Introduction
Sick sinus syndrome (SSS) is caused by lesions in
the sinoatrial node and its adjacent tissues, resulting in
sinoatrial node pacemaker function and/or sinoatrial conduction
dysfunction. SSS includes conditions such as severe sinus
bradycardia, sinus arrest, sinoatrial block, and
bradycardia-tachycardia syndrome (flutter, atrial fibrillation, and
paroxysmal supraventricular tachycardia) (1,2). SSS
may lead to insufficient blood supply to the heart, brain, kidney,
and other organs, and cause sudden cardiac death. Symptomatic SSS
requires the implantation of an electronic pacemaker. SSS accounts
for about half of all pacemaker implantations in the United States
(3). Given the low survival rate
(less than 10%) (4), the
identification of patients with high-risk SSS is of importance for
the prevention of sudden cardiac death.
Although a variety of pathological factors leading
to sinus node or cardiac nerve dysfunction may cause SSS, a
positive correlation was observed between SSS and age (5). Increasing evidences suggest a
distinct genetic background in SSS, including some genetic
mutations in genes encoding cytoskeletal proteins and ion channels
that may result in familial sick sinus syndrome (FSSS). The L-type
voltage-gated calcium channel (I-caL) is one of the significant ion
channels involved in the automatic cell depolarization in
sinoatrial node. Loss of I-cal in some hereditary SSS is of great
significance. For instance, the deletion of L-calcium currents in
CaV1.3 knockout mice (CaV1.3−/−) may contribute to
reduced heart rate and sinus arrhythmia (6). The presence of CaV1.3-mediated I-caL
dysfunction may clinically lead to sinus node dysfunction and
deafness syndrome (SANDD). Individuals affected with SANDD present
with bradycardia, profound deafness, and dysfunction of
atrioventricular conduction (7).
However, no study has reported the association between sinus
bradycardia or SSS and the variant of calcium voltage-gated channel
subunit alpha1 C (CACNA1C).
The early repolarization (ER) pattern displays two
or more continuous wall or side wall leads with a J point elevation
above 0.1 mV in the standard 12-lead electrocardiogram (ECG). The
ST elevation is above 0.1 mV and exhibits concave upward or oblique
type elevation. ER is often inherited and several studies have
shown its relationship with malignant arrhythmias and sudden death
(8–10). It is known that ER syndrome is a
polygenic inherited disease, similar to hereditary sinus
bradycardia, caused by mutations in cardiomyocyte genes encoding
ion channels. The most common pathogenic genes are encoded by
cardiac electrical ion channels, including calcium channels
(CACNA1C, CACNB2B, and CACNA2D1) (11), ATP-sensitive potassium channels
(KCNJ8) (12), and sodium channel
α subunit 5 (SCN5A) (13).
Functional studies have revealed its association with ‘enhanced
function’ or ‘loss of function’ of ion channels. A change of 1–5%
was reported in the incidence of ER in the general population
(14) and is most commonly seen in
young men (15).
We applied next generation sequencing (NGS) for the
identification of a genetically arrhythmic family of SSS and ER
characterized by complex bradycardia to further understand the
interaction between the complex genotype and phenotype in this
family.
Materials and methods
Subjects
The proband of this family is a 76-year-old male
patient from Fujian with Han nationality and clinical manifestation
of recurrent palpitation, flustered, and bradycardia with
irregularity. Multiple electrocardiographic examinations indicated
that the patient had slow atrial fibrillation (the slowest heart
rate 29 beats per min). According to the family data provided by
the proband, medical records, routine physical examination, ECG,
and cardiac ultrasound examination were performed for other 11
family members. In addition, blood, urine, biochemical kit,
troponin, pro-BNP, etc., were analyzed and the family genetic map
was recorded. The study was approved by the medical ethics
committee of the hospital and all subjects signed the informed
consent.
Genomic DNA extraction
Genomic DNA was extracted with Tiangen blood genomic
DNA extraction kit (DP348) as per the manufacturer's
instructions.
Illumina sequencing (16–18)
and bioinformatic analysis (19)
DNA samples of the proband were detected by Nanodrop
2000 and over 3 µg sample was used for DNA fragmentation. DNA
fragments were interrupted to about 250 bp by Covaris method,
followed by their recovery. The fragments were subjected to the
end-repair reaction and the sequence-specific attachment (adapter)
was linked with the end-repair products. The product was amplified
and purified based on the universal primer-binding site on the
adapter. For the capture and enrichment of the target, multiple
gene fragments, including target genes CACNA1C and TTN, were
enriched by DNA capture chip with multiple genes and the enrichment
products were sequenced by Illumina (Illumina, Inc., San Diego, CA,
USA) (5). For data analysis, the
original file was subjected to base reading to obtain double
terminal sequence of 90 bp reads. For the removal of the
low-quality and polluted reads, the adapter sequence was removed
and the purification data analyzed by sequence alignment. Soap
software (SOAPdenovo V2.04; SOAP3/GPU V0.01beta; SOAPaligner/soap2
V2.20; SOAPsplice V1.1; SOAPsnp V1.03; SOAPindel V1.0; SOAPsv
V1.02) was used to analyze copy number, single nucleotide
polymorphism (SNP), and insertion/deletion (INDEL). Annotations
were used to screen suspected pathogenic variants. The polymorphism
phenotyping version 2 (Polyphen2) software was applied (genetices.bwh.harvard.edu/pph2/) for the
prediction of protein functions. The process was completed by
Shenzhen Huada Genomics Institute in accordance with its operating
standards.
Sanger sequencing
Variants were confirmed using Sanger DNA sequencing.
The primers targeting the target sequence were designed using
Primer Premier 5 software and synthesized by Thermo Fisher
Scientific, Inc. (Waltham, MA, USA). CACNA1C gene sequence was
derived from GenBank (NM_001129843) and the target sequence was 230
bp. The primers used were as follows: Forward, GCTCGGATCTCATCCCTCTC
and reverse, GACGCATCTGAGCACGGA. Another target sequence of CACNA1C
was 260 bp long and the specific primers used were as follows:
Forward, TTCACCCCGAGCAGCTAC and reverse, TCCACTGTCTCCTGAGGGTT. TTN
gene sequence was obtained from GenBank (NM_003319) and the target
sequence was 270 bp. The primers used were as follows: Forward,
CATTGTCAAGAACAAGAGAGGTGAAAC and reverse,
CGTATCTGTGCTATTAATAAAGCTGGAGT. Amplification of polymerase chain
reaction (PCR) products: The reaction was carried out in a final
volume of 25 µl and comprised 2.5 µl of 10X Ex Taq buffer, 2 µl
dNTP (2.5 mmol/l), 3 µl of each of forward and reverse primer (3
mmol/l), 1 µl DNA template, 0.2 µl Ex Taq, and 18.3 µl water. PCR
products were amplified on PCR machine (PTC-200, PCR; Bio-Rad
Laboratories, Inc., Hercules, CA, USA) under following conditions:
Initial denaturation at 94°C for 5 min, 35 cycles of denaturation
at 94°C for 40 sec, annealing at 58–62°C for 40 sec, extension at
72°C for 60 sec, followed by the final extension at 72°C for 10
min. Purification and sequencing of PCR products: PCR products were
obtained from Omega corporation E.Z.N.A.™ Gel Extraction
kit. Sequencing was performed according to the standard procedure
of BigDye Terminator v1.1 kit PCR products. Sequencing results were
obtained by comparing DNAMAN version 5.2.2 with the normal
sequence.
Results
Clinical report
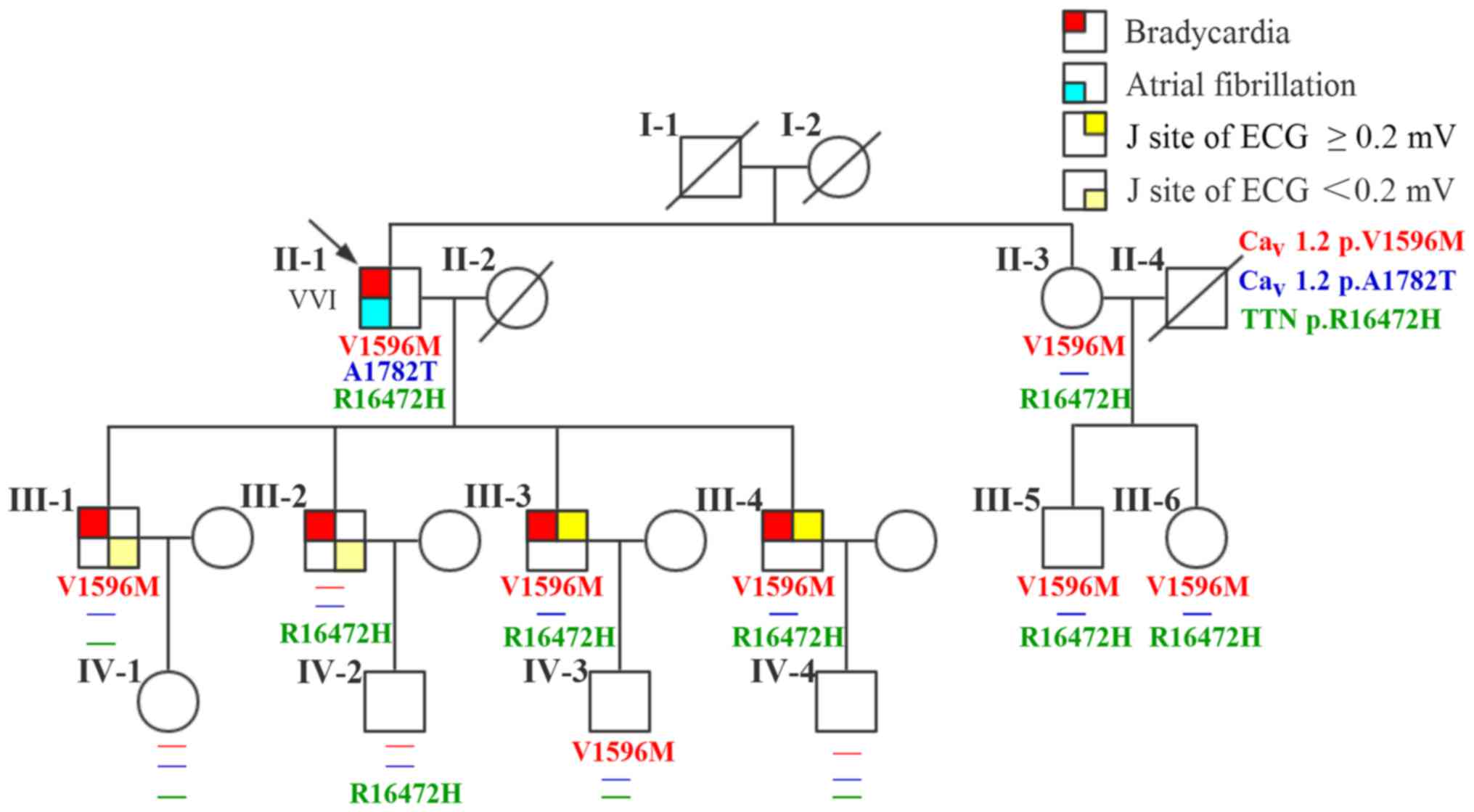
There are 20 members of the family, with 5 diseases
(Fig. 1). A 76-year-old male
(II-1; the proband) patient presented with a 40-year history of
palpitation, chest tightness, and stress sweating and was
repeatedly admitted to our hospital. The patient had an episode of
syncope 8 years ago following progressive palpitation, fatigue, and
dizziness. He received no treatment, although his ECG suggested
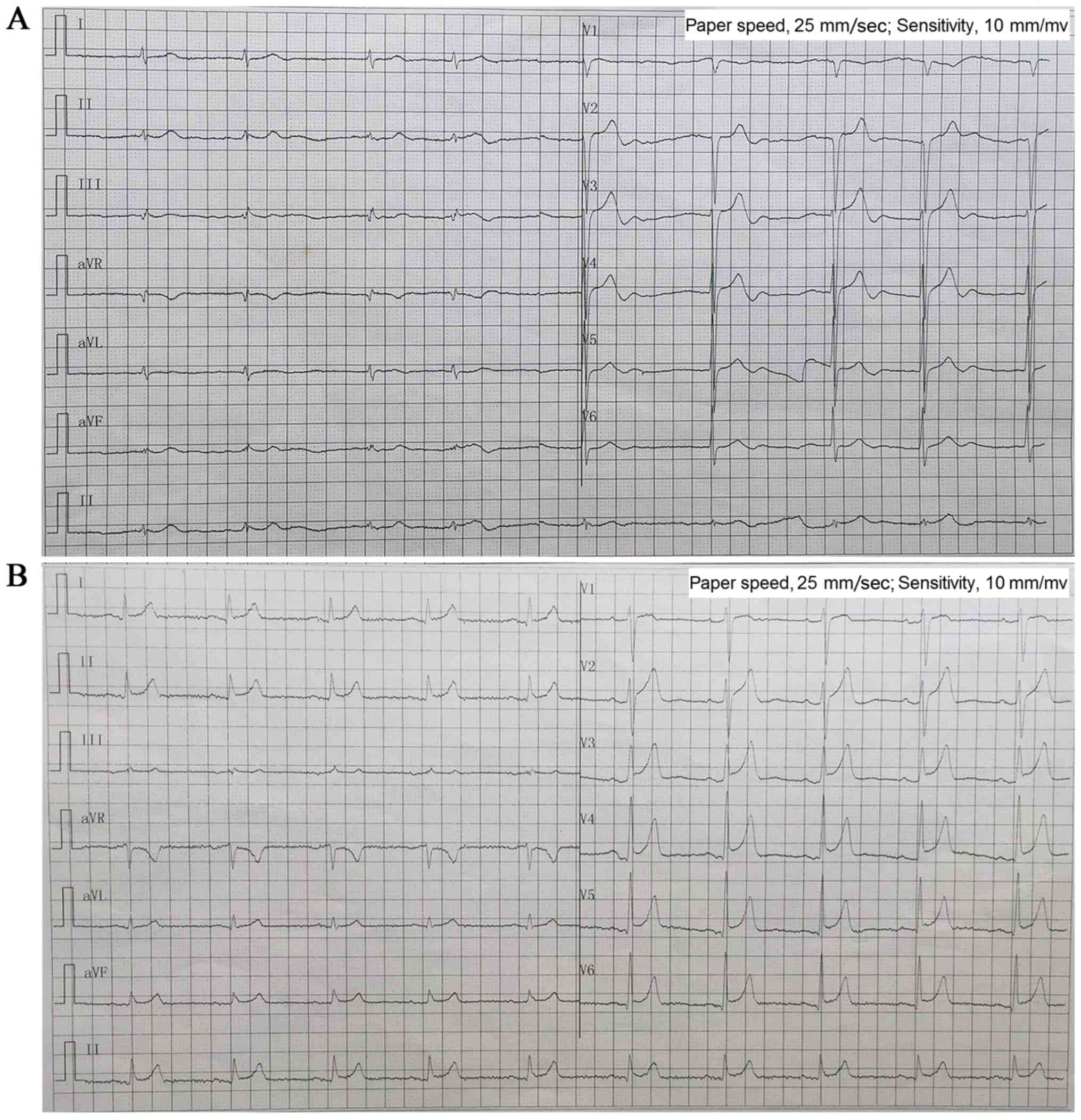
bradycardia and atrial fibrillation. In 2013, a 24-h Holter ECG
monitoring (Fig. 2A) confirmed
bradycardia (average heart rate, 49 bpm; range, 29–89 bpm) and
atrial fibrillation. The echocardiogram showed moderate mitral
regurgitation, tricuspid regurgitation, and mild aortic valve
regurgitation. He was diagnosed with SSS and received a permanent
pacemaker implantation. The proband took no drugs such as
digitalis, β-blocker, or calcium blocker to slow down his heart
rate. Prior to the diagnosis, he had hypertension and an incidence
of cerebral infarction. Evaluation of the family history revealed
that all his four children (III-1, III-2, III-3, and III-4; all
male) presented with bradycardia with an average heart rate <50
bpm; two of them (III-3 and III-4) showed early onset (since their
childhood) of symptoms similar to their father's, and the other two
(III-1 and III-2) have palpitation when they are tired or emotional
swings. ECG (Fig. 2B) diagnosed
III-3 and III-4 with ER syndrome (ST elevation in V1-V6 was upward
concave, the J-point elevation ≥0.2 mV) and III-1 and III-2 with ER
pattern (ST elevation in V4-6 was upward concave, the J-point
elevation <0.2 mV).
The proband's parents (I-1 and I-2) had passed away;
his mother (I-2) reported symptoms of paroxysmal arrhythmia before
her death, but the exact diagnosis was unknown. The proband's four
grandchildren (IV-1, IV-2, IV-3, IV-4; aged 10–24 years) and
younger sister (II-2) and her two children (III-5 and III-6)
presented with no cardiac symptoms and all had normal ECG (Table I). No other cutaneous, retinal,
neurologic, and somatic abnormalities were reported or observed in
this family.
 | Table I.Clinical data of the family
members. |
Table I.
Clinical data of the family
members.
| ID | Age (years) | Sex | Onset age
(years) | Symptoms | ECG/HOLTER | Echocardiogram |
|---|
| II-1a | 76 | Male | Twenties | Palpitation, chest
tightness, stress sweating, emotional, and syncope | Bradycardia, atrial
fibrillation | Mitral, tricuspid,
and aortic valve regurgitation |
| II-3 | 73 | Female | Twenties | Palpitation, chest
tightness, stress sweating | Normal | Normal |
| III-1 | 52 | Male | Twenties | Palpitation | Bradycardia,
J-point elevation <0.2 mV | Normal |
| III-2 | 51 | Male | Twenties | Palpitation | Bradycardia,
J-point elevation <0.2 mV | Normal |
| III-3 | 49 | Male | Twenties | Palpitation, chest
tightness, stress sweating, and emotional | Bradycardia,
J-point elevation ≥0.2 mV | Normal |
| III-4 | 43 | Male | Twenties | Palpitation, chest
tightness, stress sweating, and emotional | Bradycardia,
J-point elevation ≥0.2 mV | Normal |
| III-5 | 48 | Male | NA | Normal | Normal | Normal |
| III-6 | 44 | Female | NA | Normal | Normal | Normal |
| IV-1 | 23 | Female | NA | Normal | Normal | Normal |
| IV-2 | 22 | Male | NA | Normal | Normal | Normal |
| IV-3 | 21 | Male | NA | Normal | Normal | Normal |
| IV-4 | 9 | Male | NA | Normal | Normal | Normal |
Genetic analysis
Twelve members in this family were enrolled into the
genetic analysis study. First, we conducted a targeted exome
sequencing on the proband for 61 genes, which are related with
arrhythmia. The enriched and purified targeted regions were
sequenced on an Illumina HiSeq 2000 sequencer (Illumina) for 90-bp
reads; the mean read depth was 243X and >97% bases were >30X.
A total of 123 variants (including point variant and
insertion/deletion) were detected, and mutant polymorphism (SNP)
loci greater than 1% were filtered out through 1000 Genomes MAF
(population frequency information from 1000 genomes project).
Values of dbSNP and the remaining nine variants were found in
clinvar database, excluding synonymous and intron variants (out of
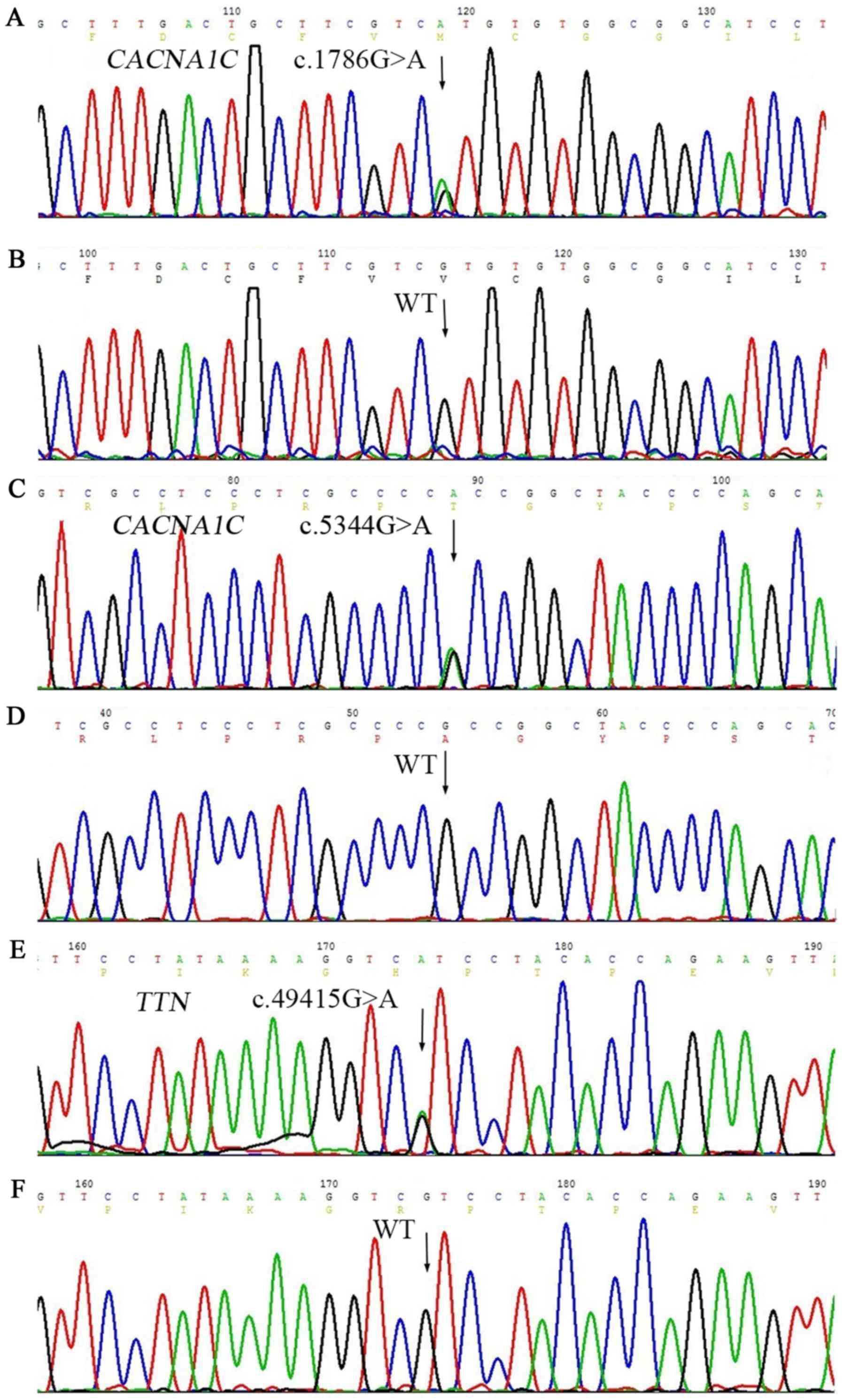
exon over 2 bp), to obtain three variants (Table II). No rare nonsynonymous variants
were found except heterozygous c.1786G>A (rs768034509) in
CACNA1C, c.5344G>A (rs750078053) in CACNA1C, and c.49415G>A
(rs561977468) in TTN, predicting valine to methionine substitution
at the amino acid 596 (p. V596M) in CACNA1C (Fig. 3A), alanine to threonine
substitution at the amino acid 1782 (p. A1782T) in CACNA1C
(Fig. 3C), and arginine to
histidine substitution at the amino acid 16472 (p. R16472H) in TTN
(Fig. 3E). In addition,
c.3979-8delC was present in the intron of MYH6, and c.68-5C>T
was present in the intron region of TNNT2. Three synonymous
variants were detected on TTN (p.Ala3897=p.Tyr6529=p.Gly12282)
(Table II). These variants were
in the non-coding region, suggestive of the absence of any effect
of this family with sinus bradycardia on ER.
| Table II.Nine mutations found in Clinvar
database of a propositus in a family with hereditary complex sinus
bradycardia (MAF ≤0.01). |
Table II.
Nine mutations found in Clinvar
database of a propositus in a family with hereditary complex sinus
bradycardia (MAF ≤0.01).
| Genes | RefSeq | Nucleic acid
alternation | Amino acid
alternation | Mutation
location | Zygosity | Chr:location | RS-ID | MAF | Mutation type |
|---|
| CACNA1C | NM_001129843 | c.1786G>A | p.Val596Met | EX13 | Het | chr12:2676851 | rs768034509 | 0 | Missense |
| CACNA1C | NM_001129843 | c.5344G>A | p.Ala1782Thr | EX42 | Het | chr12:2788862 | rs750078053 | 0 | Missense |
| MYH6 | NM_002471 | c.3979-8delC | – | IN28 | Het | chr14:23858272 | rs193922652 | 0 | – |
| TNNC1 | NM_003280 | c.108C>A | p.Ile36Ile | EX3 | Het | chr3:52486216 | rs202000367 | 0.0027 | Synonymous |
| TNNT2 | NM_000364 | c.68-5C>T | – | IN4 | Het | chr1:201338978 | rs540630390 | 0 | – |
| TTN | NM_003319 | c.11691G>T | p.Ala3897Ala | EX45 | Het | chr2:179605180 | rs746578 | 0 | Synonymous |
| TTN | NM_003319 | c.19587C>T | p.Tyr6529Tyr | EX79 | Het | chr2:179483495 | rs397517587 | 0 | Synonymous |
| TTN | NM_003319 | c.36846C>A | p.Gly12282Gly | EX135 | Het | chr2:179451897 | – | 0 | Synonymous |
| TTN | NM_003319 | c.49415G>A | p.Arg16472His | EX154 | Het | chr2:179434249 | rs561977468 | 0 | Missense |
These three mutant sites (V596M and A1782T on
CACNA1C and R16472H on TTN) are heterozygous
variants. The frequency of the variant in 1000 genomes project was
0, while clinvar database had no relevant report at these sites
(http://www.ncbi.nlm.nih.gov/clinvar/?term=CACNA1C
[gene] and http://www.ncbi.nlm.nih.gov/clinvar/?term=TTN [gene]).
V596M variant in CACNA1C was predicted to be damaging with a
score of 1.000 (sensitivity, 0.00; specificity, 1.00) by PolyPhen-2
software. The variant A1782T was predicted to be benign, with a
score of 0.010 (sensitivity, 0.96; specificity, 0.77). We speculate
that these two variants are likely to affect the function of
CACNA1C-encoded ion channels. R16472H variant in TTN
was predicted to be probably damaging with a score of 0.743
(sensitivity, 0.85; specificity, 0.92). These three variant sites
are highly conserved among many species (Fig. 4).
Genetic analysis showed that none of the eleven
family members carried c.5344G>A variant in CACNA1C gene.
On the other hand, c.1786G>A in CACNA1C gene and
c.49415G>A in TTN gene were detected in five family
members, including the proband's two sons III-3 and III-4, and
sister II-3 and her two children III-5 and III-6. Other six family
members only had either of them.
Discussion
Cardiomyopathy and ion channel dysfunction are two
major causes for inherited cardiac arrhythmias (20). In this study, we identified two
novel missense variants in CACNA1C gene (calcium channel
gene) and one novel missense variant in TTN gene
(cardiomyocyte gene) in a family with symptomatic bradycardia,
which eventually progressed into SSS in the proband.
CACNA1C gene encodes an alpha-1 subunit of a
voltage-dependent calcium channel. I-caL plays a key role in the
generation of spontaneous action potential in pacemaker (e.g.,
sinoatrial node) cells through the conduction of inward calcium
currents. Any dysfunction in I-caL leads to significantly
attenuated automaticity of these cells (21). Variants in CACNA1C gene have
been associated with cardiac diseases such as Long-QT syndrome
(22), Timothy syndrome (23), and Brugada syndrome (24). However, none has been reported in
SSS. This is the first study to report missense variants of
CACNA1C gene in patients with SSS. The two SNPs in
CACNA1C gene detected in this family result in two amino
acid replacements, localized at the third transmembrane segment of
domain II (DIIS3) (p.596V>M) and carboxyl (C)-terminus
(p.1782A>T) of CACNA1C, respectively. However, only p.596V>M
variant was thought to be damaging, while p.1782A>T variant was
neutral. Regardless, the proband was the only carrier of
p.1782A>T variant; therefore, it is unlikely that this variant
contributes to the inheritance of bradycardia and ER in the family.
Although the proband is also the only one diagnosed with SSS, this
diagnosis is unlikely to be associated with p.1782A>T variant
but more of a reflection of his disease progression. A previous
study showed that the expression of CACNA1C in the
sinoatrial node decreases with aging (25); this observation may explain the
deterioration of cardiac symptoms in the proband as he aged. It
should be alerted that the cardiac symptoms displayed by the
proband's sons may eventually develop into SSS. Furthermore, the
proband's four sons exhibited ER, an ECG pattern that was
unobserved in the proband. This observation may be due to different
manifestations of the disease at different stages. We estimate that
p.596V>M variant in CACNA1C may partly explain ER
observed in the proband's three sons because CACNA1C gene
has been identified as a susceptibility gene for ER syndrome
(11).
A variant in the gene encoding for the SCN5A,
c.2365G>A (p.789V>I), was a disease-causing missense variant
for Brugada syndrome (26) and
identified as a paralogue variant for CACNA1C variant
c.1786G>A (p.596V>M). According to the Paralogue Annotation
theory (www.cardiodb.org/paralogue_annotation/), known
disease-causing paralogue variants (e.g., SCN5A variant
c.2365G>A in this case) may be transferred to equivalent amino
acids in the protein of interest (e.g., CACNA1C variant
c.1786G>A in this case) to identify residues likely to be
intolerant to the variation (27).
Furthermore, CACNA1C variant c.1786 G allele, similar to
SCN5A variant c.2365 G allele, may be intolerant to genetic
variant; therefore, G>A variant at this position is very likely
to induce arrhythmias.
The gene TTN encodes for titin (connectin),
the largest known protein, which spans half of a sarcomere and
plays critical roles in cardiac (and skeletal) muscle function.
Mutations in TTN gene have been associated with cardiac
diseases such as dilated cardiomyopathy (28), familial hypertrophic cardiomyopathy
(29), early-onset myopathy with
fatal cardiomyopathy (30), and
proximal myopathy with early respiratory muscle involvement
(31). The variant c.49415G>A
in TTN gene detected in this family results in an amino acid
replacement, which localizes at an immunoglobulin (Ig)-like and Fn3
domains super-repeat segment in the A-band region. This region was
identified as a hot spot for inherited dilated cardiomyopathy, a
major cause of heart failure and premature death (32); however, the study only included
truncation mutations. The missense variant c.49415G>A
(p.16472R>H) in this region is likely to be damaging, although
it may not directly cause the malfunction of sinoatrial node. A
recent study showed that patients with arrhythmogenic right
ventricular cardiomyopathy and variants in TTN gene were
more likely to experience supraventricular arrhythmia such as
atrial fibrillation than those without TTN variant (33). Likewise, the presence of TTN
c.49415G>A variant may exacerbate cardiac symptoms accompanied
by bradycardia in the proband (atrial fibrillation) and his two
sons III3 and III4 (more severe ER).
Genetic mutations in genes encoding ankyrin-B
(ANK2) (34),
hyperpolarization-activated channel (HCN4) (35), and SCN5A (36) have been associated with sinoatrial
dysfunction. However, we failed to observe any clinically
meaningful variants in these genes. We cannot rule out genes that
were not covered by our genetic analyses and the contribution of
their variants to disease development. First, six family members
carried both CACNA1C c.1786G>A and TTN
c.49415G>A variants, but three of them (II3, III5, and III6)
failed to show any cardiac symptoms observed in the proband and his
two sons III3 and III4. Second, the proband's son III2 carried only
TTN c.49415G>A variant, which is unlikely to cause
bradycardia alone, but he showed symptoms of bradycardia. However,
the discrepancies between phenotypes and genotypes are very common
in genetics; for instance, phenotypes may not always manifest in
all individuals carrying the same genetic variants (incomplete
penetrance), and the type and severity of phenotypes vary between
genotype-positive individuals (variable expressivity) (37). Factors such as age, sex, and
nutrition may also contribute to different manifestations of
genetic diseases. In rare diseases such as SSS, finding
disease-causing variants by looking non-exhaustedly in a
genome-wide range is almost impossible; hence, it is more realistic
to focus on target genes to identify disease-causing variants based
on the knowledge from databases and previous studies despite the
limitation that the target gene list may always be incomplete.
In conclusion, our study suggests the involvement of
the novel missense CACNA1C c.1786G>A and TTN
c.49415G>A variants in the inheritance of symptomatic
bradycardia and development of SSS. Future studies are required to
understand the etiology at the molecular level.
Acknowledgements
This study was supported by Financial scheme for
young talents training program of Fujian Health industry (grant no.
2015-ZQN-ZD-7) and the Science and Technology Project of Fujian
Province (grant no. 2014Y0007), China.
References
|
1
|
Sanders P, Lau DH and Kalman JK: Sinus
node abnormalitiesCardiac Electrophysiology: From Cell to Bedside.
Zipes D and Jalife J: 6th. Elsevier Saunders; Philadelphia: pp.
691–696. 2014, View Article : Google Scholar
|
|
2
|
Semelka M, Gera J and Usman S: Sick sinus
syndrome: A review. Am Fam Physician. 87:691–696. 2013.PubMed/NCBI
|
|
3
|
Mond HG and Proclemer A: The 11th world
survey of cardiac pacing and implantable
cardioverter-defibrillators: Calendar year 2009-a World Society of
Arrhythmia's project. Pacing Clin Electrophysiol. 34:1013–1027.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hayashi M, Shimizu W and Albert CM: The
spectrum of epidemiology underlying sudden cardiac death. Circ Res.
116:1887–1906. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Monfredi O and Boyett MR: Sick sinus
syndrome and atrial fibrillation in older persons-A view from the
sinoatrial nodal myocyte. J Mol Cell Cardiol. 83:88–100. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Platzer J, Engel J, Schrott-Fischer A,
Stephan K, Bova S, Chen H, Zheng H and Striessnig J: Congenital
deafness and sinoatrial node dysfunction in mice lacking class D
L-type Ca2+ channels. Cell. 102:89–97. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Baig SM, Koschak A, Lieb A, Gebhart M,
Dafinger C, Nürnberg G, Ali A, Ahmad I, Sinnegger-Brauns MJ, Brandt
N, et al: Loss of Ca(v)1.3 (CACNA1D) function in a human
channelopathy with bradycardia and congenital deafness. Nat
Neurosci. 14:77–84. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rosso R, Kogan E, Belhassen B, Rozovski U,
Scheinman MM, Zeltser D, Halkin A, Steinvil A, Heller K, Glikson M,
et al: J-point elevation in survivors of primary ventricular
fibrillation and matched control subjects: Incidence and clinical
significance. J Am Coll Cardiol. 52:1231–1238. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Derval N, Simpson CS, Birnie DH, Healey
JS, Chauhan V, Champagne J, Gardner M, Sanatani S, Yee R, Skanes
AC, et al: Prevalence and characteristics of early repolarization
in the CASPER registry: Cardiac arrest survivors with preserved
ejection fraction registry. J Am Coll Cardiol. 58:722–728. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mahida S, Derval N, Sacher F, Berte B,
Yamashita S, Hooks DA, Denis A, Lim H, Amraoui S, Aljefairi N, et
al: History and clinical significance of early repolarization
syndrome. Heart Rhythm. 12:242–249. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Burashnikov E, Pfeiffer R,
Barajas-Martinez H, Delpón E, Hu D, Desai M, Borggrefe M,
Häissaguerre M, Kanter R, Pollevick GD, et al: Mutations in the
cardiac L-type calcium channel associated with inherited J-wave
syndromes and sudden cardiac death. Heart Rhythm. 7:1872–1882.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Haïssaguerre M, Chatel S, Sacher F,
Weerasooriya R, Probst V, Loussouarn G, Horlitz M, Liersch R,
Schulze-Bahr E, Wilde A, et al: Ventricular fibrillation with
prominent early repolarization associated with a rare variant of
KCNJ8/KATP channel. J Cardiovasc Electrophysiol. 20:93–98. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guo Q, Ren L, Chen X, Hou C, Chu J, Pu J
and Zhang S: A novel mutation in the SCN5A gene contributes to
arrhythmogenic characteristics of early repolarization syndrome.
Int J Mol Med. 37:727–733. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Haïssaguerre M, Derval N, Sacher F, Jesel
L, Deisenhofer I, de Roy L, Pasquié JL, Nogami A, Babuty D,
Yli-Mayry S, et al: Sudden cardiac arrest associated with early
repolarization. N Engl J Med. 358:2016–2023. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gussak I and Antzelevith C: Early
repolarization syndrome: Clinical characteristics and possible
cellular and ionic mechanisms. J Electrocardiol. 33:299–309. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
He J, Wu J, Jiao Y, Wagner-Johnston N,
Ambinder RF, Diaz LA Jr, Kinzler KW, Vogelstein B and Papadopoulos
N: IgH gene rearrangements as plasma biomarkers in Non-Hodgkin's
lymphoma patients. Oncotarget. 2:178–185. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu J, Matthaei H, Maitra A, Dal Molin M,
Wood LD, Eshleman JR, Goggins M, Canto MI, Schulick RD, Edil BH, et
al: Recurrent GNAS mutations define an unexpected pathway for
pancreatic cyst development. Sci Transl Med. 3:92ra662011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhu YB, Gan JH, Luo JW, Zheng XY, Wei SC
and Hu D: Splicing mutation of a gene within the Duchenne muscular
dystrophy family. Genet Mol Res. 15:Jul 14–2016.doi:
10.4238/gmr.15028258. View Article : Google Scholar
|
|
19
|
Li R, Li Y, Kristiansen K and Wang J:
SOAP: Short oligonucleotide alignment program. Bioinformatics.
24:713–714. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mizusawa Y: Recent advances in genetic
testing and counseling for inherited arrhythmias. J Arrhythm.
32:389–397. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Verheijck EE, van Ginneken AC, Wilders R
and Bouman LN: Contribution of L-type Ca2+ current to
electrical activity in sinoatrial nodal myocytes of rabbits. Am J
Physiol. 276:H1064–H1077. 1999.PubMed/NCBI
|
|
22
|
Crotti L, Celano G, Dagradi F and Schwartz
PJ: Congenital long QT syndrome. Orphanet J Rare Dis. 3:182008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Splawski I, Timothy KW, Sharpe LM, Decher
N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM,
Condouris K, et al: Ca(V)1.2 calcium channel dysfunction causes a
multisystem disorder including arrhythmia and autism. Cell.
119:19–31. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Antzelevitch C, Pollevick GD, Cordeiro JM,
Casis O, Sanguinetti MC, Aizawa Y, Guerchicoff A, Pfeiffer R, Oliva
A, Wollnik B, et al: Loss-of-function mutations in the cardiac
calcium channel underlie a new clinical entity characterized by
ST-segment elevation, short QT intervals, and sudden cardiac death.
Circulation. 115:442–449. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jones SA, Boyett MR and Lancaster MK:
Declining into failure: The age-dependent loss of the L-type
calcium channel within the sinoatrial node. Circulation.
115:1183–1190. 2007.PubMed/NCBI
|
|
26
|
Kapplinger JD, Tester DJ, Alders M, Benito
B, Berthet M, Brugada J, Brugada P, Fressart V, Guerchicoff A,
Harris-Kerr C, et al: An international compendium of mutations in
the SCN5A-encoded cardiac sodium channel in patients referred for
Brugada syndrome genetic testing. Heart Rhythm. 7:33–46. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Walsh R, Peters NS, Cook SA and Ware JS:
Paralogue annotation identifies novel pathogenic variants in
patients with Brugada syndrome and catecholaminergic polymorphic
ventricular tachycardia. J Med Genet. 51:35–44. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gerull B, Gramlich M, Atherton J, McNabb
M, Trombitás K, Sasse-Klaassen S, Seidman JG, Seidman C, Granzier
H, Labeit S, et al: Mutations of TTN, encoding the giant muscle
filament titin, cause familial dilated cardiomyopathy. Nat Genet.
30:201–204. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Satoh M, Takahashi M, Sakamoto T, Hiroe M,
Marumo F and Kimura A: Structural analysis of the titin gene in
hypertrophic cardiomyopathy: Identification of a novel disease
gene. Biochem Biophys Res Commun. 262:411–417. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Carmignac V, Salih MA, Quijano-Roy S,
Marchand S, Al Rayess MM, Mukhtar MM, Urtizberea JA, Labeit S,
Guicheney P, Leturcq F, et al: C-terminal titin deletions cause a
novel early-onset myopathy with fatal cardiomyopathy. Ann Neurol.
61:340–351. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nicolao P, Xiang F, Gunnarsson LG,
Giometto B, Edström L, Anvret M and Zhang Z: Autosomal dominant
myopathy with proximal weakness and early respiratory muscle
involvement maps to chromosome 2q. Am J Hum Genet. 64:788–792.
1999. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Herman DS, Lam L, Taylor MR, Wang L,
Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B,
Sparks E, et al: Truncations of titin causing dilated
cardiomyopathy. N Engl J Med. 366:619–628. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Brun F, Barnes CV, Sinagra G, Slavov D,
Barbati G, Zhu X, Graw SL, Spezzacatene A, Pinamonti B, Merlo M, et
al: Titin and desmosomal genes in the natural history of
arrhythmogenic right ventricular cardiomyopathy. J Med Genet.
51:669–676. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mohler PJ, Splawski I, Napolitano C,
Bottelli G, Sharpe L, Timothy K, Priori SG, Keating MT and Bennett
V: A cardiac arrhythmia syndrome caused by loss of ankyrin-B
function. Proc Natl Acad Sci USA. 101:pp. 9137–9142. 2004;
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nof E, Luria D, Brass D, Marek D, Lahat H,
Reznik-Wolf H, Pras E, Dascal N, Eldar M and Glikson M: Point
mutation in the HCN4 cardiac ion channel pore affecting synthesis,
trafficking, and functional expression is associated with familial
asymptomatic sinus bradycardia. Circulation. 116:463–470. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Benson DW, Wang DW, Dyment M, Knilans TK,
Fish FA, Strieper MJ, Rhodes TH and George AL Jr: Congenital sick
sinus syndrome caused by recessive mutations in the cardiac sodium
channel gene (SCN5A). J Clin Invest. 112:1019–1028. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Giudicessi JR and Ackerman MJ:
Determinants of incomplete penetrance and variable expressivity in
heritable cardiac arrhythmia syndromes. Transl Res. 161:1–14. 2013.
View Article : Google Scholar : PubMed/NCBI
|