Introduction
Spinocerebellar ataxias (SCAs), a genetically
heterogeneous group of disorders, are inherited in an
autosomal-dominant pattern. Currently, over 30 different types of
SCA have been reported, however only ~50% of causative mutations
have been identified (1). The
majority of SCA cases are caused by an abnormally expanded CAG
repeat sequence that leads to the expansion of a tract of
polyglutamine residues in the encoded proteins (2). The SCAs caused by the CAG repeat
expansion mutation are classified as polyglutamine SCAs. These
include SCA1, SCA2, SCA3, SCA6, SCA7, SCA12, SCA17, and
dentatorubral-pallidoluysian atrophy (DRPLA), in which the
expansion within the glutamine-encoding CAG trinucleotide repeats
accounts for SCA (3). Healthy
individuals usually carry the heterozygous CAG repeats at a number
below the predicted pathological threshold. SCA8, probably arising
from a non-coding CTG repeat expansion is also grouped with
polyglutamine SCAs (4). The
threshold of CAG/CTG expansions that determine disease carrier
status varies between the different types of SCA (5). Several methods have been used for
detecting polyglutamine SCA mutations including fluorescence
polymerase chain reaction (PCR), denaturing polyacrylamide gel
electrophoresis of DNA, next-generation DNA sequencing and direct
sequencing (6). However, these
methods have some limitations when applied to a clinical laboratory
setting (7). Therefore, there is a
need to develop a simple and rapid method for the detection of CAG
repeat mutations. PCR amplification of the CAG repeat mutations
followed by Sanger sequencing is a simple and inexpensive method
for detection. In the current study, the authors present a
convenient method for screening causative mutations for
polyglutamine SCA using PCR amplification followed by Sanger
sequencing. A Chinese SCA1 pedigree was identified using this
method.
Materials and methods
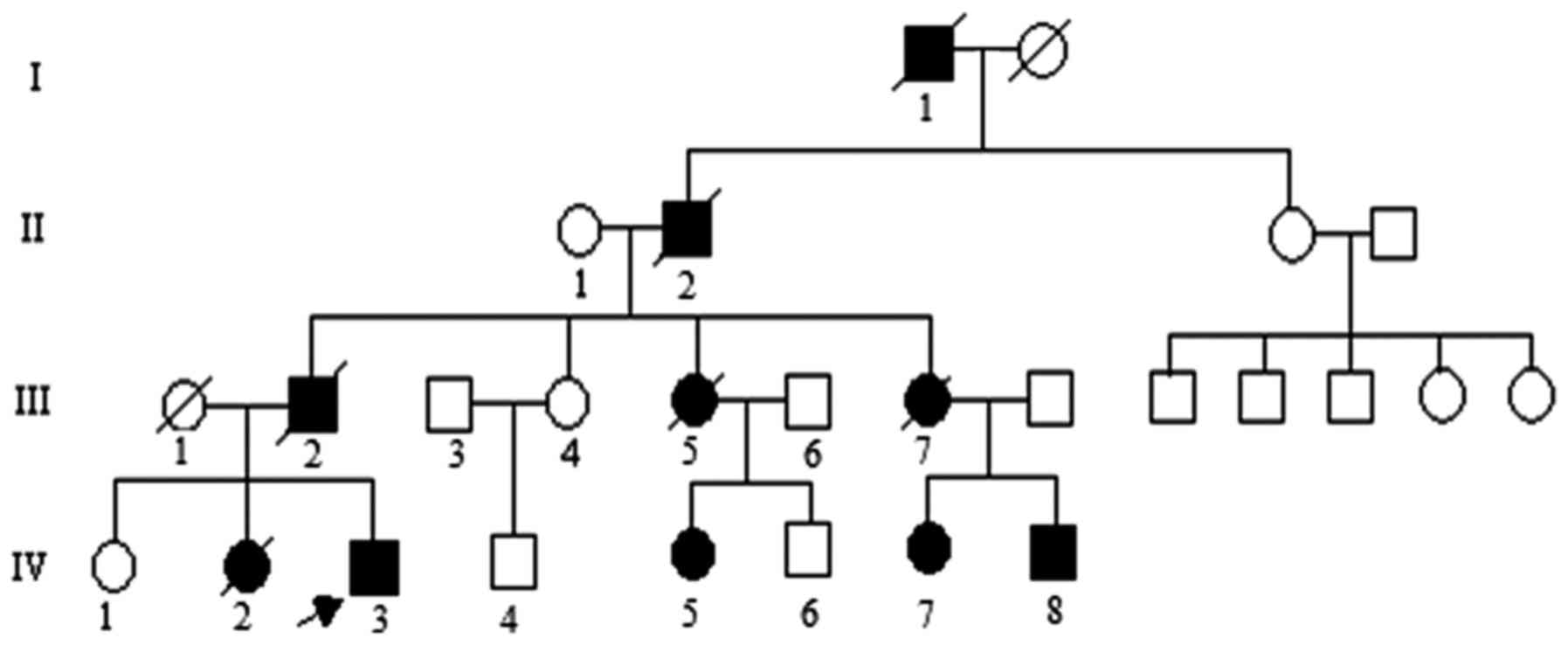
Pedigree
The study was approved by the ethics committee of
Shenzhen Baoan Hospital, Southern Medical University (Shenzhen,
China). Written informed patient consent was obtained before
initiating the study. The pedigree investigated in the present
study is shown in Fig. 1. The
propositus (IV-3) a 24-year-old male individual phenotypically
characterized by a slowly progressive incoordination of gait,
associated with poor coordination of hands, was referred to the
Department of Neurology due to primary difficulty walking. The
propositus complained of slow walking and unsteady gait from the
age of 18. The clinical features of the propositus included limb
ataxia, dysarthria, dysphagia, action tremors, increased tendon
reflexes and handwriting difficulties on examination. Brain
magnetic resonance imaging (data not shown) indicated prominent
atrophy of the cerebellum and brainstem. The family was diagnosed
with SCA by a clinical neurologist according to neuroimaging,
family history and physical examination. The age of onset was
defined according to the early signs of gait disturbance and
slurred speech described by the family members in the SCA pedigree.
Genomic DNA was extracted from the peripheral leukocytes of the 8
subjects given serial numbers in the SCA pedigree and 205 normal
controls using standard protocol (8). Three subjects (IV-5, 7 and 8) were
asymptomatic causative SCA1 gene carriers at gene diagnosis in the
pedigree.
Screening PCR
Polyglutamine SCA includes SCA1, SCA2, SCA3, SCA6,
SCA7, SCA8, SCA12, SCA17 and DRPLA. A total of 9 fragments
including CAG/CTG expansion sequences were amplified by PCR using
primer sets shown in Table I.
First, 100 ng of genomic DNA was amplified by PCR in a 50 µl
reaction volume containing 10 mM Tris-HCl, (pH 8.3), 2.5 mM
MgCl2, 50 mM KCl, 200 M of each dNTP, 2 U Taq
polymerase, and 0.5 M each primer (Table I). Following denaturation at 95°C
for 7 min, amplification was performed at 95°C for 50 seconds,
annealing for 50 sec under different temperatures as described in
Table I, and 72°C for 60 sec for
35 cycles followed by a final extension at 72°C for 7 min. After
amplification, electrophoresis was performed in a 2% agarose gel
(100 V, 45 min) to detect the amplified DNA fragments, then agarose
gel was exposed under a UV light imager.
 | Table I.Sequences of oligonucleotide primer
sets for polymerase chain reaction. |
Table I.
Sequences of oligonucleotide primer
sets for polymerase chain reaction.
| Primer | Sequence (5′-3′) | Annealing temperature
(°C) | Fragment size of the
wild-type allele |
|---|
| SCA1-F |
ggtcctcccaatacagtgga | 60 | 364–403 |
| SCA1-R |
ttctgcggagaactggaaat |
|
|
| SCA2-F |
ctccgcctcagactgttttg | 61 | 810–837 |
| SCA2-R |
ctgaccatcgccgctacc |
|
|
| SCA3-F |
tctgtatcagactaactgctcttgc | 59 | 369–438 |
| SCA3-R |
gagggaatgaagaataatgtaaagc |
|
|
| SCA6-F |
tcccgtgtctcctttgattt | 59 | 378–411 |
| SCA6-R |
gacccgcctctccatcct |
|
|
| SCA7-F |
taggagcggaaagaatgtcg | 56 | 280–310 |
| SCA7-R |
cccagcatcacttcaggact |
|
|
| SCA8-F |
gcagtatgaggaagtatggaaaaa | 59 | 230–335 |
| SCA8-R |
ttctgactcccagcttccac |
|
|
| SCA12-F |
ccactgcagcaaagagca | 59 | 280–355 |
| SCA12-R |
ggaatgagggtgctggtc |
|
|
| SCA17-F |
gaccccacagcctattcaga | 60 | 196–247 |
| SCA17-R |
gcctgaggttccctgtgtt |
|
|
| DRPLA-F |
ccaccacctcctccctatg | 60 | 209–257 |
| DRPLA-R |
agtgggtggggaaatgct |
|
|
Sanger sequencing
Following purification, amplified PCR fragments were
subjected to bidirectional direct and cycle sequencing. Direct
sequencing was performed using the ABI3730 Genetic Analyzer
(Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA,
USA). Cycle sequencing was performed using Stratagene's RoboCycler
Gradient (Stratagene; Agilent Technologies, Inc., Santa Clara, CA,
USA) 96 temperature cycler with Hot Top Assembly. The DNA
polymerase used in sequencing was Platinum SuperFi DNA Polymerase
(Invitrogen, Thermo Fisher Scientific, Inc.) which combines high
fidelity and high sequence accuracy. All sequencing of PCR
fragments was performed more than twice, the identical sequencing
results were regarded as the number of CAG triplet repeats carried
by individuals.
Results
Mutation screening
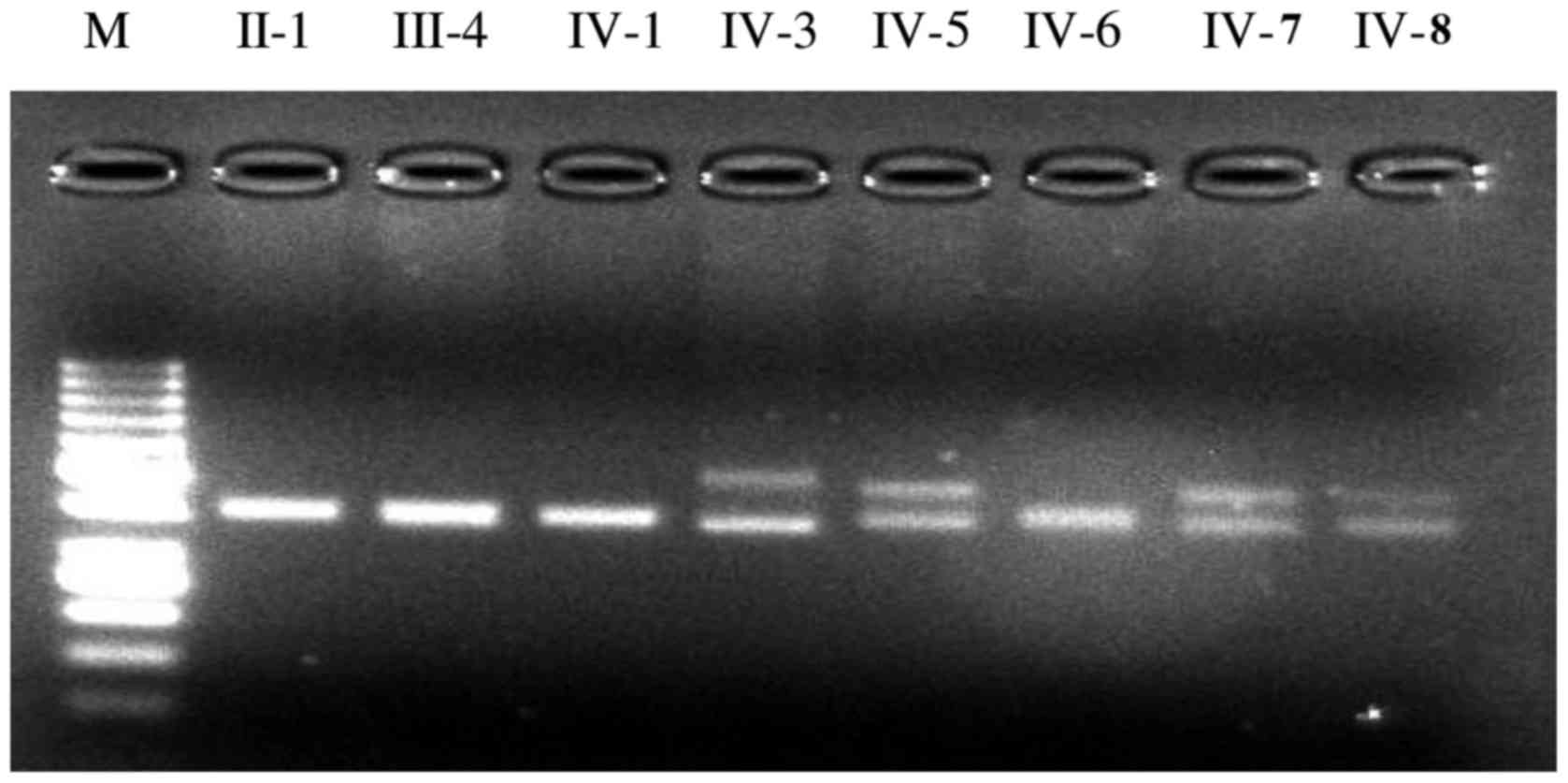
The mutation screening results of SCA1, SCA2, SCA3,
SCA6, SCA7, SCA8, SCA12, SCA17 and DRPLA are presented in Fig. 2 for the propositus in the SCA
pedigree and a healthy subject. Two amplification fragments of
distinctly different sizes were observed for the propositus using
the primer sets to amplify the CAG repeats in the ATXN1 gene
and two fragments of nearly the same size or only a fragment of
amplification were found using other primer sets. Two fragments of
nearly the same size or only a fragment of amplification were seen
for the healthy subject using each pair of PCR primers. Mutation
screening suggested that the propositus was a SCA1 patient. The
screening PCR detected two fragments of distinctly different sizes
in 4 out of the 8 tested individuals including the propositus using
the given set of primers to amplify the SCA1 CAG expansion
(Fig. 3). The three siblings of
the propositus (IV-5, IV-7 and V-8) were asymptomatic causative
SCA1 gene carriers as determined by gene mutation analysis. Sanger
sequencing was needed in order to count the number of CAG repeats
in the ATXN1 gene in the SCA pedigree for accurate
diagnosis. The other four members (II-1, III-4, IV-1 and IV-6) were
healthy subjects because only two fragments of nearly the same size
were found.
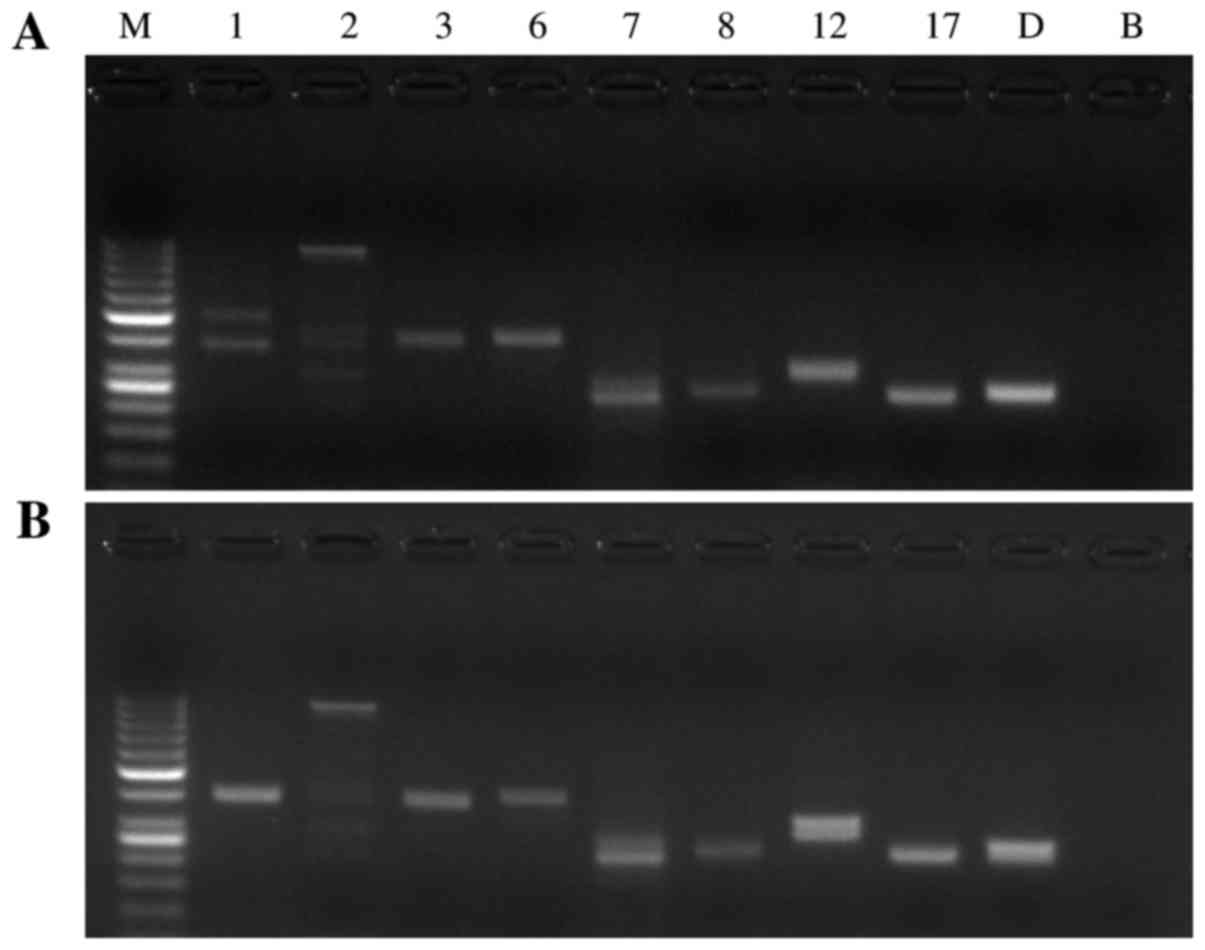 | Figure 2.Screening PCR for SCA1, SCA2, SCA3,
SCA6, SCA7, SCA8, SCA12, SCA17 and DRPLA for the (A) propositus in
the SCA pedigree and (B) the healthy subject. Lane M: Molecular
weight marker DNA ladder (1,000, 900, 800, 700, 600, 500, 400, 300,
250, 200, 150, 100 and 50 bp); lane 1-D: The amplified fragments
with primer sets for SCA1, SCA2, SCA3, SCA6, SCA7, SCA8, SCA12,
SCA17, and DRPLA, respectively; lane B: blank. PCR, polymerase
chain reaction; SCA, spinocerebellar ataxia; DRPLA,
dentatorubral-pallidoluysian atrophy. |
Sanger sequencing
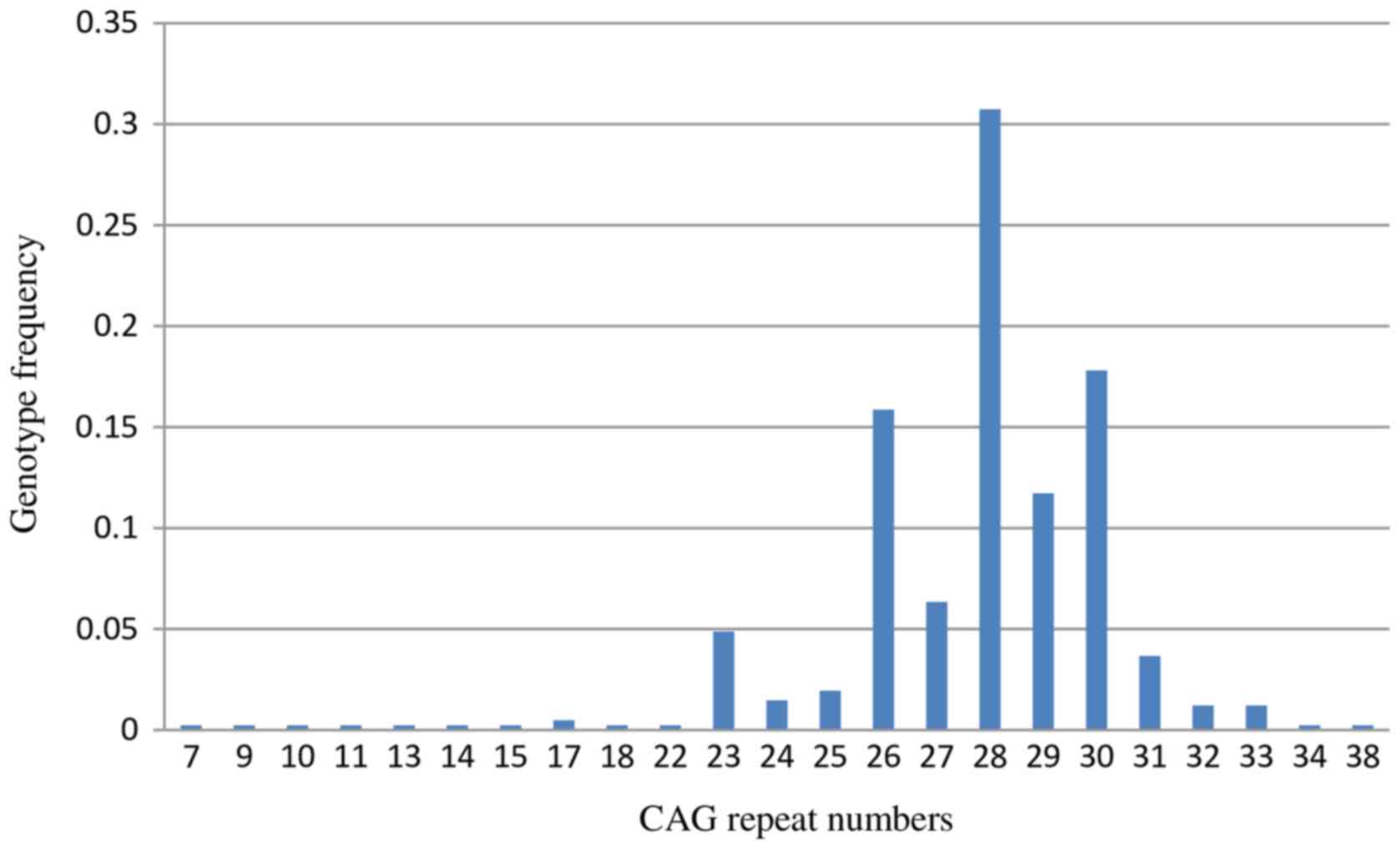
With Sanger sequencing, 67 CAG trinucleotide repeat
genotypes were detected in 205 normal healthy subjects (Table II). The normal range of the SCA1
CAG trinucleotide repeats was 7–38 repeats, with the common repeat
being 23–33 repeats in the Chinese population (Fig. 4). The normal range of CAG repeats
was similar to that reported for other ethnic populations. Usually
normal alleles containing 18–38 CAG repeats were interrupted by 1–3
CAT interruptions, however a few alleles containing 7–23 repeats
were not interrupted. In the present study, a CAG triplet repeat
(length 61) was detected for the propositus (IV-3). A repeat length
of 51 (IV-7) and 50 (in both IV-5 and IV-8) was observed in the
three younger siblings for the SCA1 pedigree. The other two
subjects (IV-1 and IV-6) carried a normal range of CAG repeats
(28–33) in the ATXN1 gene (Table III). The larger the CAG repeat,
the earlier the age of onset for SCA1 (Table IV). The three siblings with 50 or
51 triplet repeats had no SCA1 symptoms, which indicated a possible
late onset. The family members IV-5, IV-7 and IV-8 could be
diagnosed with SCA1 according to the sequencing results, while the
subjects IV-1 and IV-6 could be considered healthy individuals.
| Table II.Frequency of the CAG trinucleotide
repeat genotype in the ATXN1 gene in healthy subjects. |
Table II.
Frequency of the CAG trinucleotide
repeat genotype in the ATXN1 gene in healthy subjects.
| Allele | Number of
polyglutamines (histidines) | Frequency | Allele | Number of
polyglutamines (histidines) | Frequency |
|---|
|
(CAG)11CAT(CAG)16 | 28 | 0.282927 |
(CAG)13CATCAGCAT(CAG)15 | 31 | 0.002439 |
|
(CAG)13CATCAGCAT(CAG)10 | 26 | 0.14878 |
(CAG)12CATCAGCATCAGCAT(CAG)14 | 31 | 0.002439 |
|
(CAG)12CATCAGCAT(CAG)15 | 30 | 0.092683 |
(CAG)19CAT(CAG)10 | 30 | 0.002439 |
|
(CAG)17CATCAGCAT(CAG)10 | 30 | 0.063415 |
(CAG)13CATCAGCAT(CAG)14 | 30 | 0.002439 |
|
(CAG)16CATCAGCAT(CAG)10 | 29 | 0.053659 |
(CAG)11CATCAGCAT(CAG)14 | 28 | 0.002439 |
|
(CAG)13CATCAGCAT(CAG)13 | 29 | 0.039024 |
(CAG)10CATCAGCAGCAT(CAG)14 | 28 | 0.002439 |
|
(CAG)14CATCAGCAT(CAG)10 | 27 | 0.026829 |
(CAG)12CATCAGCATCAGCAT(CAG)10 | 27 | 0.002439 |
|
(CAG)11CAT(CAG)11 | 23 | 0.02439 |
(CAG)12CATCAGCAT(CAG)12 | 27 | 0.002439 |
|
(CAG)18CATCAGCAT(CAG)10 | 31 | 0.019512 |
(CAG)14CAT(CAG)12 | 27 | 0.002439 |
|
(CAG)11CAT(CAG)11 | 23 | 0.019512 |
(CAG)15CAT(CAG)11 | 27 | 0.002439 |
|
(CAG)11CAT(CAG)15 | 27 | 0.017073 |
(CAG)13CAT(CAG)12 | 26 | 0.002439 |
|
(CAG)15CATCAGCAT(CAG)10 | 28 | 0.014634 |
(CAG)11CATCAGCAGCAT(CAG)11 | 26 | 0.002439 |
|
(CAG)14CATCAGCAT(CAG)13 | 30 | 0.012195 |
(CAG)13CATCAGCAT(CAG)10 | 26 | 0.002439 |
|
(CAG)14CATCAGCAT(CAG)16 | 33 | 0.009756 |
(CAG)13CAT(CAG)12 | 26 | 0.002439 |
|
(CAG)12CATCAGCAT(CAG)14 | 29 | 0.009756 |
(CAG)13CATCAGCAT(CAG)9 | 25 | 0.002439 |
|
(CAG)12CATCAGCAT(CAG)10 | 25 | 0.009756 |
(CAG)8CAT(CAG)16 | 25 | 0.002439 |
|
(CAG)19CATCAGCAT(CAG)10 | 32 | 0.007317 |
(CAG)12CAT(CAG)12 | 25 | 0.002439 |
|
(CAG)12CATCAGCAT(CAG)16 | 31 | 0.007317 |
(CAG)8CAT(CAG)16 | 25 | 0.002439 |
|
(CAG)12CATCAGCAGCAT(CAG)14 | 30 | 0.004878 |
(CAG)12CATCAGCAT(CAG)9 | 24 | 0.002439 |
|
(CAG)11CATCAGCAT(CAG)15 | 29 | 0.004878 |
(CAG)11CATCAGCAT(CAG)10 | 24 | 0.002439 |
|
(CAG)11CAT(CAG)17 | 29 | 0.004878 |
(CAG)7CAT(CAG)16 | 24 | 0.002439 |
|
(CAG)11CATCAGCAGCAT(CAG)14 | 29 | 0.004878 |
(CAG)11CAT(CAG)12 | 24 | 0.002439 |
|
(CAG)13CATCAGCAT(CAG)12 | 28 | 0.004878 |
(CAG)8CAT(CAG)14 | 23 | 0.002439 |
|
(CAG)10CAT(CAG)16 | 27 | 0.004878 |
(CAG)23 | 23 | 0.002439 |
|
(CAG)11CAT(CAG)15 | 27 | 0.004878 |
(CAG)11CAT(CAG)10 | 22 | 0.002439 |
|
(CAG)11CAT(CAG)12 | 24 | 0.004878 |
(CAG)5CATCAGCAT(CAG)10 | 18 | 0.002439 |
|
(CAG)17 | 17 | 0.004878 |
(CAG)15 | 15 | 0.002439 |
|
(CAG)11CAT(CAG)9CAT(CAG)16 | 38 | 0.002439 |
(CAG)14 | 14 | 0.002439 |
|
(CAG)21CATCAGCAT(CAG)10 | 34 | 0.002439 |
(CAG)13 | 13 | 0.002439 |
|
(CAG)12CAT(CAG)20 | 33 | 0.002439 |
(CAG)11 | 11 | 0.002439 |
|
(CAG)13CATCAGCAT(CAG)16 | 32 | 0.002439 |
(CAG)10 | 10 | 0.002439 |
|
(CAG)11CAT(CAG)20 | 32 | 0.002439 |
(CAG)9 | 9 | 0.002439 |
|
(CAG)12CATCAGCAGCAT(CAG)15 | 31 | 0.002439 |
(CAG)7 | 7 | 0.002439 |
|
(CAG)15CATCAGCAT(CAG)13 | 31 | 0.002439 |
|
|
|
| Table III.CAG repeats in the ATXN1 gene in the
spinocerebellar ataxia pedigree. |
Table III.
CAG repeats in the ATXN1 gene in the
spinocerebellar ataxia pedigree.
| Subjects | Allele | Number of
polyglutamines (histidines) |
|---|
| II-1 |
(CAG)16CATCAGCAT(CAG)10/(CAG)17CATCAGCAT(CAG)10 | 29/30 |
| III-4 |
(CAG)16CATCAGCAT(CAG)10/(CAG)13CATCAGCAT(CAG)10 | 29/26 |
| IV-1 |
(CAG)16CATCAGCAT(CAG)10/(CAG)11CAT
(CAG)16 | 29/28 |
| IV-3 |
(CAG)13CATCAGCAT(CAG)10/(CAG)61 | 26/61 |
| IV-5 |
(CAG)12CATCAGCAT(CAG)15/(CAG)50 | 30/50 |
| IV-6 |
(CAG)15CATCAGCAT(CAG)15/(CAG)17CATCAGCAT(CAG)10 | 33/30 |
| IV-7 |
(CAG)12CATCAGCAT(CAG)15/(CAG)51 | 30/51 |
| IV-8 |
(CAG)12CATCAGCAT(CAG)15/(CAG)50 | 30/50 |
| Table IV.Age of the onset in the
spinocerebellar ataxia 1 pedigree. |
Table IV.
Age of the onset in the
spinocerebellar ataxia 1 pedigree.
| Subjects | Age of the onset
(Years) | Number of
polyglutamines (histidines) |
|---|
| I-1 | 37 | N/A |
| II-2 | 34 | N/A |
| III-2 | 28 | N/A |
| III-5 | 26 | N/A |
| III-7 | 29 | N/A |
| IV-2 | 22 | N/A |
| IV-3 | 18 | 26/61 |
| IV-5 | Asymptomatic | 30/50 |
| IV-7 | Asymptomatic | 30/51 |
| IV-8 | Asymptomatic | 30/50 |
Discussion
Polyglutamine SCAs represent a genetically
heterogeneous group of disorders involving the nervous system with
a dominant pattern of inheritance. These SCAs are caused by the
expansion of a CAG/CTG triplet repeat (9). Diagnosis of polyglutamine SCA
requires clinical assessment for the presence of ataxia and onset
age, a family history, and the clinical characteristics. The
diagnosis is only the beginning; a confirmative genetic study
should be performed to classify the type of SCA for genetic
counseling and family planning (10). This would gradually reduce the
incidence of the SCA. Healthy subjects often carry the heterozygous
CAG repeats below the pathological threshold, therefore, two PCR
fragments of nearly the same size are usually found using agarose
gel electrophoresis. Two fragments of distinctly different sizes
indicate the pathologically expanded CAG repeat sequence is
present. The present study demonstrated a convenient molecular
analysis technique for accurate diagnosis of polyglutamine SCA by
PCR amplification and Sanger sequencing. The diagnosis of SCA1 was
suspected for the family of the propositus with the CAG expansion
in the ATXN1 gene by PCR amplification. The CAG triplet
repeat expansion was found in the propositus and his three younger
siblings in the ATXN1 gene. PCR is a relatively simple and
inexpensive tool that can be used in most clinical laboratories,
and agarose gel electrophoresis of the PCR products is easy to
perform. Screening for polyglutamine SCAs by PCR is a rapid and
efficient method compared with other mutation detection methods.
The definitive diagnosis of SCA1 depended on the results of Sanger
sequencing of the CAG repeat expansion in the ATXN1 gene. A
total of 61 CAG triplet repeats were detected for the propositus
who presented with initial ataxia symptoms and signs at the age of
18. The three younger siblings of the propositus carrying 51 or 50
CAG repeats in the ATXN1 gene showed no symptoms or signs of
ataxia at the age of 22–28 years for the SCA1 pedigree. Three
subjects carrying the CAG repeat expansion were diagnosed with SCA1
according to the sequencing results.
The onset age is inversely correlated with the
number of CAG triplet repeats, the triplet repeat number is also
associated with the severity of the disease. The larger the CAG
triplet repeat number, the more serious the disease (11). In the present study, a pedigree
showing genetic anticipation in SCA was investigated. Genetic
anticipation in the age at which onset was observed in the past
three generations for the family according to the families'
recollection (Table IV). The
mechanism of genetic anticipation in SCA is the intergenerational
unstable amplification of CAG triplet repeat sequences. The greater
the CAG repeat number increases with the generation, the more the
age-at-onset decreases in successive generations (12). Genetic anticipation tends to be
more prominent when the CAG expansion comes from paternal
inheritance compared with maternal inheritance (13). The different onset age between the
propositus and his three younger siblings should be caused by a
distinctive inheritance pattern. The propositus obtained the
causative mutation of CAG repeat expansion from his father who died
of SCA1 at the age of 35. Three siblings of the propositus carrying
50–51 CAG repeats showed no symptoms or signs of SCA1 at the age of
22–28 years as a result of maternal inheritance. The phenotype of
the disease may be present ~10 years later for the three younger
siblings carrying 50–51 CAG repeats, according to the correlation
between the length of CAG repeat expansion and age at disease
onset, although statistical analyses were not performed. Certainly,
the age-at-onset and phenotype of the disease can be affected by
regulating genetic and environmental factors (14). Thus, Sanger sequencing of the
fragments of the trinucleotide repeat sequence is helpful to
predict the age-at-onset and phenotype of SCA. In the Chinese
population, gene mutations in ATXN3 are the most common,
followed by SCA2, SCA1, SCA7, SCA6, and SCA12 (15). In the present study, there was
evidence of genetic anticipation, which could be caused by an
abnormally expanded CAG repeat sequence in the pedigree. The family
was affected by a polyglutamine SCA. The SCA1 was a preliminary
diagnosis for the family by PCR amplification. Accurate diagnosis
of 4 cases with SCA1 was performed using Sanger sequencing of the
CAG repeat expansion in the ATXN1 gene, which was helpful to
infer the age-at-onset. The results of the present study suggest
that PCR amplification followed by Sanger sequencing is a useful
tool to diagnose polyglutamine SCA.
Acknowledgments
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (31571294).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
CC analyzed the patient data and performed screening
PCR. XF performed Sanger sequencing. SS designed the study and was
a major contributor in writing the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The study was approved by the ethics committee of
Shenzhen Baoan Hospital, Southern Medical University. Written
informed patient consent was obtained before initiating the
study.
Consent for publication
The patient has provided written informed consent
for the publication of any associated data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nibbeling EAR, Duarri A,
Verschuuren-Bemelmans CC, Fokkens MR, Karjalainen JM, Smeets CJLM,
de Boer-Bergsma JJ, van der Vries G, Dooijes D, Bampi GB, et al:
Exome sequencing and network analysis identifies shared mechanisms
underlying spinocerebellar ataxia. Brain. 140:2860–2878. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dell'Orco JM, Pulst SM and Shakkottai VG:
Potassium channel dysfunction underlies Purkinje neuron spiking
abnormalities in spinocerebellar ataxia type 2. Hum Mol Genet.
26:3935–3945. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sutton JR, Blount JR, Libohova K, Tsou WL,
Joshi GS, Paulson HL, Costa MDC, Scaglione KM and Todi SV:
Interaction of the polyglutamine protein ataxin-3 with Rad23
regulates toxicity in drosophila models of spinocerebellar ataxia
type 3. Hum Mol Genet. 26:1419–1431. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Terada T, Kono S, Konishi T, Miyajima H
and Ouchi Y: Altered GABAergic system in the living brain of a
patient with spinocerebellar ataxia type 8. J Neurol.
260:3164–3166. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
du Montcel Tezenas S, Durr A, Bauer P,
Figueroa KP, Ichikawa Y, Brussino A, Forlani S, Rakowicz M, Schöls
L, Mariotti C, et al: Modulation of the age at onset in
spinocerebellar ataxia by CAG tracts in various genes. Brain.
137:2444–2455. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li M, Pang SY, Song Y, Kung MH, Ho SL and
Sham PC: Whole exome sequencing identifies a novel mutation in the
transglutaminase 6 gene for spinocerebellar ataxia in a Chinese
family. Clin Genet. 83:269–273. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rothberg Gould BE and Rothberg JM:
Massively parallel (‘next-generation’) DNA sequencing. Clin Chem.
61:997–998. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Miller SA, Dykes DD and Polesky HF: A
simple salting out procedure for extracting DNA from human
nucleated cells. Nucleic Acids Res. 16:12151988. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shakkottai VG and Fogel BL: Clinical
neurogenetics: Autosomal dominant spinocerebellar ataxia. Neurol
Clin. 31:987–1007. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Melo AR, Ramos A, Kazachkova N, Raposo M,
Bettencourt BF, Rendeiro AR, Kay T, Vasconcelos J, Bruges-Armas J
and Lima M: Triplet repeat primed PCR (TP-PCR) in molecular
diagnostic testing for spinocerebellar ataxia type 3 (SCA3). Mol
Diagn Ther. 20:617–622. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Scoles DR, Meera P, Schneider MD, Paul S,
Dansithong W, Figueroa KP, Hung G, Rigo F, Bennett CF, Otis TS and
Pulst SM: Antisense oligonucleotide therapy for spinocerebellar
ataxia type 2. Nature. 544:362–366. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lai S, O'Callaghan B, Zoghbi HY and Orr
HT: 14-3-3 Binding to ataxin-1(ATXN1) regulates its
dephosphorylation at Ser-776 and transport to the nucleus. J Biol
Chem. 286:34606–34616. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
McMurray CT: Mechanisms of trinucleotide
repeat instability during human development. Nat Rev Genet.
11:786–799. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chatterjee N, Lin Y, Santillan BA, Yotnda
P and Wilson JH: Environmental stress induces trinucleotide repeat
mutagenesis in human cells. Proc Natl Acad Sci USA. 112:3764–3769.
2015.PubMed/NCBI
|
|
15
|
Wang J, Shen L, Lei L, Xu Q, Zhou J, Liu
Y, Guan W, Pan Q, Xia K, Tang B and Jiang H: Spinocerebellar
ataxias in mainland China: an updated genetic analysis among a
large cohort of familial and sporadic cases. Zhong Nan Da Xue Xue
Bao Yi Xue Ban. 36:482–489. 2011.PubMed/NCBI
|