Introduction
Diabetic encephalopathy (DE) is one of the most
prevalent chronic complications of diabetes mellitus (DM) and is
characterized by degeneration and dysfunction of the central
nervous system. Numerous studies have reported that chronic
hyperglycemia not only leads to inhibition of glucose metabolism in
the brain (1), however also
results in neuronal damage, leading to cognitive function
impairment in rats (2).
Diabetes-associated cognitive decline is a central nervous system
complication induced by DM (3). DE
increases the probability of cognitive decline, as well as
acceleration of Alzheimer's disease and other forms of dementia
(4,5).
The neuronal cytoskeleton is a fibrous protein
filament structure responsible for information transmission, cell
shape maintenance, energy conversion, cell movement and other
cellular functions. The neuronal cytoskeleton is primarily composed
of microtubules (MT), microfilaments (MF) and neurofilaments. The
primary component of MF is filamentous actin (F-actin). F-actin
polymerization and depolymerization take place during cytoskeletal
remodeling and may to a certain extent, reflect the functional
status of cells (6,7). Previous studies identified certain
tau species to be associated with compromised MT integrity and
impaired neuronal function, leading to long-lasting cellular
degradation and cognitive deficits (8–10).
McLean et al (11) revealed
a number of post-translational modifications of neuronal
cytoskeletal proteins that may contribute to the altered axonal
transport and subsequent nerve dysfunction in experimental
diabetes. Phosphorylation of microtubule-associated protein (MAP) 2
induced by glycogen synthase kinase 3 modulates its association
with MT and regulates MT stability (12). Chen et al (13) observed that mice with DM exhibited
a decrease in MAP 2 protein expression in the hippocampus and
cerebral cortex. Advanced glycation end-products (AGEs)/AGEs
receptor promote MT stabilization via the suppression of the
NAD-dependent protein deacetylase sirtuin-2/acetylated α-tubulin
signaling pathway during development of diabetic cardiomyopathy
(14).
The authors previously demonstrated that myosin
light chain kinase (MLCK) is upregulated in the brain tissues and
vascular walls of diabetic rats using two-dimensional
electrophoresis and mass spectrometry detection technology
(7). The results demonstrated a
significant positive association between MLCK and development of DM
(7). These results also indicate
that MLCK may be associated with the occurrence and development of
cognitive dysfunction in diabetes (7). As a serine/threonine kinase, MLCK
modulates the phosphorylation of myosin light chain (p-MLC),
induces rearrangement of the hippocampal neuronal cytoskeleton and
is involved in cell movement, migration and apoptosis (15). MLCK may affect material transport,
energy conversion, information transfer and cell differentiation of
vascular smooth muscle cells (16–18).
In the present study, the primary hippocampal
neurons were cultured in a high-glucose environment and the
alterations in cellular morphology and microfilament cytoskeleton
were evaluated. Furthermore, MLCK and p-MLC protein expression
levels were investigated to determine how MLCK affects the
hippocampal neuronal microfilament structure in a high-glucose
environment, with the aim to elucidate the pathogenesis of diabetic
cognitive dysfunction and offer a novel approach to clinical
treatment.
Materials and methods
Experimental animals
A total of 100 healthy newborn (24 h-old)
Sprague-Dawley rats (5–10 g) were purchased from the Experimental
Animal Center of Guizhou Medical University (Guiyang, China). Prior
to the experiments, the animals were acclimatized in a temperature-
and humidity-controlled environment with food and water (ad
libitum) under a 12 h light/dark cycle. Of the 100 rats, 44 were
female and 56 were male. All experiments were approved by the
Ethics Committee of Guizhou Medical University (approval no.
1702093).
Hippocampal neuronal cell preparation
and culture
Hippocampal neurons were isolated from newborn SD
rats and digested with 0.125% trypsin, as previously described
(19). The hippocampal neurons
were subsequently cultured in basal cell culture medium [0.5 ml
penicillin-streptomycin solution, 0.5 ml L-glutamine, 2.5 ml fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA), 5 ml horse serum (Gibco; Thermo Fisher Scientific, Inc.) and
41.5 ml Dulbecco's modified Eagle medium (Gibco; Thermo Fisher
Scientific, Inc.)]. The cells were incubated at 37°C with 5%
CO2. After 6 h the medium was changed to maintenance
culture medium [0.5 ml penicillin-streptomycin solution, 0.5 ml
L-glutamine, 1 ml B27 and 48 ml Neurobasal-A culture medium (Gibco;
Thermo Fisher Scientific, Inc.)]. Every 2 days, half of the culture
medium was replaced with fresh medium. The cells were incubated at
37°C with 5% CO2.
Identification of hippocampal
neurons
Hippocampal neuronal cells were cultured using the
aforementioned procedure. After 7 days, the cells were rinsed three
times with PBS, fixed for 30 min with 4% paraformaldehyde at room
temperature, incubated for 5–10 min with 3%
H2O2 deionized water at room temperature and
immuno-stained with anti-neuron-specific enolase (NSE) antibody
(1:100; M02930; Wuhan Boster Biological Technology, Ltd., Wuhan,
China) at room temperature, followed by the addition of goat
anti-rabbit immunoglobulin G (IgG) secondary antibody (1:200,
BA1001; Wuhan Boster Biological Technology, Ltd.) and visualized
using fluorescence microscopy (magnification, ×400). ImageJ 1.45s
(National Institutes of Health, USA) was used to estimate the
purity of neurons.
Transmission electron microscopy (TEM)
of hippocampal neuronal cells
The primary cultured hippocampal neurons were
divided into the control (untreated), high-glucose (45 mmol/l), and
high-glucose+MLCK inhibitor (ML-7; 10 mmol/l Sigma-Aldrich, Merck
KGaA, Darmstadt, Germany) groups. A previous study evaluated the
effects of MLCK under conditions of differing glucose
concentrations (20). The preset
study reported that the exposure of hippocampal neurons to 45
mmol/l glucose did not affect cell viability, however increased the
expression of MLCK; therefore, 45 mmol/l glucose was selected as
the high-glucose group (data not shown).
After 5 days, 45 mmol/l glucose was added to the
high-glucose group, and 45 mmol/l glucose with 10 mmol/l ML-7 was
added to the high-glucose + ML-7 group. After 2 days,
differentially treated hippocampal neuronal cells were fixed with
2.5% glutaraldehyde in 0.2 M cacodylate buffer (pH 7.0) for 2 h at
4°C, and subsequently washed in a solution of 0.2 M cacodylate
buffer, sucrose and distilled water overnight at 4°C. The samples
were postfixed in 1% osmium tetroxide in 0.2 M cacodylate buffer
for 2 h at 4°C, washed with distilled water and stained with 2%
aqueous uranyl acetate for 1 h at 4°C. Subsequently, the samples
were washed with distilled water and stained with 2% aqueous uranyl
acetate for 20 min at room temperature, dehydrated through a graded
ethanol series (30, 50, 70, 80, 96 and 99%; 15 min each), then
incubated twice with propylene oxide for 15 min at room
temperature. Finally, the cell pellets were embedded in epoxy resin
for 24 h at 60°C, and ultra-thin sections (70 nm) were cut by a
Leica UC6 ultramicrotome (Leica Microsystems GmbH, Wetzlar,
Germany) and transferred to copper grids (200 mesh). Following
staining with uranyl acetate for 5 min and lead citrate for 1 min
at room temperature, the sections were examined with a transmission
electron microscope (JEOL, Ltd., Tokyo, Japan; 80 kV) and the
images were captured with a digital camera (BioScan model 792;
Gatan, Inc., Pleasanton, CA, USA).
Cell apoptosis analysis
Differentially treated hippocampal neuronal cells
were cultured in serum-free medium (Gibco; Thermo Fisher
Scientific, Inc.) for 48 h. Subsequently, apoptosis was assessed
with the Annexin V-7AAD apoptosis detection kit (Nanjing KeyGen
Biotech Co., Ltd., Nanjing, China) according to the manufacturer's
protocol. Cells were analyzed using fluorescence-activated cell
sorting cytometry (BD Biosciences, Franklin Lakes, NJ, USA).
GraphPad Prism 5 (GraphpPad Software, Inc., La Jolla, CA, USA) was
used to analyze the results.
Immunofluorescence study
Glass slides (14×14-mm) were placed into 24-well
plates (Corning Life Sciences, Corning, NY, USA). Differentially
treated hippocampal neuronal cells were cultured on 14×14-mm glass
slides for 48 h using the aforementioned procedure. After 48 h, the
cells were fixed on 14×14-mm glass slides with 4% paraformaldehyde
for 30 min at room temperature then washed with PBS. Following
blocking with 3% goat serum (Wuhan Boster Biological Technology,
Ltd.) for 30 min at room temperature. The cells were incubated with
antibodies specific for F-actin (1:100; ab205; Abcam, Cambridge,
UK) overnight at 4°C. Following three washes with PBS, the cells
were incubated with goat anti-mouse IgG antibodies (Alexa Fluor488;
cat. no. 4408S, Cell Signaling Technology, Inc., Danvers, MA, USA)
for 1 h at 37°C and then the slides were mounted by adding
DAPI-Fluoromount-G (1:1,000; Wuhan Sanying Biotechnology, Inc.,
Wuhan, China) for 5 min at room temperature. Subsequently, cells
were examined under a confocal laser scanning microscope
(magnification, ×630). ImageJ software 1.45s (National Institutes
of Health) was used to analyze the results.
Western blot analysis
Total proteins were extracted from differentially
treated hippocampal neuronal cells using radioimmunoprecipitation
assay buffer (Beyotime Institute of Biotechnology) on ice. The
protein concentration was determined with BCA Protein Assay kit
(Beyotime Institute of Biotechnology). The proteins (50 µg) were
isolated by 12% SDS-PAGE at room temperature. Following
electrophoresis, the proteins were transferred onto polyvinylidene
membranes (Thermo Fisher Scientific, Inc.) for 1.5 h. The membranes
were incubated with 5% non-fat milk in TBS for 1.5 h at room
temperature, followed by incubation with primary antibodies against
MLCK (1:5,000; ab76092; Abcam), p-MLC (1:1,000; cat. no. 3675; Cell
Signaling Technology, Inc.) and β-actin (1:2,000; BS6007MH; Biogot
Technology Co., Ltd., Nanjing, China) overnight at 4°C.
Subsequently, the membranes were incubated with horseradish
peroxidase-conjugated goat anti-mouse IgG secondary antibody
(1:2,000; Bioworld Technology, China) for 2 h at room temperature.
Bands were visualized with enhanced chemiluminescence western blot
detection reagents (Advansta, Inc., Menlo Park, CA, USA) and
analyzed using the Image-Pro Plus V6.0 software (Media Cybernetics,
Inc., Rockville, MD, USA).
Statistical analysis
Statistical analyses were performed using SPSS
software, version 17.0 (SPSS, Inc., Chicago, IL, USA). Data were
analyzed using one-way analysis of variance with the Bonferroni
post hoc test. Each experiment was performed three times and the
results are presented as the mean ± standard deviation. P<0.05
was considered to indicate a statistically significant
difference.
Results
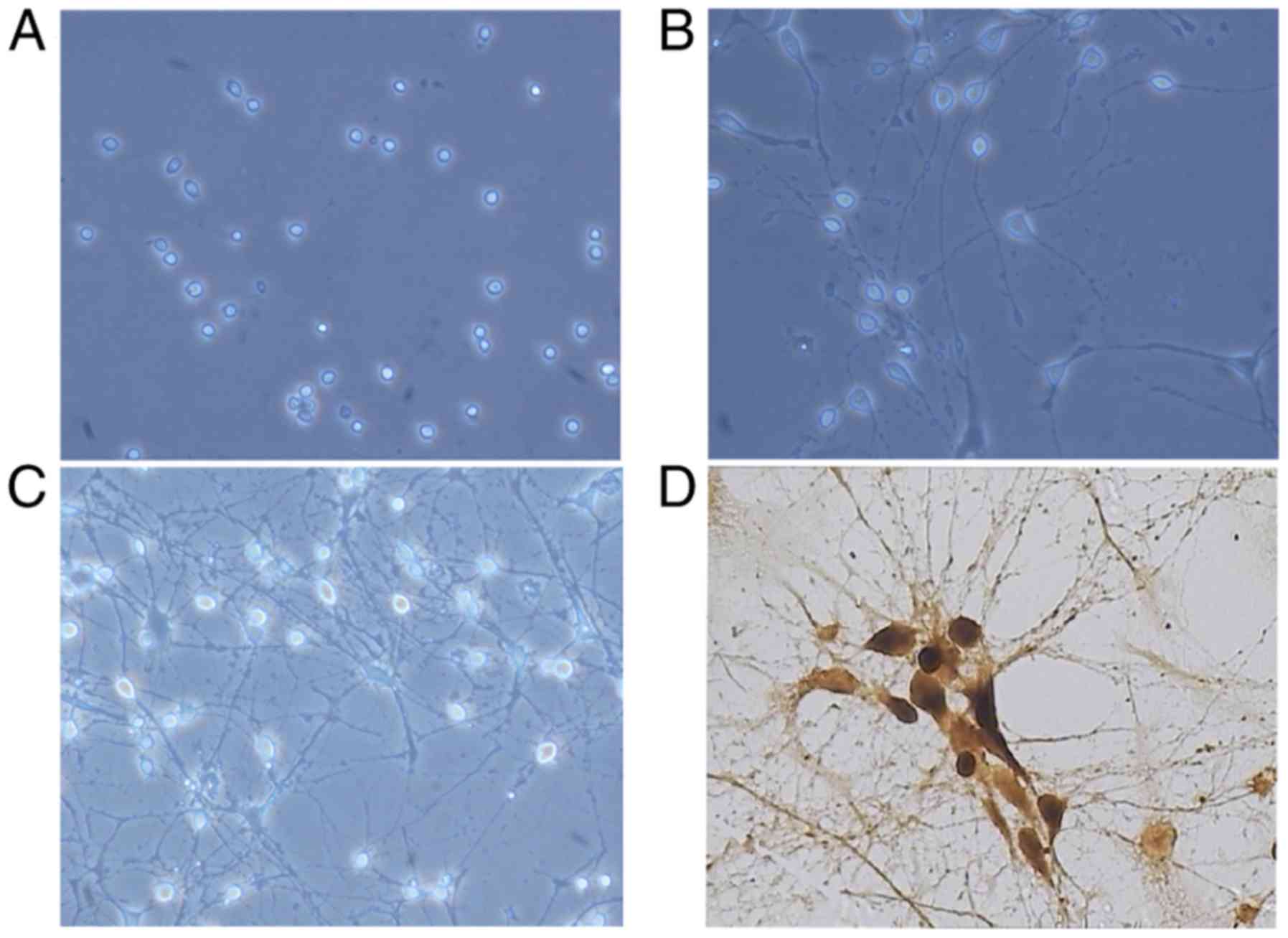
Morphology and identification of
hippocampal neurons
The hippocampal neuronal cells were successfully
isolated from newborn SD rats and grown in culture. After 6 h, 90%
of the cells had attached to the dish. The cells exhibited a round,
oval or tapered shape, and the cell bodies were clearly visible and
had a three-dimensional appearance with strong refraction (Fig. 1A). After 4 days, the volume of the
cells had distinctly increased and the neurites were branched
(Fig. 1B). After 7 days, the
neurites were branched and appeared to be overlapping (Fig. 1C). The purity of hippocampal
neurons was measured by immunohistochemical staining with anti-NSE
antibody and was determined to be >90% (Fig. 1D).
Ultrastructure of hippocampal
neurons
To determine whether the ultrastructure of
hippocampal neurons was altered when they were cultured in a
high-glucose environment, the neurons were examined using TEM. In
the control group, the nuclear morphology was regular and the
chromatins in the nucleus were abundant and homogeneously
distributed. The cell morphology was normal and the cell organelles
were abundant. The endoplasmic reticulum and mitochondria were
normal in morphology, and there were large number of microfilaments
in the cytoplasm (Fig. 2A). In the
high-glucose group, the nuclear membranes were shrunk, the
morphology of the nucleus was irregular, and the chromatins were
heterogeneously distributed, granular and aggregated into blocks.
The cytoplasm was fragmented, the endoplasmic reticulum appeared
edematous and the mitochondria were markedly enlarged. These
alterations were generally accompanied by dilatation of the Golgi
complex and were frequently accompanied by autophagy and appearance
of dense bodies (Fig. 2B). In the
high-glucose + ML-7 group, the cell morphology was almost normal.
The nucleus was normal, with a round or oval shape and the
chromatins were abundant. The endoplasmic reticulum and Golgi
complex were only partly edematous. The mitochondria were abundant
and their morphology was normal. The cytoplasmic volume of these
neurons was increased. Compared with the high-glucose group, the
damaged structural elements had mostly recovered in the high
glucose + ML-7 group (Fig.
2C).
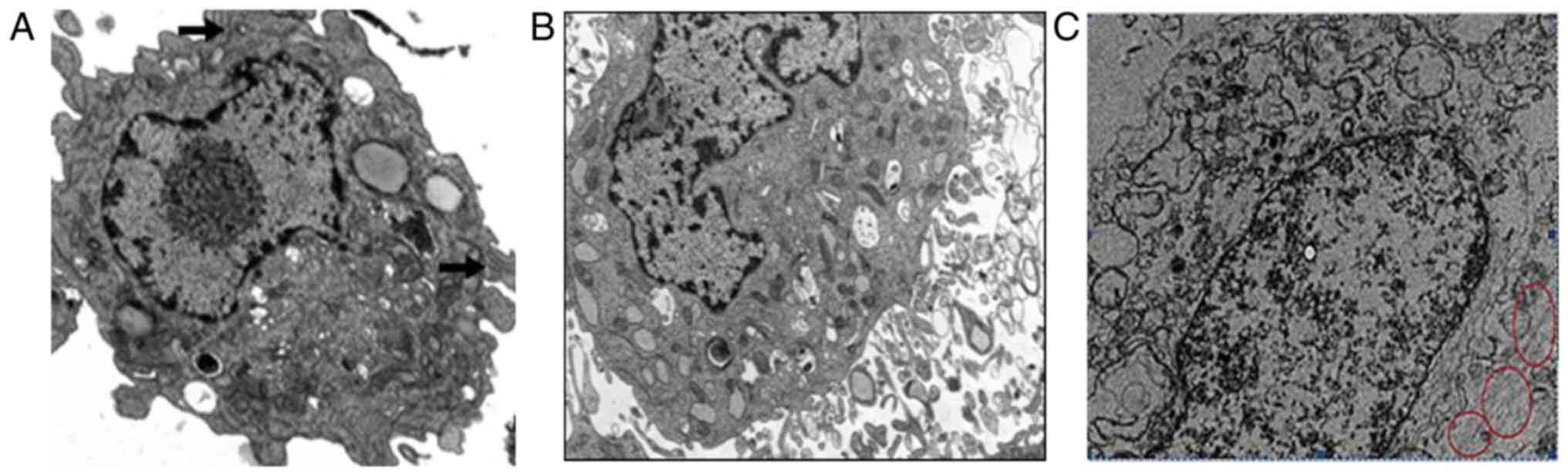 | Figure 2.Ultrastructure of hippocampal neurons
in culture. (A) In the control group, the morphology of the nucleus
was regular, and the chromatins in the nucleus were abundant and
homogeneously distributed. The cell organelles were abundant, the
endoplasmic reticulum and mitochondria were normal in morphology,
and there were large numbers of microfilaments in the cytoplasm
(magnification, ×10,000). Black arrow, microfilament structural
integrity. (B) In the high-glucose group, a large fragment of a
relatively well-preserved perikaryon of the pyramidal neuron was
observed. Certain mitochondria appeared to be enlarged. The nuclear
membrane appeared to have shrunk (magnification, ×10,000). (C) In
the high glucose+ML-7 group the nuclei were normal, round or oval,
with abundant chromatin. The endoplasmic reticulum and Golgi
complex were only partly edematous. The mitochondria were abundant
and their morphology was normal (magnification, ×10,000). Red
circles, abundant microfilament. |
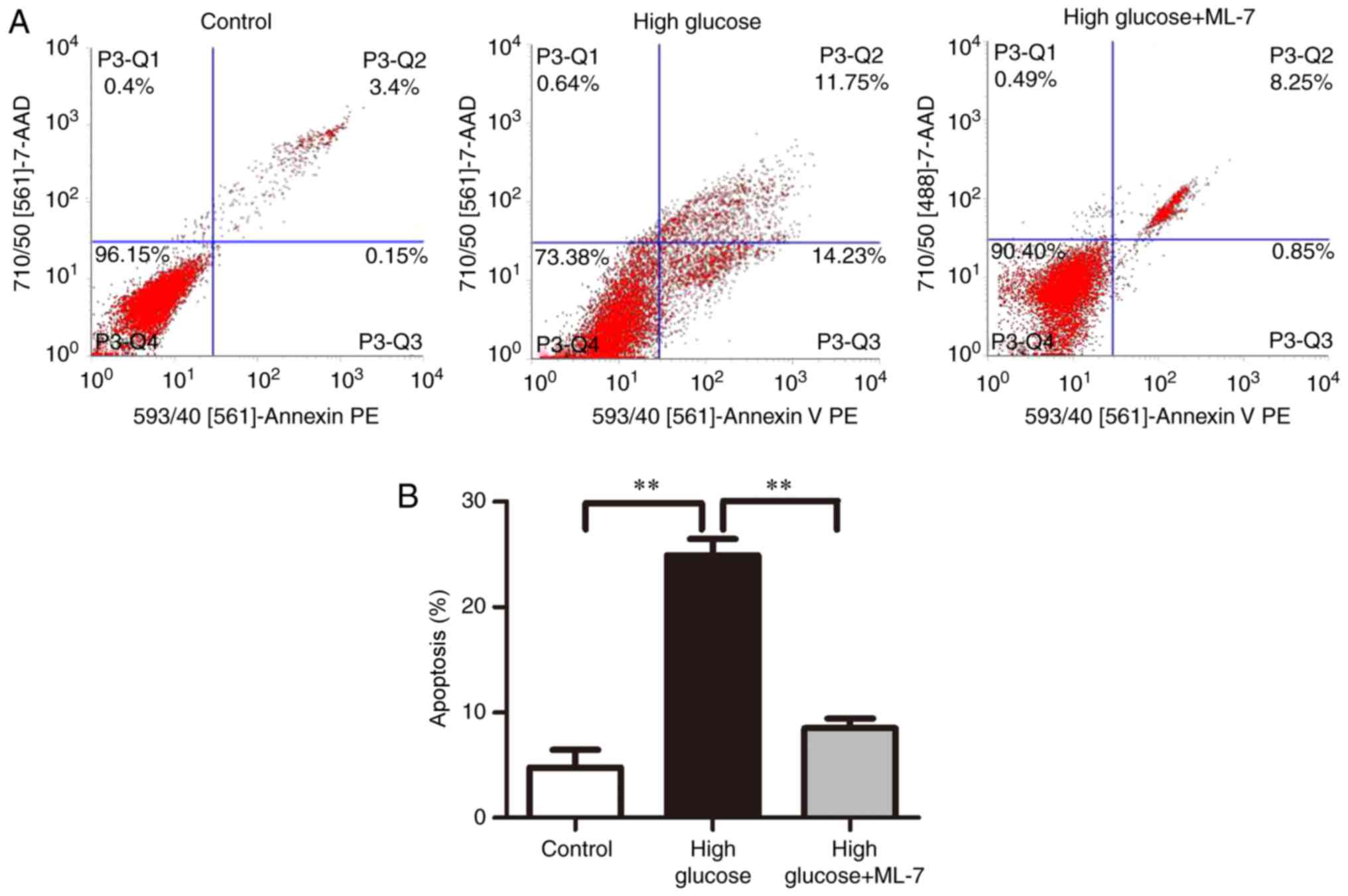
Apoptosis rate in different
groups
In order to determine the cause of cell death
following the ultrastructural alterations of the hippocampal
neurons, flow cytometry was performed to examine cell apoptosis
among different groups. The results revealed that the apoptosis
rates were 4.75±1.20, 24.91±1.07 and 8.5±0.63% in the control,
high-glucose and high glucose + ML-7 groups, respectively (Fig. 3A and B), suggesting that high
glucose induced hippocampal neuron apoptosis.
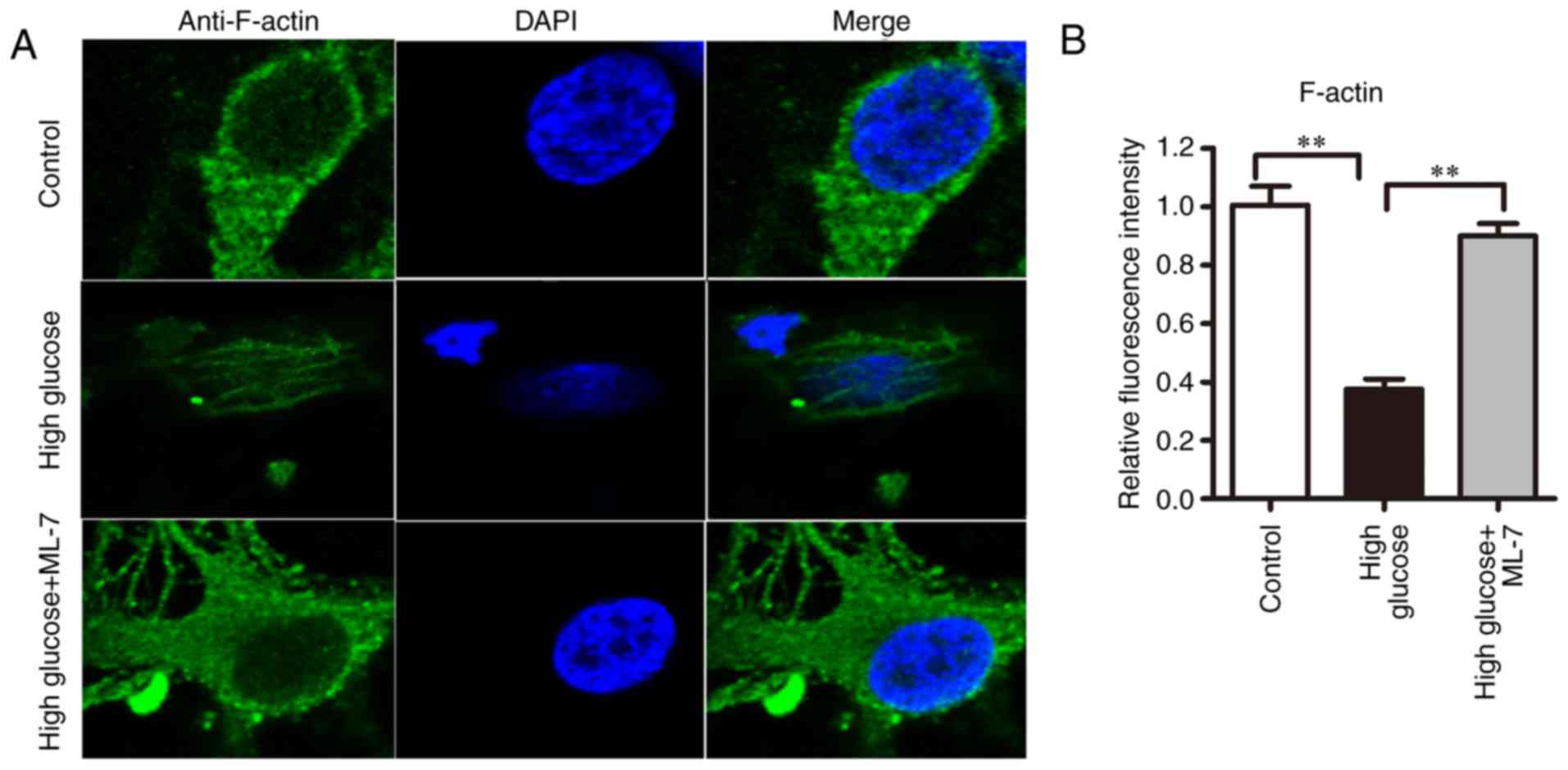
F-actin cytoskeleton organization
In the control group, F-actin was distributed
homogeneously, surrounding the cell body and dendritic spines and
forming a complete and continuous actin band (Fig. 4A). In the high-glucose group, the
actin bands had disappeared and the F-actin was depolymerized or
rearranged. In addition, the dendrites and axons were shrunk.
Compared with the high-glucose group, in the high-glucose + ML-7
group the F-actin bands were recovered and F-actin was apparent in
dendritic spines, although it was abnormally arranged. To calculate
the F-actin density in different groups, the fluorescence intensity
was calculated in the cell area excluding the nucleus. Compared
with the control group, the density of F-actin was lower in the
high-glucose group (from three cell preparations; P<0.01).
Compared with the high-glucose group, the density of F-actin
increased in the high-glucose + ML-7 group (P<0.01; Fig. 4B). The aforementioned results
indicated that ML-7 may induce F-actin polymerization.
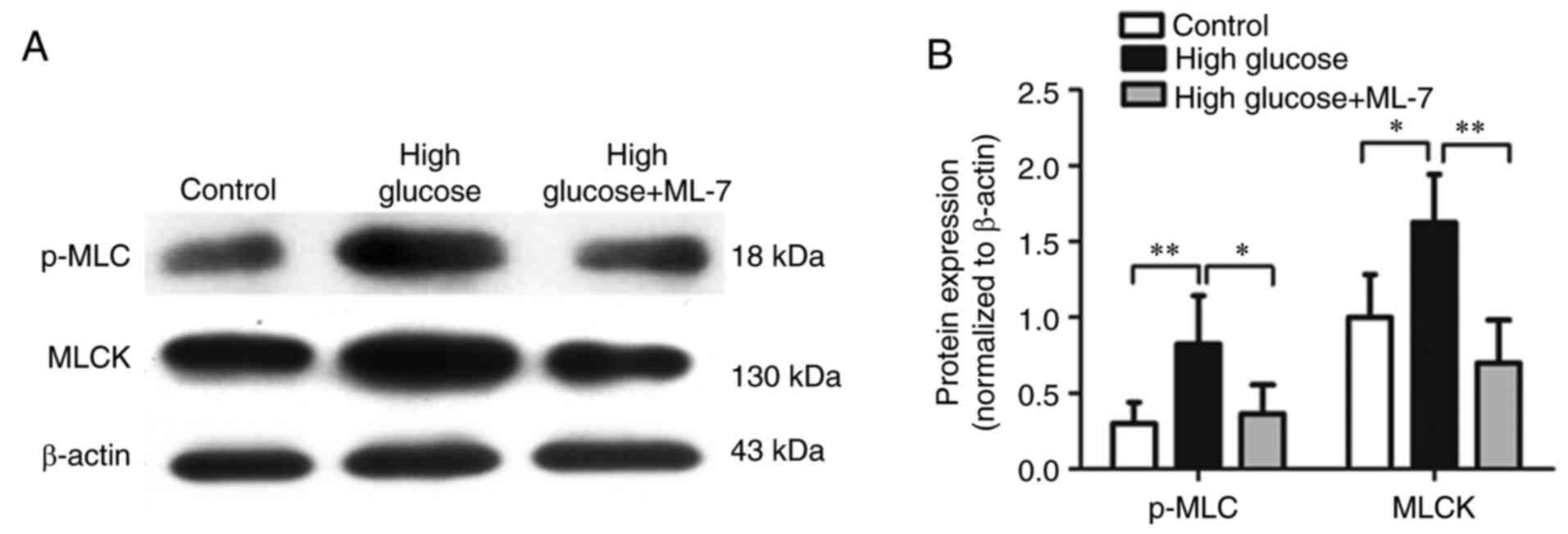
Protein expression of MLCK and p-MLC
in hippocampal neurons
To investigate whether high glucose affects MLCK,
the protein expression of MLCK and p-MLC were measured by western
blot analysis. Compared with the control group, the protein
expression of MLCK and p-MLC were upregulated in the high-glucose
group. By contrast, compared with the high-glucose group, the
expression of MLCK and p-MLC were downregulated in the high-glucose
+ ML-7 group (Fig. 5A and B).
Discussion
DE is characterized by degeneration and dysfunction
of the central nervous system. Neuronal plasticity serves a role in
the brain and refers to morphological, biochemical and
physiological alterations in the developing nervous system.
Accumulating evidence indicates that alterations in synaptic
contacts modulate the function of the nervous system (21,22).
In recent years, studies of the cytoplasm have
focused on the organizational properties and functional
significance of actin-containing microfilaments. Actin is the
primary component of microfilaments, whereas actin and myosin
molecules are the main components of non-muscle cells (23–25).
In non-muscle cells, actin is present in the form of polymerized
filaments (F-actin) and in the globular form of G-actin (23). In order to be physiologically
active, actin must be polymerized in filaments (23). The viscosity of the cytoplasm is
determined by the state of actin polymerization (23). The presence or absence of myosin
molecules determines whether actin filaments will be involved in
movement generation (23). Myosin
has been identified in neurons and non-muscle cells in biochemical
experiments. The contractile mechanism involves associations
between the myosin heads and actin filaments in non-muscle cells
(23). Electron microscopy studies
have established that myosin and actin filament co-localization is
not uniform throughout the cell. Experiments with the isolated
brush border have demonstrated that conformation of myosin is
determined by the functional state of the tissue at the time of
fixation (23). When myosin is
phosphorylated, it is released from the actin cytoskeleton and is
free to assemble into bipolar filaments (23). Following contraction, myosin is
dephosphorylated by a phosphatase that restores myosin to its
largely unpolymerized form and it becomes again attached to the
actin cytoskeleton (26).
MLCK is the primary regulator of various forms of
eukaryotic motility. MLCK serves a role in numerous functions of
non-muscle cells, including cytoskeletal clustering, platelet shape
alterations, transepithelial permeability and cytoskeletal
arrangements, stress fiber formation, cell spreading and migration,
cytokinesis, secretion and neurite growth cone advancement, which
affect ion exchange or ion currents at the plasma membrane
(27–29). In synapses, MLCK and its downstream
substrate myosin, regulate different stages of neurotransmitter
release (30), including refilling
of readily releasable pools following tetanus stimulation, vesicle
mobility (31,32), supply of fast-releasing vesicles
(33) and vesicle mobilization
(34). MLCK is required for
activity-dependent functions of myosin in hippocampal neurons
(35). Certain cellular effects of
MLCK result from its phosphorylation of MLC (36).
The molecular mechanism underlying neuronal damage
in hyperglycemia has not yet been fully elucidated. In the authors'
previous study, a proteomic approach, reverse transcription PCR,
western blot and immunohistochemistry assays were used to identify
differential proteins expressed in the brain of rats with type 1
diabetes mellitus in vivo, and it was determined that MLCK
is upregulated in rats with type 1 DM (7). The authors also previously
demonstrated that the degree of cognitive dysfunction in diabetic
rats is aggravated in weeks 6–12 following induction of type 1 DM
(37). The ultrastructure of
hippocampal neurons is markedly altered and rate of cell apoptosis
increases (37). The number of
organelles decreases and nuclei exhibit increased intensity of
staining (37). In week 12
following induction of the model of type 1 DM, microscopic
observation revealed that the number of hippocampal neurons
decreases markedly and the morphology of hippocampal neurons is
abnormal (37). The authors
previous study also demonstrated that the structure of the cerebral
neurons distinctly degenerates and the expression of MLCK is
upregulated in diabetic rats, and MLCK may serve a role in neuronal
plasticity (38). The present
study analyzed the ultrastructure of primary cultured hippocampal
neurons treated with high glucose (45 mmol/l) by TEM and it was
demonstrated that the nuclear membranes had shrunk, the morphology
of the nucleus was irregular, and the chromatins were
heterogeneously distributed, granular and aggregated into blocks.
Cytoplasm fragmentation, distinctly edematous endoplasmic reticulum
and markedly enlarged mitochondria appeared, however the MLCK
inhibitor ML-7 was able to reverse these alterations. Exposure to
high-glucose conditions resulted in F-actin depolymerization and
rearrangement, however ML-7 reversed this effect. Protein
expression of MLCK and p-MLC was also determined in differentially
treated hippocampal neurons and it was demonstrated that MLCK and
p-MLC were upregulated in the high-glucose environment. Following
addition of ML-7, the expression of MLCK and p-MLC was
downregulated.
In conclusion, the present study demonstrated that
high glucose upregulated the expression of MLCK to promote F-actin
depolymerization, which, in turn, induced microfilament
cytoskeleton rearrangement in hippocampal neurons. It is necessary
to elucidate the pathogenesis of diabetic cognitive dysfunction in
order to design novel modalities for clinical treatment, and the
present study contributed to these efforts.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Funding (grant nos. 81560720 and
81560138).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
All authors take public responsibility for the
integrity of the data and the accuracy of analysis in the study. WP
and XL made substantial contributions to the concept and design of
the present study. LZ and CL made substantial contributions to the
acquisition, analysis and interpretation of data. LZ drafted the
manuscript. LZ conducted statistical analysis. GD bred the mice,
and prepared and cultured hippocampal neuronal cells. MP identified
the hippocampal neurons and carried out Western blot analysis. GL
carried out immunofluorescence study and cell apoptosis
analysis.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Guizhou Medical University (approval no. 1702093).
Consent for publication
No applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Duelli R, Maurer MH, Staudt R, Heiland S,
Duembgen L and Kuschinsky W: Increased cerebral glucose utilization
and decreased glucose transporter Glut1 during chronic
hyperglycemia in rat brain. Brain Res. 858:338–347. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Malone JI, Hanna S, Saporta S, Mervis RF,
Park CR, Chong L and Diamond DM: Hyperglycemia not hypoglycemia
alters neuronal dendrites and impairs spatial memory. Pediatr
Diabetes. 9:531–539. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ahtiluoto S, Polvikoski T, Peltonen M,
Solomon A, Tuomilehto J, Winblad B, Sulkava R and Kivipelto M:
Diabetes, Alzheimer disease, and vascular dementia: A
population-based neuropathologic study. Neurology. 75:1195–1202.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Summers WK: Alzheimer's disease, oxidative
injury, and cytokines. J Alzheimers Dis. 6:651–657; discussion
673–681. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pasquier F, Boulogne A, Leys D and
Fontaine P: Diabetes mellitus and dementia. Diabetes Metab.
32:403–414. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee A, Fischer RS and Fowler VM:
Stabilization and remodeling of the membrane skeleton during lens
fiber cell differentiation and maturation. Dev Dyn. 217:257–270.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li X, Pan W, Yang GZ, Di YN, Zhao F, Zhu
LY and Jiang ZH: Proteome analysis of differential protein
expression in brain of rats with type 1 diabetes mellitus. Exp Clin
Endocrinol Diabetes. 119:265–270. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang Y, Tian Q, Zhang Q, Zhou X, Liu S
and Wang JZ: Hyperphosphorylation of microtubule-associated tau
protein plays dual role in neurodegeneration and neuroprotection.
Pathophysiology. 16:311–316. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kruger L and Mandelkow EM: Tau
neurotoxicity and rescue in animal models of human tauopathies.
Curr Opin Neurobiol. 36:52–58. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Santacruz K, Lewis J, Spires T, Paulson J,
Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan
E, et al: Tau suppression in a neurodegenerative mouse model
improves memory function. Science. 309:476–481. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
McLean WG, Pekiner C, Cullum NA and Casson
IF: Posttranslational modifications of nerve cytoskeletal proteins
in experimental diabetes. Mol Neurobiol. 6:225–237. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang J and Dong XP: Dysfunction of
microtubule-associated proteins of MAP2/tau family in prion
disease. Prion. 6:334–338. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen J, Liang L, Zhan L, Zhou Y, Zheng L,
Sun X, Gong J, Sui H, Jiang R, Zhang F and Zhang L: ZiBuPiYin
recipe protects db/db mice from diabetes-associated cognitive
decline through improving multiple pathological changes. PLoS One.
9:e916802014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yuan Q, Zhan L, Zhou QY, Zhang LL, Chen
XM, Hu XM and Yuan XC: SIRT2 regulates microtubule stabilization in
diabetic cardiomyopathy. Eur J Pharmacol. 764:554–561. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Stull JT, Kamm KE and Vandenboom R: Myosin
light chain kinase and the role of myosin light chain
phosphorylation in skeletal muscle. Arch Biochem Biophys.
510:120–128. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gao N, Tsai MH, Chang AN, He W, Chen CP,
Zhu M, Kamm KE and Stull JT: Physiological vs. pharmacological
signalling to myosin phosphorylation in airway smooth muscle. J
Physiol. 595:6231–6247. 2017. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Khapchaev AY and Shirinsky VP: Myosin
light chain kinase MYLK1: Anatomy, interactions, functions, and
regulation. Biochemistry (Mosc). 81:1676–1697. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Martinsen A, Dessy C and Morel N:
Regulation of calcium channels in smooth muscle: New insights into
the role of myosin light chain kinase. Channels (Austin).
8:402–413. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wilcox KS, Buchhalter J and Dichter MA:
Properties of inhibitory and excitatory synapses between
hippocampal neurons in very low density cultures. Synapse.
18:128–151. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gaspar JM, Castilho A, Baptista FI,
Liberal J and Ambrosio AF: Long-term exposure to high glucose
induces changes in the content and distribution of some exocytotic
proteins in cultured hippocampal neurons. Neuroscience.
171:981–992. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pardal R and Barneo Lopez J: Mature
neurons modulate neurogenesis through chemical signals acting on
neural stem cells. Dev Growth Differ. 58:456–462. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yarden-Rabinowitz Y and Yarom Y: In vivo
analysis of synaptic activity in cerebellar nuclei neurons unravels
the efficacy of excitatory inputs. J Physiol. 595:5945–5963. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fifkova E: Actin in the nervous system.
Brain Res. 356:187–215. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
He Q and Roblodowski C: Functional
analysis of actin-binding proteins in the central nervous system of
drosophila. Methods Mol Biol. 1365:349–355. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu L, Luo M, Yang B, Wu X, Zhu W, Guan Y,
Cai W, Troidl K, Schaper W and Schaper J: Actin-binding rho
activating protein is expressed in the central nervous system of
normal adult rats. Neural Regen Res. 7:965–970. 2012.PubMed/NCBI
|
|
26
|
Broschat KO, Stidwill RP and Burgess DR:
Phosphorylation controls brush border motility by regulating myosin
structure and association with the cytoskeleton. Cell. 35:561–571.
1983. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bresnick AR: Molecular mechanisms of
nonmuscle myosin-II regulation. Curr Opin Cell Biol. 11:26–33.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Aromolaran AS, Albert AP and Large WA:
Evidence for myosin light chain kinase mediating
noradrenaline-evoked cation current in rabbit portal vein myocytes.
J Physiol. 3:853–863. 2000. View Article : Google Scholar
|
|
29
|
Yang X, Wang JG, Ma DB, Ma XF, Zhu GJ,
Zhou H, Yu CJ, Qian XY and Gao X: Myosin light chain kinase
regulates hearing in mice by influencing the F-actin cytoskeleton
of outer hair cells and cochleae. Int J Mol Med. 33:905–912. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mochida S, Kobayashi H, Matsuda Y, Yuda Y,
Muramoto K and Nonomura Y: Myosin II is involved in transmitter
release at synapses formed between rat sympathetic neurons in
culture. Neuron. 13:1131–1142. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee JS, Kim MH, Ho WK and Lee SH:
Presynaptic release probability and readily releasable pool size
are regulated by two independent mechanisms during posttetanic
potentiation at the calyx of Held synapse. J Neurosci.
28:7945–7953. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Peng A, Rotman Z, Deng PY and Klyachko VA:
Differential motion dynamics of synaptic vesicles undergoing
spontaneous and activity-evoked endocytosis. Neuron. 73:1108–1115.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Garcia-Morales V, Montero F,
Gonzalez-Forero D, Rodriguez-Bey G, Gomez-Perez L,
Medialdea-Wandossell MJ, Dominguez-Vias G, Garcia-Verdugo JM and
Moreno-Lopez B: Membrane-derived phospholipids control synaptic
neurotransmission and plasticity. PLoS Biol. 13:e10021532015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Seabrooke S and Stewart BA: Synaptic
transmission and plasticity are modulated by nonmuscle myosin ii at
the neuromuscular junction of drosophila. J Neurophysiol.
105:1966–1976. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li L, Wu X, Yue HY, Zhu YC and Xu J:
Myosin light chain kinase facilitates endocytosis of synaptic
vesicles at hippocampal boutons. J Neurochem. 138:60–73. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Somlyo AP and Somlyo AV: Ca2+ sensitivity
of smooth muscle and nonmuscle myosin II: Modulated by G proteins,
kinases, and myosin phosphatase. Physiol Rev. 83:1325–1358. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang Feng, He Yu, Liu Mi, Chang Xiao, Ren
Zhijing, Pan Wei and Li Xing: The establishment of the diabetic rat
model with cognitive dysfunction and the observation of cognitive
dysfunction in the diabetic rats. Chongqing Medicine. 16:2234–2236.
2015. View Article : Google Scholar
|
|
38
|
He Y, Wang F, Chen S, Liu M, Pan W and Li
X: The protective effect of radix polygoni multiflori on
diabetic encephalopathy via regulating myosin light chain kinase
expression. J Diabetes Res. 2015:4847212015. View Article : Google Scholar : PubMed/NCBI
|