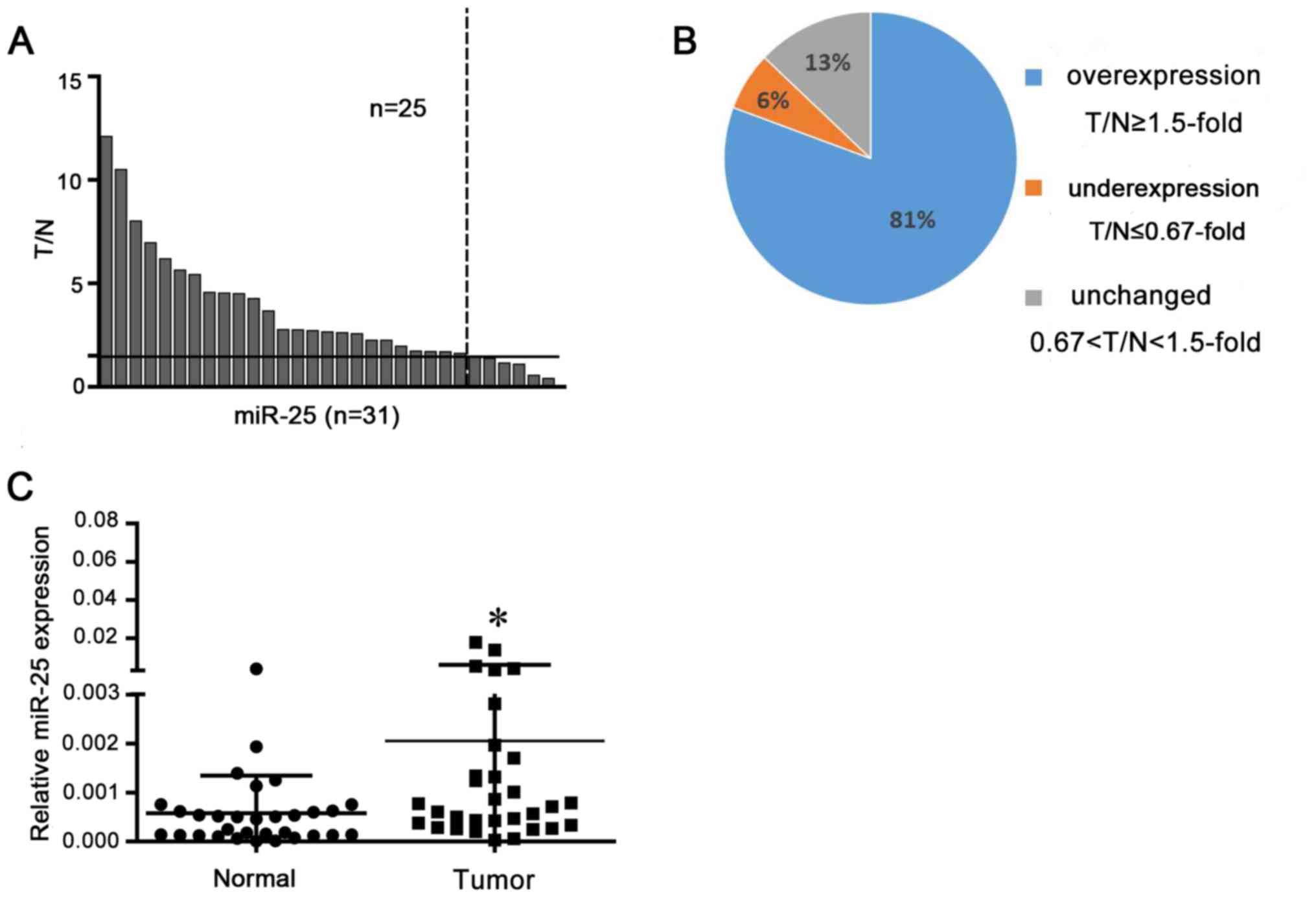
miR-25 is overexpressed in NSCLC
tissues
miR-25 expression levels were analyzed in NSCLC
tissues and matched normal tissue by RT-qPCR. Overexpression was
defined as a >1.5 fold increase and underexpression a <0.67
fold decrease. Analysis demonstrated that 25 of the 31 tissues
exhibited miR-25 overexpression compared with in corresponding
normal tissues (P<0.05; Fig.
1).
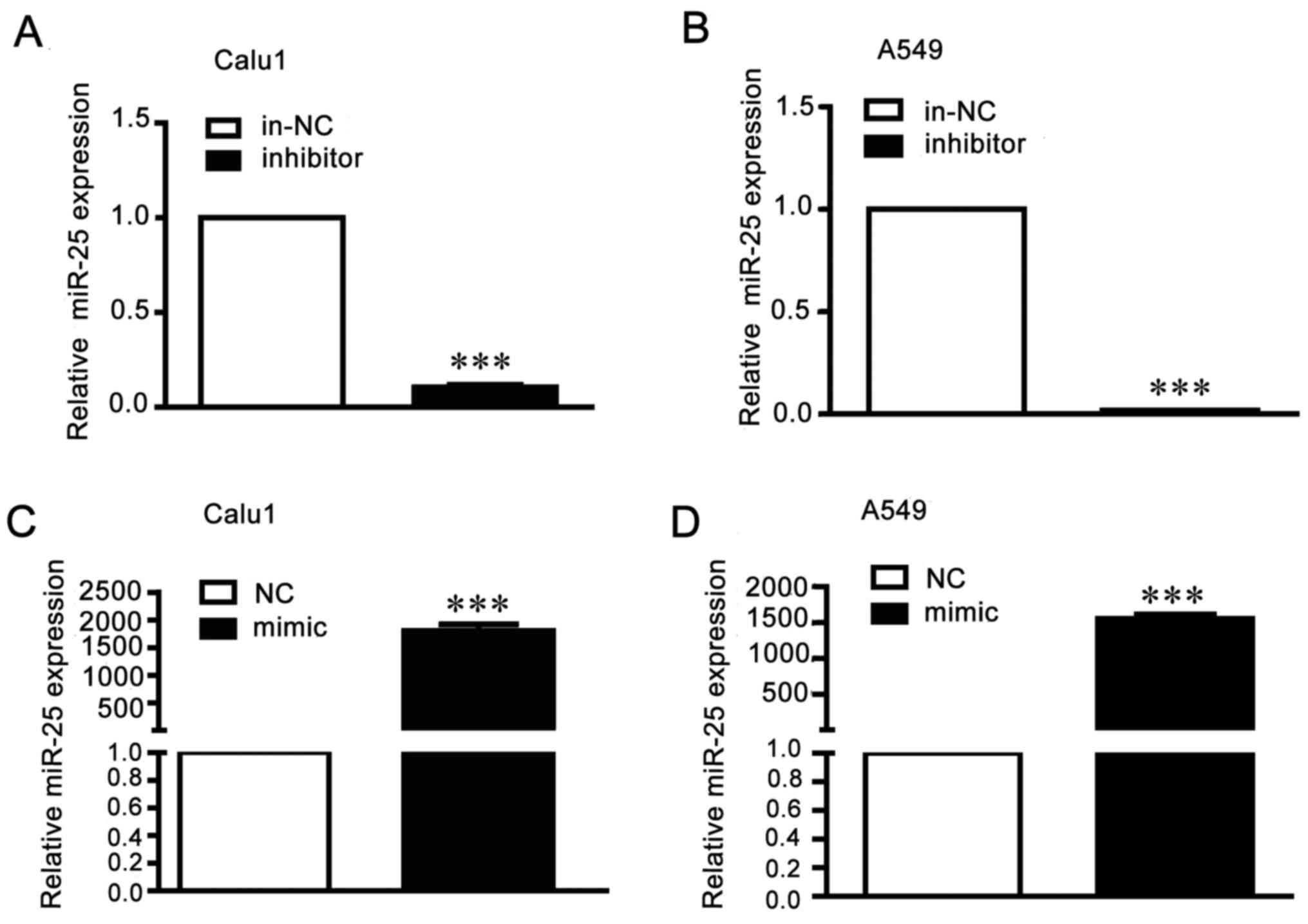
miR-25 regulates invasion and
migration of NSCLC cells
To research the function of miR-25 in migration and
invasion in NSCLC cells, A549 and Calu1 cells were transfected with
miR-25 mimics, inhibitor or the negative controls. The transfection
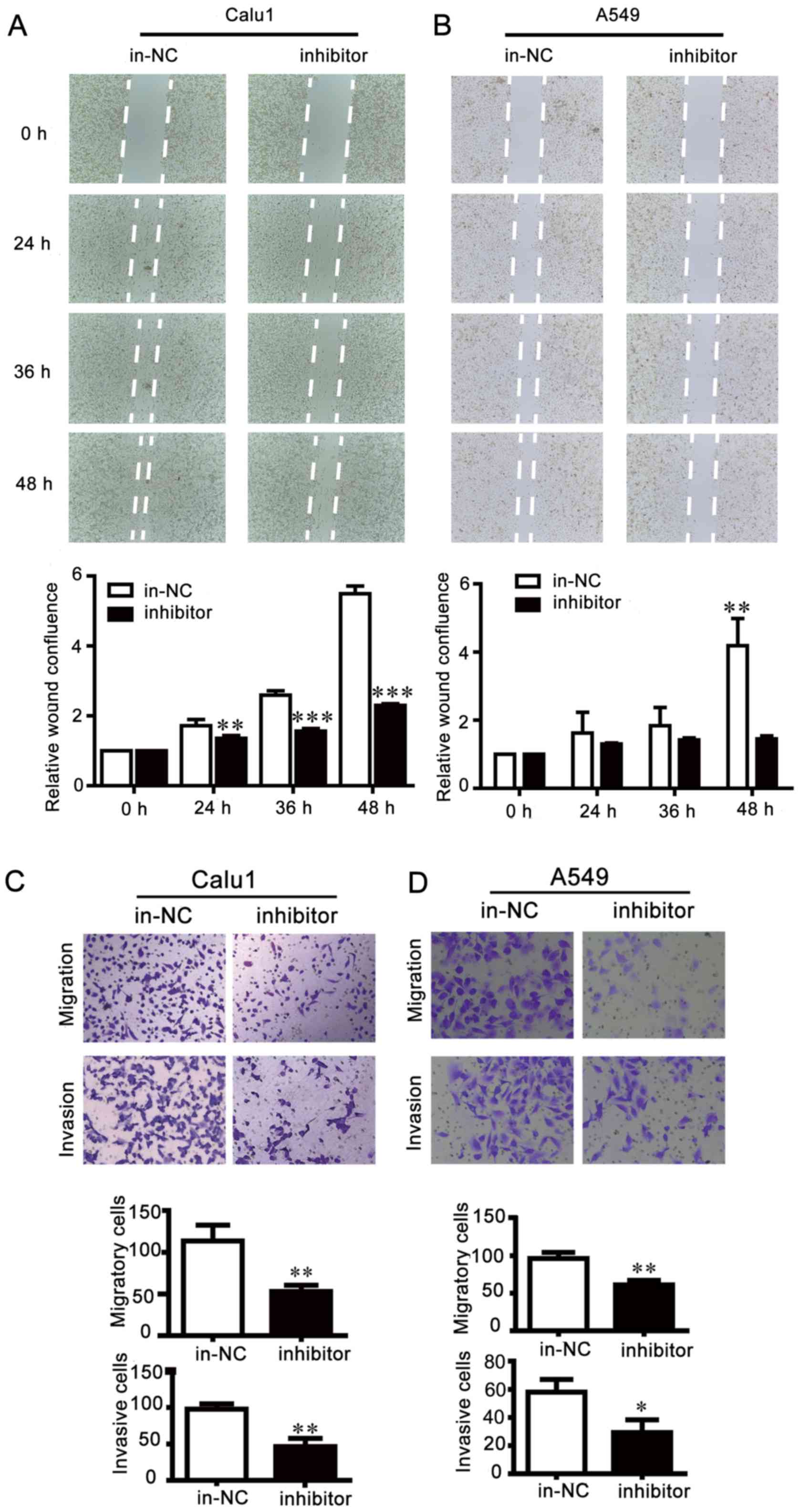
efficiency was detected by RT-qPCR (Fig. 2). Results of wound healing assays
showed that miR-25 inhibition reduced cell migration compared with
control cells, leading to larger scratch areas than control cells
(Fig. 3A and B). Cell motility was
then assessed via Transwell migration and invasion assays, which
indicated that miR-25 inhibition effectively repressed the invasion
and migration of NSCLC cells (Fig. 3C
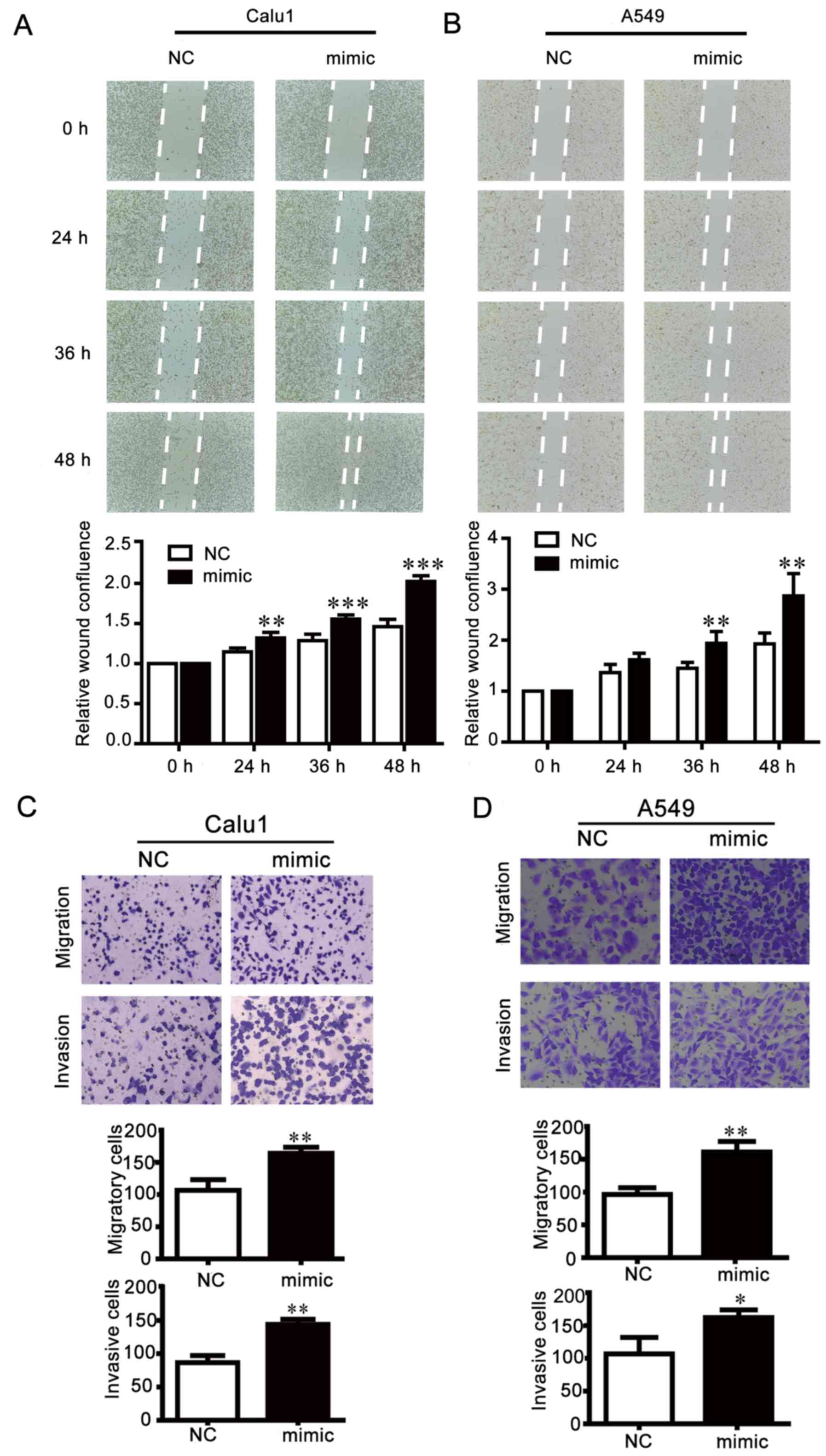
and D). By contrast, the scratch gap in the miR-25
overexpression group was smaller than the NC group (Fig. 4A and B), and in the Transwell
assays, the number of cells crossing through the membrane was
increased by miR-25 mimics compared the NC group (Fig. 4C and D). These results indicated
that the inhibition of miR-25 weakened NSCLC cell motility, while
overexpression of miR-25 exhibited opposite effects.
Identification of KLF4 as an
endogenous target gene of miR-25 in NSCLC cells
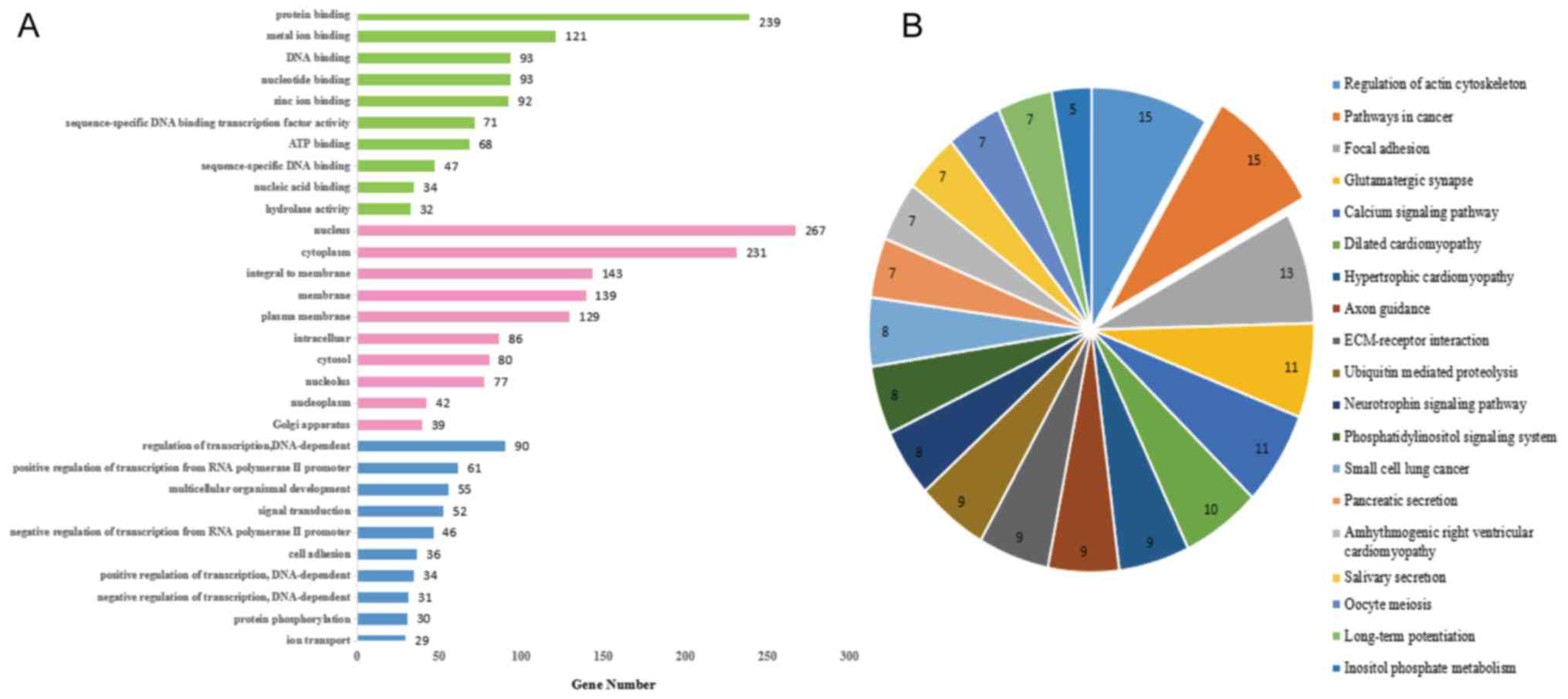
GO analysis of miR-25 targets demonstrated that the
molecular function of the target genes was predominantly
concentrated in ‘protein binding’ (Fig. 5A). In addition, KEGG functional
annotation of the target genes in cancer pathways is presented in
Fig. 5B. In comprehensive
consideration of functional targets and literature reports, KLF4
was eventually identified as a potential target gene of miR-25
(Fig. 6A) and further verified by
a dual luciferase assay and western blotting. It was observed that
overexpression of miR-25 significantly inhibited the relative
luciferase value of psi-CHECK2-KLF4-3′UTR-WT; however, it did not
alter psi-CHECK2-KLF4-3′UTR-MUT (Fig.
6B). Western blot analysis demonstrated that the miR-25
inhibition promoted KLF4 protein expression, whereas the miR-25
mimics reduced KLF4 protein expression levels (Fig. 6C and D). In addition, RT-qPCR
revealed that 28 of 31 pairs of lung cancer tissues exhibited a
decrease in KLF4 expression levels (Fig. 6E and F), indicating that KLF4
expression was decreased in NSCLC tissues compared with adjacent
control tissues. These data suggested that KLF4 may be a target
gene of miR-25.
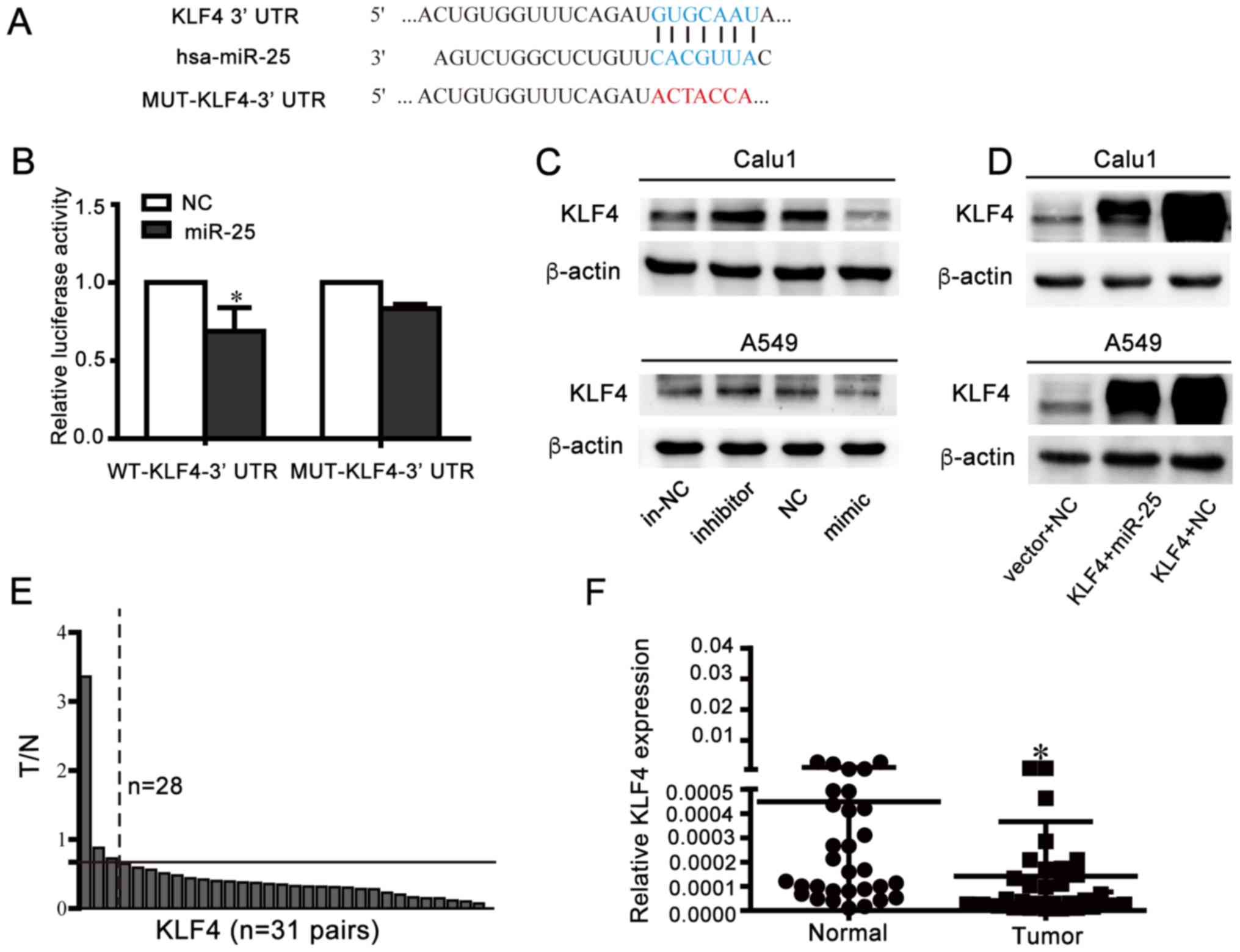 | Figure 6.Identification of KLF4 as an
endogenous target gene of miR-25 in NSCLC cells. (A) Wild-type or
mutant psi-CHECK2-KLF4-3′-UTR vector sequence and the binding site
(blue sequence) between wild-type KLF4-3′-UTR and miR-25 and
wild-type mutation location (red sequence) were presented. (B) 293T
cells transfected with luciferase vectors containing wide type or
mutant KLF4-3′-UTR and NC or miR-25, respectively were used for the
luciferase assay. Luciferase activity was normalized to firefly
luciferase activity. *P<0.05 vs. NC. Western blotting analysis
demonstrated expression KLF4 post-transfection with (C) miR-25
mimic/inhibitor or (D) co-transfection of miR-25 and KLF4. (E)
Relative expression alterations of KLF4 (T/N) were measured by
RT-qPCR in 31 pairs of human NSCLC samples and their corresponding
control tissues. The relative expression levels of KLF4 in each
tissue were calculated using the 2−ΔΔCq method using 18S
as the endogenous reference gene. In each pair of clinical samples,
the expression of KLF4 in the tumor tissues (T) was referenced to
corresponding normal tissues (N). The settings presented in the
figure were as follows: The graph is divided into high expression
and low expression sections by black solid lines of T/N=0.67. The
expression of KLF4 in 28 pairs cancerous tissues samples 0.67-fold
lower than in adjacent tissues (right of the black dashed line, T/N
<0.67-fold), and the remaining 3 pairs of samples (left of black
dashed lines) with T/N >0.67-fold. (F) Relative expression
levels of KLF4 in 31 pairs NSCLC tissues and adjacent normal
tissues samples measured by RT-qPCR and using 18S as reference
genes. *P<0.05 vs. normal. RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; NSCLC,
non-small-cell lung cancer; KLF4, Krüppel-like factor 4; UTR,
untranslated region; MUT, mutated; NC, negative control; miR,
microRNA; in-NC, inhibitor negative control; T/N, tumor/normal. |
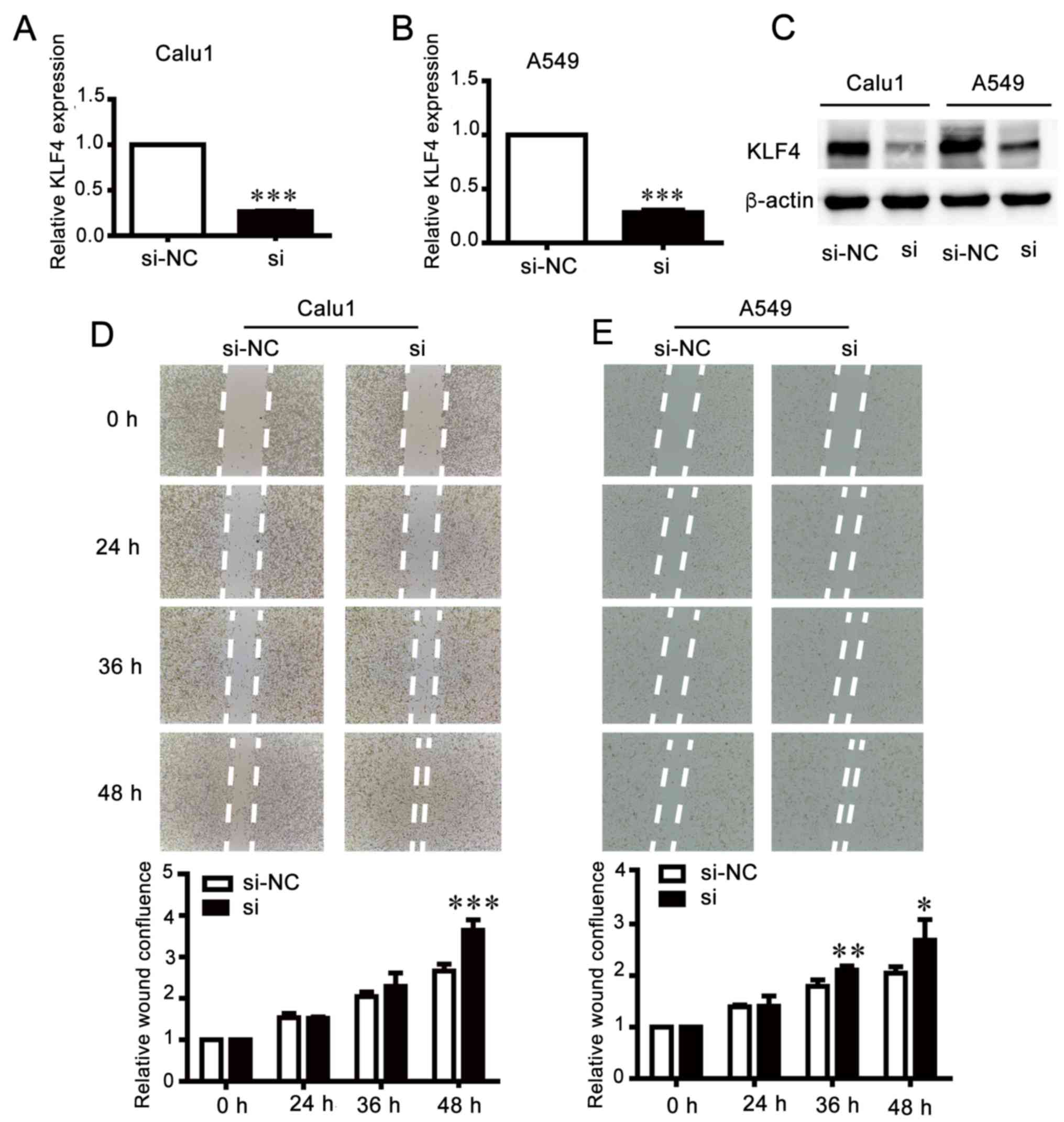
Knockdown of KLF4 increases invasion
and migration of NSCLC cells
In order to investigate the biological function of
KLF4, KLF4 expression was inhibited by transfection with siRNA
(Fig. 7A-C). A wound healing assay
demonstrated that knockdown of KLF4 accelerated the migration of
cells compared with the control groups (Fig. 7D and E). The results of the
migration and invasion assays were consistent with those of the
wound healing assay (Fig. 7F and
G), which suggested that the effects of KLF4 knockdown were
similar to those of miR-25 overexpression.
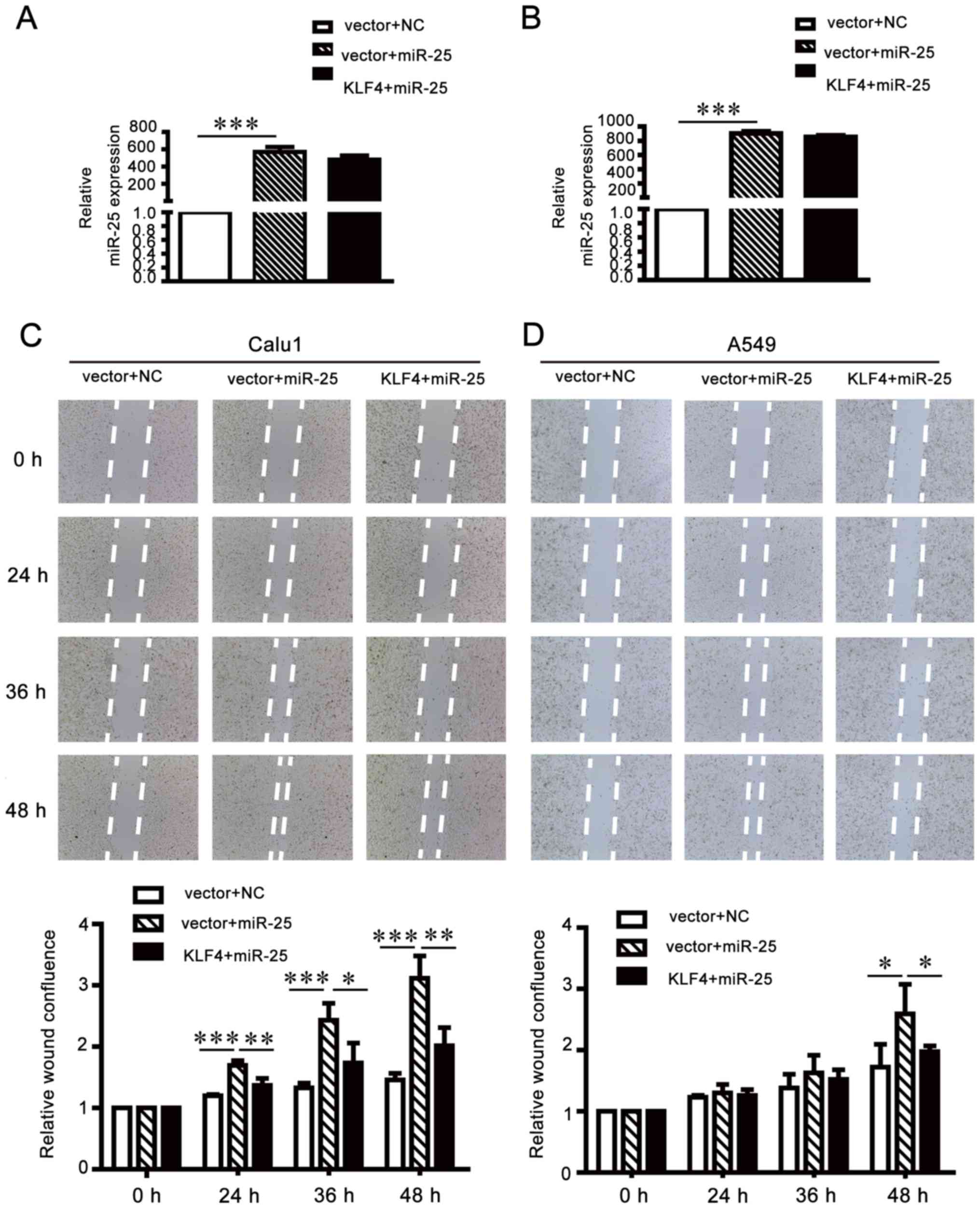
Migration and invasion-promoting
effects of miR-25 are partially attenuated by KLF4
overexpression
To further investigate whether KLF4 upregulation may
attenuate the oncogenic effects of miR-25 on NCSLC cells, the
pCMV-N-flag-KLF4 plasmid was transfected into miR-25-overexpressing
A549 and Calu1 cells (Fig. 8A and
B). Wound healing assays (Fig. 8C
and D) and Transwell invasion and migration assays (Fig. 8E and F) demonstrated that the
exogenous supplementation of KLF4 may weaken the oncogenic function
of miR-25 in NSCLC cells. The results of the present study
suggested that miR-25 promoted NSCLC cell migration and invasion by
inhibiting KLF4 expression.
miR-25 promotes cells invasion and
migration via the ERK signaling pathway
To clarify the mechanism of miR-25 in regulating
cell motility, the downstream signaling pathways of miR-25 were
investigated. Considering that KLF4 inhibited the role of miR-25 in
suppressing NSCLC cell invasion and migration, and KLF4 is involved
in the ERK signaling pathway, this suggests that miR-25 may
regulate the ERK1/2 pathway by inhibiting the expression of KLF4.
As presented in Fig. 9, the level
of p-ERK1/2 and the markers of invasion and metastasis (vimentin,
N-cadherin and MMP11) were reduced, and expression of E-cadherin
was increased by miR-25 inhibition. Consistent with these findings,
the overexpression of miR-25 in NSCLC cells activated the ERK1/2
signaling pathway, which was reversed by overexpression of KLF4 via
co-transfection with pCMV-N-flag-KLF4. The findings of the present
study demonstrated that miR-25, which inhibited KLF4 expression,
promoted cell invasion and migration by activating the ERK
signaling pathway.
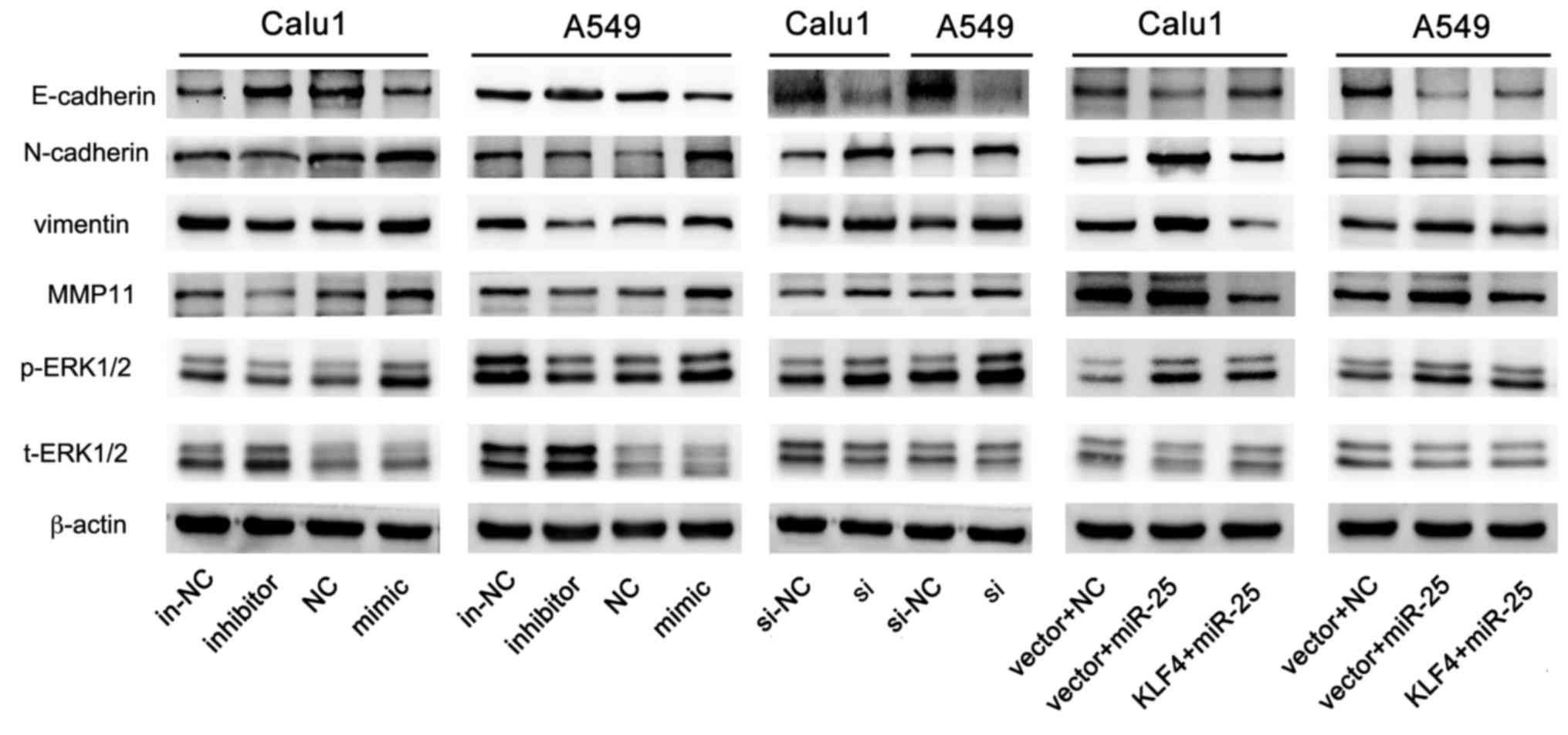 | Figure 9.miR-25 promotes cells invasion and
migration via the ERK signaling pathway. The expression of
E-cadherin, N-cadherin, vimentin, MMP11, p-ERK1/2, t-ERK1/2 and
β-actin in A549 and Calu1 post-transfection with miR-25 inhibitor,
miR-25 mimic, KLF4 si or KLF4 + miR-25. MMP11, matrix
metalloproteinase; p-, phospho-; ERK, extracellular
signal-regulated kinase; t-, total-; in-NC, inhibitor negative
control; NC, mimic negative control; si-NC, si negative control;
si, KLF4 small interfering RNA; miR, microRNA; KLF4, Krüppel-like
factor 4. |
Discussion
NSCLC, the most common lung cancer type, is studied
by researchers worldwide. As invasion and metastasis facilitate
NSCLC progression, it is important to further clarify the
mechanisms of pathogenesis and metastasis of NSCLC.
The results of the present study indicated that
miR-25 promoted the invasion and migration of NSCLC cells via the
ERK1/2 signaling pathway by targeting KLF4. In the present study,
the detailed experiments revealed that miR-25 was upregulated
within NSCLC tumor tissue samples compared with in the
corresponding paracancerous tissues by RT-qPCR analysis.
Downregulation of miR-25 using miR inhibitor repressed the
migration and invasion of NSCLC cells. By contrast, overexpression
of miR-25 by transfection with miR-25 mimic accelerated the
motility of NSCLC cells in vitro. The results of the present
study suggested that miR-25 promoted the invasion and migration of
NSCLC cells.
In addition, using the prediction software,
TargetScan 7.1, KLF4 was identified as a potential target of
miR-25, which was verified by luciferase reporter assay and western
blot analysis. Subsequently, the clinical expression of miR-25 and
KLF4 was investigated. Previous studies have reported the role of
KLF4 on inducible senescence in the process of the generation of
induced pluripotent stem cells (32–34).
Xu et al (35) clarified
that cell senescence may be promoted by KLF4 via the
miR-203-survivin-p21 pathway. Studies have indicated that KLF4 may
act as an anti-oncogene in lung cancer (36,37).
Exogenous KLF4 may reduce the colony formation and viability
ability of cells in in vitro assays. Additionally,
overexpressing KLF4 in lung cancer cells also inhibits tumor
proliferation in vivo (38). The findings of the present study
demonstrated that the knockdown of KLF4 promoted cell invasion and
migration, similar to the effects of miR-25 overexpression;
However, overexpressing KLF4 induced the opposite effects.
Providing that the restoration of KLF4 partly
reversed the effect of miR-25 on inhibiting lung cancer cell
invasion and migration, the present study aimed to identify the
downstream cascade effectors. Based on previous work, particular
attention given to ERK1/2, a promoter of cell invasion progression.
Zheng et al (39)
demonstrated that KLF4 was involved in antiproliferative functions
in vascular smooth muscle cells in culture, and knockdown of KLF4
slightly increased p-ERK1/2 levels. Thus, the present study aimed
to verify whether miR-25 regulated the ERK signaling pathway by
altering the expression of KLF4. Markers of invasion (MMP11,
vimentin) and epithelial-to-mesenchymal transition (E- and
N-cadherin) were detected at the protein level. The results of the
present study demonstrated that the downregulation of KLF4
activated the ERK signaling pathway, increasing the expression of
N-cadherin, MMP11 and vimentin, with reduced E-cadherin
expression.
Currently, few reports have investigated the
downstream signaling pathway of miR-25, or the association between
miR-25 and ERK signaling. Additionally, few studies have assessed
the expression of miR-25 and KLF4 in clinical samples. The limited
number of clinical sample abundance and lack of further research
regarding how KLF4 affects the ERK pathway were limitations of the
present study.
In summary, miR-25 was significantly overexpressed
in NSCLC tissues compared with paracancerous tissue in the present
study, and the exogenous addition of miR-25 significantly promoted
cell motility of NSCLC cells by reducing the expression of KLF4. In
addition, miR-25 was observed to induce migration and invasion of
NSCLC cells by regulating the ERK signaling pathway, particularly
by affecting the phosphorylation status of molecules upstream of
ERK1/2. The results of the present study indicated that miR-25 may
be a potential therapeutic target for future NSCLC treatment.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Torre LA, Siegel RL and Jemal A: Lung
cancer statistics. Adv Exp Med Biol. 893:1–19. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen Z, Fillmore CM, Hammerman PS, Kim CF
and Wong KK: Non-small-cell lung cancers: A heterogeneous set of
diseases. Nat Rev Cancer. 14:535–546. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Moreira AL and Eng J: Personalized therapy
for lung cancer. Chest. 146:1649–1657. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Preiss A, Rosenberg UB, Kienlin A, Seifert
E and Jäckle H: Molecular genetics of Kruppel, a gene required for
segmentation of the drosophila embryo. Nature. 313:27–32. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shields JM, Christy RJ and Yang VW:
Identification and characterization of a gene encoding a
gut-enriched Kruppel-like factor expressed during growth arrest. J
Biol Chem. 271:20009–20017. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pandya AY, Talley LI, Frost AR, Fitzgerald
TJ, Trivedi V, Chakravarthy M, Chhieng DC, Grizzle WE, Engler JA,
Krontiras H, et al: Nuclear localization of KLF4 is associated with
an aggressive phenotype in early-stage breast cancer. Clin Cancer
Res. 10:2709–2719. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen YJ, Wu CY, Chang CC, Ma CJ, Li MC and
Chen CM: Nuclear Kruppel-like factor 4 expression is associated
with human skin squamous cell carcinoma progression and metastasis.
Cancer Biol Ther. 7:777–782. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang N, Liu ZH, Ding F, Wang XQ, Zhou CN
and Wu M: Down-regulation of gut-enriched Kruppel-like factor
expression in esophageal cancer. World J Gastroenterol. 8:966–970.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wei D, Gong W, Kanai M, Schlunk C, Wang L,
Yao JC, Wu TT, Huang S and Xie K: Drastic down-regulation of
Kruppel-like factor 4 expression is critical in human gastric
cancer development and progression. Cancer Res. 65:2746–2754. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wei D, Kanai M, Huang S and Xie K:
Emerging role of KLF4 in human gastrointestinal cancer.
Carcinogenesis. 27:23–31. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Friedman RC, Farh KK, Burge CB and Bartel
DP: Most mammalian mRNAs are conserved targets of microRNAs. Genome
Res. 19:92–105. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li X, Li H, Zhang R and Liu J and Liu J:
MicroRNA-449a inhibits proliferation and induces apoptosis by
directly repressing E2F3 in gastric cancer. Cell Physiol Biochem.
35:2033–2042. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang C, Ning S, Li Z, Qin X and Xu W:
miR-22 is down-regulated in esophageal squamous cell carcinoma and
inhibits cell migration and invasion. Cancer Cell Int. 14:1382014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang D, Qiu C, Zhang H, Wang J, Cui Q and
Yin Y: Human microRNA oncogenes and tumor suppressors show
significantly different biological patterns: From functions to
targets. PLoS One. 5:pii: e13067. 2010. View Article : Google Scholar
|
|
17
|
Xiao P, Liu W and Zhou H: miR-429 promotes
the proliferation of non-small cell lung cancer cells via targeting
DLC-1. Oncol Lett. 12:2163–2168. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu W, Jin P, Ding C and Liu W: miR-19a/b
modulates lung cancer cells metastasis through suppression of MXD1
expression. Oncol Lett. 12:1901–1905. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang WM and Liu JC: Effect and molecular
mechanism of mir-146a on proliferation of lung cancer cells by
targeting and regulating MIF gene. Asian Pac J Trop Med. 9:806–811.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang HB, Sun LC, Ling L, Cong LH and Lian
R: miR-143 suppresses the proliferation of NSCLC cells by
inhibiting the epidermal growth factor receptor. Exp Ther Med.
12:1795–1802. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shi H, Ji Y, Zhang D, Liu Y and Fang P:
MicroRNA-3666-induced suppression of SIRT7 inhibits the growth of
non-small cell lung cancer cells. Oncol Rep. 36:3051–3057. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Perepelyuk M, Maher C, Lakshmikuttyamma A
and Shoyele SA: Aptamer-hybrid nanoparticle bioconjugate
efficiently delivers miRNA-29b to non-small-cell lung cancer cells
and inhibits growth by downregulating essential oncoproteins. Int J
Nanomedicine. 11:3533–3544. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Petrocca F, Vecchione A and Croce CM:
Emerging role of miR-106b-25/miR-17-92 clusters in the control of
transforming growth factor beta signaling. Cancer Res.
68:8191–8194. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Savita U and Karunagaran D:
MicroRNA-106b-25 cluster targets β-TRCP2, increases the expression
of Snail and enhances cell migration and invasion in H1299 (non
small cell lung cancer) cells. Biochem Biophys Res Commun.
434:841–847. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Su ZX, Zhao J, Rong ZH, Geng WM, Wu YG and
Qin CK: Upregulation of microRNA-25 associates with prognosis in
hepatocellular carcinoma. Diagn Pathol. 9:472014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li BS, Zuo QF, Zhao YL, Xiao B, Zhuang Y,
Mao XH, Wu C, Yang SM, Zeng H, Zou QM and Guo G: MicroRNA-25
promotes gastric cancer migration, invasion and proliferation by
directly targeting transducer of ERBB2, 1 and correlates with poor
survival. Oncogene. 34:2556–2565. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou J, Wang J, Wu S, Zhu S, Wang S, Zhou
H, Tian X, Tang N and Nie S: Angiopoietin-like protein 2 negatively
regulated by microRNA-25 contributes to the malignant progression
of colorectal cancer. Int J Mol Med. 34:1286–1292. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu T, Chen W, Kong D, Li X, Lu H, Liu S,
Wang J, Du L, Kong Q, Huang X and Lu Z: miR-25 targets the
modulator of apoptosis 1 gene in lung cancer. Carcinogenesis.
36:925–935. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xiang J, Hang JB, Che JM and Li HC: MiR-25
is up-regulated in non-small cell lung cancer and promotes cell
proliferation and motility by targeting FBXW7. Int J Clin Exp
Pathol. 8:9147–9153. 2015.PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang Z, Wang J, Yang Y, Hao B, Wang R, Li
Y and Wu Q: Loss of has-miR-337-3p expression is associated with
lymph node metastasis of human gastric cancer. J Exp Clin Cancer
Res. 32:762013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen X, Zhai Y, Yu D, Cui J, Hu JF and Li
W: Valproic acid enhances iPSC induction from human bone
marrow-derived cells through the suppression of
reprogramming-induced senescence. J Cell Physiol. 231:1719–1727.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhai Y, Chen X, Yu D, Li T, Cui J, Wang G,
Hu JF and Li W: Histone deacetylase inhibitor valproic acid
promotes the induction of pluripotency in mouse fibroblasts by
suppressing reprogramming-induced senescence stress. Exp Cell Res.
337:61–67. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Patel RS, Carter G, El Bassit G, Patel AA,
Cooper DR, Murr M and Patel NA: Adipose-derived stem cells from
lean and obese humans show depot specific differences in their stem
cell markers, exosome contents and senescence: Role of protein
kinase C delta (PKCδ) in adipose stem cell niche. Stem Cell
Investig. 3:22016.PubMed/NCBI
|
|
35
|
Xu Q, Liu M, Zhang J, Xue L, Zhang G, Hu
C, Wang Z, He S, Chen L, Ma K, et al: Overexpression of KLF4
promotes cell senescence through microRNA-203-survivin-p21 pathway.
Oncotarget. 7:60290–60302. 2016.PubMed/NCBI
|
|
36
|
Yu T, Chen X, Zhang W, Liu J, Avdiushko R,
Napier DL, Liu AX, Neltner JM, Wang C, Cohen D and Liu C: KLF4
regulates adult lung tumor-initiating cells and represses
K-Ras-mediated lung cancer. Cell Death Differ. 23:207–215. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hu W, Jia Y, Xiao X, Lv K, Chen Y, Wang L,
Luo X, Liu T, Li W, Li Y, et al: KLF4 downregulates hTERT
expression and telomerase activity to inhibit lung carcinoma
growth. Oncotarget. 7:52870–52887. 2016.PubMed/NCBI
|
|
38
|
Hu W, Hofstetter WL, Li H, Zhou Y, He Y,
Pataer A, Wang L, Xie K, Swisher SG and Fang B: Putative
tumor-suppressive function of Kruppel-like factor 4 in primary lung
carcinoma. Clin Cancer Res. 15:5688–5695. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zheng B, Han M, Bernier M, Zhang XH, Meng
F, Miao SB, He M, Zhao XM and Wen JK: Kruppel-like factor 4
inhibits proliferation by platelet-derived growth factor receptor
beta-mediated, not by retinoic acid receptor alpha-mediated,
phosphatidylinositol 3-kinase and ERK signaling in vascular smooth
muscle cells. J Biol Chem. 284:22773–22785. 2009. View Article : Google Scholar : PubMed/NCBI
|