Introduction
Oxidative stress is known to be a result of
excessive levels of reactive oxygen species (ROS) including
hydroxyl radicals, superoxide anions, and hydrogen peroxide, which
are produced by cellular respiration in mitochondria and other
cellular process. ROS is known to be associated with the
pathogenesis of various diseases (1–3).
Oxidative stress is associated with cell signaling and gene
regulation systems in a number of molecular biological processes.
Excessive ROS levels have resulted in various diseases via the
dysfunction of cellular macromolecules, leading to the promotion of
lipid peroxidation, DNA fragmentation, and protein damage,
eventually resulting in cell death (4,5).
Several studies have suggested that the regulation of intracellular
ROS levels is very important to neuronal cell survival in various
diseases (6,7).
Human glutaredoxin (GLRX)-1 is a member of the
thioredoxin family, additionally termed glutathione (GSH)-dependent
thiol oxidoreductase, and is a small molecular weight (12 kDa)
protein (8). GLRX1 is distributed
in various human tissues including the brain, which are involved in
sulfhydryl homeostasis and exhibit roles in the regulation of redox
signaling, and control a variety of cellular processes (9–11). A
previous study demonstrated that GLRX1 has an important role as a
ROS scavenger and protects against protein oxidation resulting from
oxidative stress (12). However,
the exact function of GLRX1 in ischemic injury remains to be fully
elucidated.
In general, it is difficult for exogenous
macromolecules to transduce into cells. Protein transduction
domains (PTDs) or cell penetrating peptides (CPPs) consist of a
short chain of amino acids, and effectively transduce exogenous
macromolecules into cells and tissues (13). Of the various PTDs, PEP-1 peptide
has several advantages including rapid transduction of proteins,
high stability in physiological buffers, and high efficiency of
transduction (14,15). Although the precise transduction
mechanism is not clear yet, it has been demonstrated that a variety
of therapeutic PTD fused proteins were successfully delivered into
various cells in vitro and in in vivo animal models,
transduced proteins effectively prevented cell death (16–22).
The present study investigated whether PEP-1-GLRX1 protein has
protective effects against oxidative stress in HT-22 cells and
against ischemic brain injury in an animal model. It was revealed
that transduced PEP-1-GLRX1 protein has protective effects against
oxidative stress in HT-22 cells and in an animal model of
ischemia.
Materials and methods
Materials and cell culture
Ni2+-nitrilotri-acetic acid Sepharose
Superflow was purchased from Qiagen, Inc. (Valencia, CA, USA).
Histidine antibody was obtained from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). B-cell lymphoma 2 (Bcl-2) antibody was obtained
from Abcam (Cambridge, UK). Phosporylated (p)-protein kinase B
(Akt), Akt, p-mitogen-activated protein kinase 8 (JNK), JNK,
p-p44/42 mitogen-activated protein kinase (ERK), ERK, p-mitogen
activated protein kinase 14 (p38), p38, p-cellular tumor antigen
p53 (p53), p53, Bcl-2 associated protein X (Bax), caspase-9 and
β-actin primary antibodies were obtained from Cell Signaling
Technology, Inc. (Danvers, MA, USA). 2′,7′-Dichlorofluorescein
diacetate (DCF-DA) was purchased from Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany). 8-hydroxy-2-deoxyguanosine (8-OHdG) antibodies
were purchased from Santa Cruz Biotechnology, Inc. The polymerase
chain reaction (PCR) technique was applied to isolate human GLRX1
cDNA. All remaining chemicals and reagents used in this experiment
were of the highest available commercial grade.
Cell culture
HT-22 murine hippocampal neuronal cells were
cultured in Dulbecco's modified Eagle's medium (Lonza Group, Ltd.,
Basel, Switzerland) supplemented with 10% fetal bovine serum (Lonza
Group, Ltd.) and antibiotics (100 µg/ml streptomycin, 100 U/ml
penicillin) at 37°C, under humidified conditions of 95% air and 5%
CO2.
Expression and purification of
PEP-1-GLRX1 proteins
A cell permeable PEP-1 expression vector was
prepared in the present laboratory as previously described
(19,20). The cDNA sequence for human GLRX1
was amplified by PCR using the following primer sequences: Forward,
5′-GGTCTCCTCGAGATGGCTCAAGAGTTTG-3′ and reverse,
5′-GGATCCTTACTGCAGAGCTCCAATCTG-3′. PCR products were excised,
eluted (Expin Gel; GeneAll Biotechnology Co., Ltd., Seoul, Korea),
and ligated into a TA cloning vector (pGEM®-T easy
vector; Promega Corporation, Madison, WI, USA) according to the
manufacture's protocol. The purified TA vector containing human
GLRX1 cDNA was ligated into the PEP-1 expression vector to produce
a PEP-1-GLRX1 fusion protein. In a similar fashion, a control GLRX1
was constructed that expressed the GLRX1 protein without PEP-1. To
produce the PEP-1-GLRX1 and control GLRX1 proteins, the plasmid was
transformed into Escherichia coli BL21 cells. The
transformed bacterial cells were grown in 100 ml of lysogeny broth
media at 37°C to a D600 value of 0.5–1.0 and induced
with 0.5 mM isopropyl β-D-1-thiogalactopyranoside at 37°C for 6 h.
Harvested cells were lysed by sonication and purified using a
Ni2+-nitrilotriacetic acid Sepharose affinity column
(Qiagen, Inc.) and PD-10 column chromatography (GE Healthcare,
Chicago, IL, USA). The purified protein concentrations were
estimated using a Bradford assay (23).
Transduction of PEP-1-GLRX1 protein
into HT-22 cells
To examine the transduction ability of PEP-1-GLRX1
protein, HT-22 cells were treated with various concentrations of
PEP-1-GLRX1 protein (0.5–1.5 µM) for 1 h or with 1.5 µM for various
time periods (10–60 min). Cells were then treated with trypsin-EDTA
and washed with phosphate-buffered saline (PBS). The cells were
harvested for the preparation of cell extracts to perform western
blot analysis. Transduced PEP-1-GLRX1 protein was detected using an
anti-histidine antibody (1:1,000; cat no. sc-804).
Western blot analysis
Following PEP-1-GLRX1 protein transduction, HT-22
cells were harvested and homogenized with NP-40 protein extraction
solution (Elpis Biotech, Inc., Daejeon, South Korea) at 4°C for 20
min. Protein concentration was determined by the Bradford assay.
Equal amounts of protein (30 µg/lane) was resolved by 15% SDS-PAGE
and the gels were subsequently transferred to a nitrocellulose
membrane. The membrane was blocked with 5% non-fat milk for 1 h at
37°C in a Tris-buffered saline buffer with 0.1% Tween 20. The
blocked membrane was incubated with histidine (1:1,000; cat no.
sc-804), p-Akt (1:1,000; cat no. 9272S), Akt (1:1,000; cat no.
4058S), p-JNK (1:1,000; cat no. 9251S), JNK (1:1,000; cat no.
9258S), p-ERK (1:1,000; cat no. 4376S), ERK (1:1,000; cat no.
9102S), p-p38 (1:1,000; cat no. 4631S), p38 (1:1,000; cat no.
9212S), p-p53 (1:1,000; cat no. 9284S), p53 (1:1,000; cat no.
9282S), Bax (1:1,000; cat no. 2772S), Bcl-2 (1:1,000; cat no.
ab59348), capase-9 (1:1,000; cat no. 9504S) and β-actin (1:1,000;
cat no. 4967S) primary antibodies overnight at 4°C, followed by
incubation with horseradish peroxidase-conjugated secondary
antibodies (1:10,000; cat no. 7074S) for 1 h at 37°C. Bands were
visualized with a Chemidoc imaging system (version 5.2; Bio-Rad
Laboratories, Inc., Hercules, CA, USA) and an enhanced
chemiluminescence kit according to the manufacturer's protocol (GE
Healthcare) (24). Bands were
quantified with ImageJ software (version 1.48; National Institutes
of Health, Bethesda, MD, USA).
Fluorescence confocal microscopy
analysis
The distribution of transduced proteins was assessed
using fluorescence microscopy as previously described (20,22).
HT-22 cells were grown on coverslips and treated with PEP-1-GLRX1
protein (1.5 µM) for 1 h and then washed twice with PBS, fixed with
4% paraformaldehyde at 37°C for 5 min, permeabilized with 0.1%
Triton X-100 and blocked at 37°C for 40 min with 3% bovine serum
albumin (Sigma-Aldrich; Merck KGaA) in PBS (PBS-BT) and washed with
PBS-BT. The cells were incubated with an anti-histidine antibody
(1:2,000; Santa Cruz Biotechnology, Inc.) for 1 h at 37°C, followed
by incubation with Alexa Fluor 488-conjugated secondary antibody
(1:15,000; Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) in the dark for 1 h at 37°C. Nuclei were then stained for 3
min with 1 µg/ml DAPI diluted 1:3,000 (Roche Diagnostics GmbH,
Mannheim, Germany) At each step, the cells were washed with PBS-BT
three times. Coverslips were mounted onto glass slides with Dako
fluorescent mounting solution (Agilent Technologies, Inc., Santa
Clara, CA, USA) The cells were analyzed by confocal microscopy
using a model FV-300 microscope (magnification, ×630; Olympus
Corporation, Tokyo, Japan).
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
Briefly, HT-22 cells were pretreated with
PEP-1-GLRX1 protein (0.5–1.5 µM) for 1 h and then treated with
hydrogen peroxide (H2O2, 1 mM) for 3 h. A
cell viability assay was performed using MTT as previously
described (20,22). The absorbance was read at a
wavelength of 570 nm using an ELISA microplate reader (Labsystems
Multiskan MCC/340) and cell viability was defined as the % of
untreated control cells.
Measurement of oxidative
stress-induced intracellular ROS levels
DCF-DA dye was used to measure intracellular ROS
levels, which was converted into fluorescent
2′7′-dichlorofluorescein (DCF) by ROS (22). HT-22 cells were incubated in the
absence or presence of PEP-1-GLRX1 (1.5 µM) for 1 h prior to
treatment with H2O2 (1 mM) for 10 min. Those
cells were washed twice with PBS and then incubated at 37°C for 30
min using DCF-DA (10 µM). The image was produced at 485 nm
excitation and 538 nm emission using a Fluoroskan ELISA plate
reader (Labsystems Oy, Helsinki, Finland).
Terminal deoxynucleotidyl transferase
dUTP nick end labeling (TUNEL) assay
To investigate DNA fragmentation, TUNEL staining was
performed using a Cell Death Detection Kit (Roche Applied Science,
Penzberg, Germany) according to the manufacturer's protocol
(22). Briefly, HT-22 cells were
incubated in the absence or presence of PEP-1-GLRX1 (1.5 µM) for 1
h, and then treated with H2O2 (1 mM) for 5 h.
Next, nuclei were stained at 37°C for 3 min with 1 µg/ml DAPI and
washed with PBS. Coverslips were mounted onto glass slides using
Dako fluorescent mounting solution (CA, USA). Images were taken
using a fluorescence microscope (Nikon Eclipse 80i; Nikon
Corporation, Tokyo, Japan). TUNEL-positive cells were counted by
phase-contrast microscopy in at least 5 random fields at ×200
magnification.
Measurement of apoptotic protein
expression
HT-22 cells were incubated in the absence or
presence of PEP-1-GLRX1 (1–1.5 µM) for 1 h, and then treated with
H2O2 for various time periods. The expression
levels of Bcl-2 (1 h), Bax (2 h), and pro-caspase-9 (4 h) in whole
cell lysates were analyzed by western blotting using respective
antibodies as described above. The bands were quantified by ImageJ
software (version 1.4.8; National Institutes of Health).
Experimental animals and induction of
cerebral forebrain ischemia
Male Mongolian gerbils (70–80 g; 6 months old; n=40)
were obtained from the Experimental Animal Center of Soonchunhyang
University (Cheonan, Korea). The animals were housed at an adequate
temperature (23°C) and humidity (60%) with a 12 h light/12 h dark
cycle, and free access to food and water. All experimental
procedures involving animals and their care conformed to the Guide
for the Care and Use of Laboratory Animals of the National
Veterinary Research & Quarantine Service of Korea and were
approved by the Institutional Animal Care and Use Committee of
Soonchunhyang University (Cheonan-Si, Korea; SCH16-0024).
Cerebral forebrain ischemia damage was induced as
previously described (25,26). Briefly, the animals were
anesthetized with a mixture of 2.5% isoflurane (Baxtor Healthcare,
Deerfield, IL, USA) in 33% oxygen and 67% nitrous oxide. Bilateral
common carotid arteries were isolated and occluded using
nontraumatic aneurysm clips. The complete interruption of blood
flow was verified by observing the central retinal artery using an
ophthalmoscope. Following 5 min of occlusion, the aneurysm clips
were removed from the common carotid arteries. The body temperature
under free-regulating or normothermic (37±0.5°C) conditions was
monitored with a rectal temperature probe (TR-100; Fine Science
Tools, Foster City, CA, USA) and maintained using a thermometric
blanket prior to, during, and following surgery until the animals
completely recovered from anesthesia. Thereafter, the animals were
kept on the thermal incubator (Mirae Medical Industry, Seoul, South
Korea) to maintain body temperature until the animals were
euthanized.
Treatment of ischemic animals with
PEP-1-GLRX1 and immunohistochemistry
To explore the protective effects of PEP-1-GLRX1
protein against ischemic damage, the animals were divided into 4
groups (n=10 per group); control sham group (normal control), model
group (ischemia control), control GLRX1-treated group, and
PEP-1-GLRX1-treated group (each 2 mg/kg). The control GLRX1 and
PEP-1-GLRX1 proteins were administered intraperitoneally 30 min
following ischemia-reperfusion.
Immunohistochemistry was performed as described in
previous studies (20,22). The brain tissue samples were
obtained 7 days following ischemia-reperfusion. To examine the
protective effects of PEP-1-GLRX1 protein against ischemic damage,
the sections were stained overnight at 4°C with histidine (1:500;
cat no. sc-804), neuronal nuclei (NeuN; 1:100; cat no. MAB377; EMD
Millipore, Billerica, MA, USA), 0.5% cresyl violet acetate (CV; cat
no. 10510-54-0; Sigma-Aldrich; Merck KGaA), Fluoro-Jade B (FJB;
1:500; cat no. AG310-30MG; EMD Millipore), glial fibrillary acidic
protein (GFAP; 1:500; cat no. AB5804; EMD Millipore) and ionized
calcium-binding adapter molecule 1 (Iba-1; 1:500; cat no.
019-19741; Wako Pure Chemical Industries, Ltd., Osaka, Japan).
Subsequently, sections were incubated with biotinylated goat
anti-mouse IgG antibody (1:200; cat no. BA-9200; Vector
Laboratories, Inc. Burlingame, CA, USA) or biotinylated goat
anti-rabbit IgG antibody (1:200: cat no. BA-1,000; Vector
Laboratories, Inc.) for 1 h at 25°C. A cell count was conducted in
the hippocampal area to quantify immunostaining as described in a
previous study (20,22).
Statistical analysis
Data are expressed as the mean ± standard error of
the mean of three experiments. Differences between groups were
analyzed by one-way analysis of variance followed by a Bonferroni's
post-hoc test using GraphPad Prism software (version 5.01; GraphPad
Software Inc., San Diego, CA, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
Construction and purification of
PEP-1-GLRX1 protein
The present study constructed a cell permeable
PEP-1-GLRX1 protein expression vector. As presented in Fig. 1A, the vector contained a cDNA
sequence with an amino-terminal tag containing human GLRX1, PEP-1
peptide and 6 histidine residues. A control GLRX1 expression vector
without the PEP-1 peptide was also constructed. PEP-1-GLRX1 and
control GLRX1 proteins were overexpressed in bacterial cells and
purified by using Ni2+-NTA and PD-10 column
chromatography. The purified PEP-1-GLRX1 and control GLRX1 proteins
were verified using SDS-PAGE and western blot analysis (Fig. 1B and C).
Transduction of PEP-1-GLRX1 protein
into HT-22 cells
To examine of transduction ability of PEP-1-GLRX1
protein, HT-22 cells were treated with PEP-1-GLRX1 proteins at
various concentrations (0.5–1.5 µM) for 1 h. Cells were also
treated with the same concentration of PEP-1-GLRX1 protein (1.5 µM)
for various times (10–60 min). The transduction of PEP-1-GLRX1
protein was verified by western blotting. As presented in Fig. 2A and B, PEP-1-GLRX1 protein
concentration- and time-dependently transduced into HT-22 cells.
However, control GLRX1 protein did not transduce into the cells.
Furthermore, transduced PEP-1-GLRX1 protein was examined using DAPI
and histidine immunostaining. Through immunostaining signals, it
was verified that PEP-GLRX1 protein efficiently transduced into
HT-22 cells (Fig. 2C). Since the
stability of protein is a significant factor in the development of
therapeutic agents, the stability of transduced PEP-1-GLRX1 protein
was measured. As presented in Fig.
2D, the concentration of transduced PEP-1-GLRX1 protein in the
cells decreased following 24 h, however remained present for up to
a maximum of 60 h.
Effects of PEP-1-GLRX1 on cell
survival from oxidative stress
In order to examine whether transduced PEP-1-GLRX1
protein has protective effects against
H2O2-induced HT-22 cell death, the present
study measured cell viability using an MTT assay. As presented in
Fig. 3A, when cells were treated
with H2O2, cell survival was ~50%.
PEP-1-GLRX1 protein markedly increased cell viability to up to 73%
dose-dependently. However, control GLRX1 protein did not
demonstrate the protective effects in H2O2
treated cells. There was no protective effect on cell viability
when the PEP-1 peptide alone was used for treatment under the same
conditions (data not shown).
In addition, intracellular ROS production levels
were investigated. In H2O2 exposed cells,
intracellular ROS levels were markedly increased, whereas ROS
levels were significantly reduced by PEP-1-GLRX1 protein. However,
control GLRX1 protein did not affect ROS levels (Fig. 3B). It was also investigated whether
transduced PEP-1-GLRX1 protein inhibits DNA damage resulting from
H2O2 using TUNEL and 8-OHdG staining
(Fig. 3C and D). In the
H2O2 exposed cells, TUNEL or 8-OHdG-positive
cells were markedly increased compared with the non-treated control
cells. However, TUNEL and 8-OHdG-positive cells were significantly
decreased by PEP-1-GLRX1 protein. Conversely, there was no
protective effect against H2O2-induced DNA
damage in the control GLRX1 protein treated cells. These results
indicated that transduced PEP-1-GLRX1 protein protected against
H2O2-induced HT-22 cell death via suppression
of intracellular ROS production and DNA damage.
PEP-1-GLRX1 prevents
H2O2-induced RAC-a serine/threonine-protein
kinase (Akt) and mitogen activated protein kinase (MAPK)
activation
Excessive intracellular ROS production leads to the
activation of MAPK pathways in various cell types.
H2O2 is known as an oxidizing agent which
induces the activation of MAPK pathways (5). Therefore, the present study
investigated whether the role of PEP-1-GLRX1 protein is in response
to the activation of Akt and MAPK [JUN N-terminal kinase (JNK),
extracellular signal-regulated kinase (ERK)1/2, and p38) in
H2O2 exposed HT-22 cells (Fig. 4). When cells were exposed
H2O2, the activation levels of Akt and MAPK
[phosphorylated (p)-Akt and p-MAPK] were markedly increased,
however PEP-1-GLRX1 protein significantly inhibited p-Akt and
p-MAPK levels in a concentration-dependent manner. However, levels
of p-Akt and p-MAPK were not significantly altered in control GLRX1
protein and non-treated control cells. The results provided
evidence that transduced PEP-1-GLRX1 inhibited the activation of
Akt and MAPK in H2O2 exposed HT-22 cells.
PEP-1-GLRX1 protects HT-22 cells
against H2O2-mediated apoptosis
It is well known that oxidative stress promotes
activation of apoptotic signaling pathways (27,28).
Therefore, the present study determined the ability of PEP-1-GLRX1
protein to inhibit H2O2-induced apoptotic
pathways. Firstly, the expression levels of p-p53 were verified as
oxidative stress increases expression levels of p-p53. As presented
in Fig. 5A, the levels of p-p53
were significantly increased in the H2O2
exposed cells, whereas the levels were markedly inhibited in cells
treated with PEP-1-GLRX1 protein in a concentration-dependent
manner. However, control GLRX1 protein did not alter the expression
level of p-p53 in H2O2 exposed cells.
The mechanism of the anti- or pro-apoptotic effects
of PEP-1-GLRX1 protein were additionally investigated. As presented
in Fig. 5B, increased Bax
expression levels resulting from H2O2 were
significantly reduced in the cells treated with PEP-1-GLRX1
protein. Conversely, Bax expression levels were not reduced in the
control GLRX1 protein treated cells.
As presented in Fig.
6, Bcl-2 and pro-caspase-9 expression levels were significantly
decreased in the H2O2 exposed cells compared
with the non-treated control cells, whereas Bcl-2 and pro-caspase-9
expression levels were markedly recovered in a
concentration-dependent manner in cells treated with PEP-1-GLRX1
protein. However, the alterations of Bcl-2 and pro-caspase-9
expression levels in control GLRX1 protein treated cells were
similar to the alterations induced by H2O2,
indicating that PEP-1-GLRX1 efficiently inhibited
H2O2-induced HT-22 cell death by preventing
the activation of apoptotic signaling pathways.
PEP-1-GLRX1 protects against neuronal
cell death in ischemic animal model
PTD fusion proteins are well known for their ability
to transduce into the animal brain, crossing the blood-brain
barrier (BBB) and improving neuronal cell survival (13). Therefore, the present study
determined whether PEP-1-GLRX1 protein transduced into the animal
brain and the neuroprotective effects of PEP-1-GLRX1 in an animal
model of ischemic injury. PEP-1-GLRX1 protein (2 mg/kg) was
intraperitoneally injected into gerbils 30 min following
ischemia-reperfusion. A total of 7 days following ischemic brain
injury, the transduction of PEP-1-GLRX1 protein into the Cornu
Ammonis (CA)1 region was verified, and cell viability determined.
As presented in Fig. 7, in the
sham control-, vehicle-, and control GLRX1 protein-treated groups,
there were no differences in the Histidine antibody staining.
However, in the PEP-1-GLRX1 protein treated groups, fluorescent
stained signals were markedly increased compared with the sham
control groups. In addition, through NeuN-immunostaining it was
revealed that PEP-1-GLRX1 protein increased neuronal cell survival
in the CA1 region.
The protective effect of PEP-1-GLRX1 proteins
against ischemic brain injury were investigated by CV and FJB
staining, which are known to be sensitive markers for the detection
of neuronal injury (Fig. 8). In
the PEP-1-GLRX1 protein treated groups, CV-positive neurons were
significantly increased compared with the vehicle-treated groups.
However, the number of CV-positive neurons were similar in the
control GLRX1 protein-treated and vehicle-treated groups. In
addition, FJB positive neuronal cells were not evident in the sham
control group, whereas FJB positive neuronal cells were observed in
the vehicle- and control GLRX1 protein treated group of the CA1
regions. FJB positive neuronal cells were significantly reduced in
the PEP-1-GLRX1 protein-treated group compared with the
vehicle-treated group.
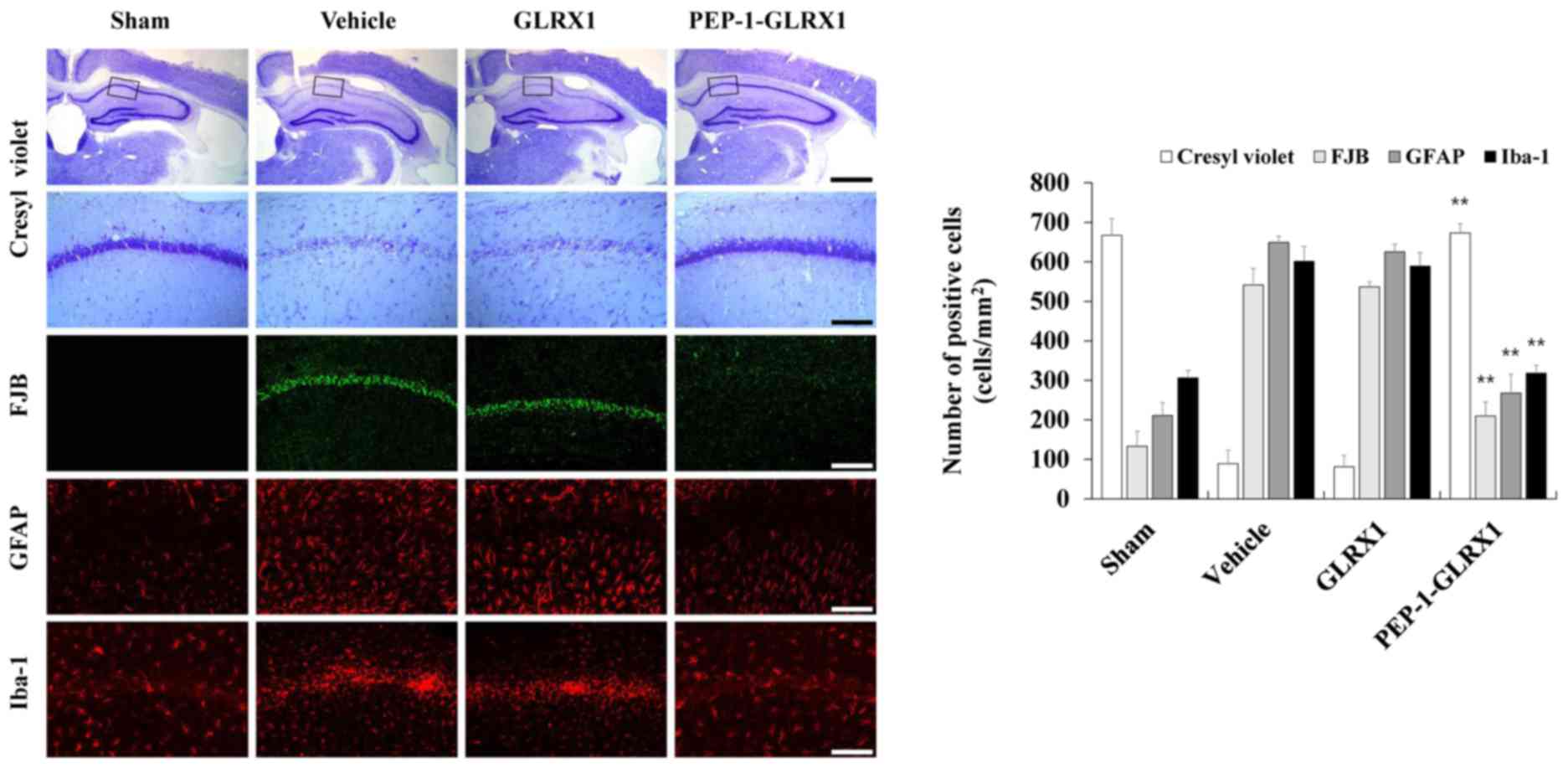 | Figure 8.Effects of transduced PEP-1-GLRX1
protein against neuronal cell death in an animal model of ischemia.
Neuroprotective effects of transduced PEP-1-GLRX1 protein were
analyzed by CV, FJB, GFAP, and Iba-1 immunohistochemistry in the
Cornu Ammonis 1 region of each group. Immunohistochemistry was
performed in each of the control sham-, vehicle-, control GLRX1-,
and PEP-1-GLRX1-treated groups 7 days following
ischemia-reperfusion. Scale bars, 50 µm (FJB, GFAP, Iba-1 and
bottom CV panel) and 400 µm (top CV panel). **P<0.01 vs. vehicle
group. GLRX1, glutaredoxin; CV, cresyl violet; FJB, Fluoro-Jade B;
GFAP, glial fibrillary acidic protein; Iba-1, ionized
calcium-binding adapter molecule 1. |
Since the activation of astrocytes and microglia are
known risk factors in ischemic injury (29), glial fibrillary acidic protein
(GFAP) and Iba-1 immunostaining were performed to examine whether
transduced PEP-1-GLRX1 protein inhibited the activation of
astrocytes and microglia in the CA1 region (Fig. 8). In the vehicle-treated group,
GFAP-immunoreactive astrocytes were significantly increased in the
CA1 regions, similar to those of control GLRX1 protein-treated
groups. However, GFAP-immunoreactive astrocytes were markedly
reduced in the PEP-1-GLRX1 protein-treated group compared with the
vehicle-treated group. In addition, Iba-1-immunoreactive microglia
in the CA1 region were significantly increased and aggregated in
the vehicle-treated group, similar to that of the control GLRX1
protein-treated group. In the PEP-1-GLRX1 protein-treated groups,
Iba-1-immunoreactive microglia aggregation recovered to a similar
level as the sham control group. Overall, the results provided
evidence that transduced PEP-1-GLRX1 protein markedly protected
against neuronal cell death and may act as a therapeutic agent
against ischemic brain injury.
Discussion
Human GLRX1, a member of GLRX family, is a
GSH-dependent thiol oxidoreductase and is a small multifunctional
protein. The two predominant forms (GLRX1 and GLRX2) exist in
mammalian systems and GLRX1 is primarily located in the cytoplasm
and GLRX2 in the mitochondria and nucleus (30). A previous study demonstrated that
overexpressed GLRX1 protects Akt from
H2O2-induced oxidation and protects cells
from apoptosis via regulation of the redox state of Akt in cardiac
H9c2 cells (31). Conversely,
other studies have demonstrated that GLRX1 has an important role in
defense against oxidative stress and cellular redox homeostasis
(32). However, the roles of the
GLRX1 protein against oxidative stress-induced neuronal cell damage
are not yet clear.
Although beneficial proteins were considered as
potential therapeutic agents, the application of these proteins has
been restricted due to their inability to transduce into cells.
PTDs, additionally termed CPPs, are small peptides consisting of 10
to 16 basic amino acids which transduce the plasma membrane either
alone or combined with various macromolecules including proteins,
without the aid of a special receptor (33). The present study used a PEP-1
peptide PTD, which has the ability to deliver a variety of proteins
into cells and tissues including the brain (34,35).
Even though numerous studies demonstrate that PEP-1 fused proteins
transduce into cells and cross the BBB, the exact transduction
mechanism is not yet fully understood.
ROS, including the superoxide anion
(·O2−), hydroxyl radicals (·OH) and
H2O2 are generated by oxidative stress and
lead to destruction of macromolecules including proteins, lipids,
and DNA (36). ROS are associated
with various diseases including ischemia, diabetes mellitus,
Parkinson's and Alzheimer's disease (37). The present study examined whether
PEP-1-GLRX1 suppressed the H2O2-induced cell
damage and elevated cellular ROS levels.
H2O2-induced cell death was significantly
decreased by PEP-1-GLRX1. Additionally, PEP-1-GLRX1 significantly
inhibited intracellular H2O2-induced ROS
production levels, which indicated that PEP-1-GLRX1 protected
against H2O2-induced cell death by inhibiting
intracellular ROS generation. These results are consistent with
other studies in which overexpressed GLRX1 protein protects H9c2
cells against H2O2 toxicity (38,39).
Previous studies have revealed that
H2O2 is the primary oxidizing agent which
induces neuronal cell death by activation of Akt and MAPK signaling
pathways, including JNK, ERK and p38 (40,41).
Conversely, other studies have indicated that activation of Akt and
MAPKs by H2O2 appears to regulate distinct
cellular responses including cell survival in different cell types
(42–44). The present study investigated
whether transduced PEP-1-GLRX1 protein inhibited the activation of
Akt and MAPK signaling induced by H2O2.
Transduced PEP-1-GLRX1 protein markedly inhibited phosphorylation
of Akt and MAPK signaling in the H2O2 treated
cells. A previous study suggested that GLRX1 protein inhibits
oxidative stress through the regulation of Akt and MAPK signaling
pathways (45). Kwon et al
(41) demonstrated that
Lonicera japonica THUNB (LJ) markedly inhibits the
activation of Akt and MAPKs, suggesting that LJ prevents
H2O2-induced apoptosis in SH-SY5Y cells by
inhibition of the activation of Akt and MAPKs (41). Studies have additionally
demonstrated that Akt and MAPKs are important in neuronal apoptosis
in response to environmental stresses and apoptotic agents
(46,47). The results of the present study are
consistent with reports that PEP-1-GLRX1 inhibits oxidative
stress-induced HT-22 cell death by suppressing the activation of
Akt and MPAK signaling.
Oxidative stress is highly associated with apoptotic
signaling pathways and p53 is known to be a key regulator of
apoptosis in neurons following ischemic injury (48–51).
PEP-1-GLRX1 protein significantly reduced activation of p53
expression levels in a dose-dependent manner. Control GLRX1 did not
affect the activation of p53 expression levels. Furthermore, the
present study examined the effects of transduced PEP-1-GLRX1
protein against H2O2-induced alterations in
Bcl-2, Bax and pro-caspase-9 expression levels in addition to
H2O2-induced DNA damage, as these proteins
and DNA damage are known to be primary markers of apoptotic
pathways (52,53). Transduced PEP-1-GLRX1 protein
significantly increased Bcl-2 and pro-caspase-9 expression levels
compared with H2O2 treated cells. Conversely,
Bax expression levels were markedly reduced. However, Bcl-2, Bax,
and pro-caspase-9 expression levels did not alter in the control
GLRX1 protein treated cells. In addition, TUNEL staining and 8-OHdG
fluorescence staining were performed, which are well-known markers
of DNA oxidation, in order to assess DNA damage (39). The results demonstrated that
PEP-1-GLRX1 protected against H2O2-induced
DNA damage in HT-22 cells, indicating that
H2O2-induced neuronal cell death is markedly
inhibited by transduced PEP-1-GLRX1 protein via regulation of
apoptotic signaling pathways.
Although the exact mechanism of ischemic injury is
not known, intracellular oxidative stress is considered to be one
of the primary factors resulting in neurodegeneration including
stroke and ischemia (28,54). To investigate the function of
PEP-1-GLRX1 against ischemic injury, the present study used a
gerbil ischemia-reperfusion model. First, immunohistochemical
staining was performed on hippocampal neuronal cells in the CA1
region to examine whether the PEP-1-GLRX1 fusion protein transduced
into the CA1 region across the BBB in an animal model of ischemia.
Crossing the BBB is a necessary step for therapeutic proteins to
treat neuronal diseases as the majority of proteins cannot easily
cross the BBB. Using His and NeuN staining, it was demonstrated
that PEP-1-GLRX1 protein transduced into the CA1 region by crossing
the BBB, and markedly protected against neuronal cell death in an
animal model of ischemia. To verify the neuronal damage, CV and
F-JB staining were conducted, which are known to be sensitive
markers for neuronal damage. As presented by CV and F-JB staining,
PEP-1-GLRX1 protected against neuronal damage in the CA1 region,
whereas the control GLRX1 protein did not affect the neuronal
damage, which indicated that PEP-1-GLRX1 protected against ischemic
injury in the animal model. These results are consistent with the
author's previous reports, where it was demonstrated that cell
permeable PTD proteins have a protective effect against neuronal
cell death in the hippocampal CA1 region (20,22).
Microglial activation is a primary cause of
degenerative brain diseases and astrocytes significantly affect the
survival of neuronal cells during brain damage including ischemia.
Therefore, the activation of microglial and astrocytes are used as
markers for the detection of ischemic neuronal injury (55,56).
Using Iba-1 and GFAP staining, which are microglia and astrocytes
markers, the present study determined the effects of PEP-1-GLRX1
protein against microglial and astrocyte activation in an animal
model of ischemia. PEP-1-GLRX1 protein significantly reduced the
levels of Iba-1 and GFAP immunoreactive cells in the animal model.
The control GLRX1 protein treated group did not demonstrate the
same activation of microglia and astrocytes compared with the
vehicle-treated group. Although the exact mechanisms need to be
examined to develop therapeutic treatments for ischemic injury
using this protein, it was demonstrated that PEP-1-GLRX1 protein
has neuroprotective effects on neuronal cell death in an ischemic
animal model and this fusion protein may be a potential therapeutic
agent for various neuronal diseases associated with to oxidative
stress.
In conclusion, cell permeable PEP-1-GLRX1 protein
was constructed and it was demonstrated that PEP-1-GLRX1 protein
efficiently transduced into HT-22 cells and inhibited intracellular
ROS production, and inhibited activation of the Akt, MAPK, and
apoptotic signaling pathways. In an ischemic animal model,
PEP-1-GLRX1 protein transduced into the animal brain following
crossing the BBB and revealed strong neuroprotective effects
against ischemic injury, suggesting that PEP-1-GLRX1 may have
potential applications for treating ischemic injury and neuronal
diseases.
Acknowledgements
Not applicable.
Funding
The present study was supported by a Priority
Research Centers Program (grant no. 2009-0093812) and Mid-Career
Researcher Program (grant no. 2016R1A2B4008356) through the
National Research Foundation of Korea funded by the Ministry of
Education, and Ministry of Science, ICT & Future Planning.
Availability of data and materials
The analyzed datasets generated during the study
are available from the corresponding author on reasonable
request.
Author's contributions
EJR, DWK and MJS designed the research and
experiments. HSJ, JHP, SBC, CHL, HJY, EJY and YJC contributed the
reagents and analytic tools and assisted with the analysis and
interpretation of the data. DSK, EJS and OS performed the cell and
animal experiments. KWL, SWC, YJC, KHH and JP analyzed and
interpreted the data. WSE and SYC designed the study and wrote the
manuscript.
Ethics approval and consent to
participate
All experimental procedures involving animals and
their care conformed to the Guide for the Care and Use of
Laboratory Animals of the National Veterinary Research &
Quarantine Service of Korea and were approved by the Institutional
Animal Care and Use Committee of Soonchunhyang University
(Cheonan-Si, Korea; SCH16-0024).
Consent for publication
Not applicable.
Competing Interests
The authors declare that they have no competing
interests.
References
|
1
|
Kirkinezos IG and Moraes CT: Reactive
oxygen species and mitochondrial diseases. Semin Cell Dev Biol.
12:449–457. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Matés JM: Effects of antioxidant enzymes
in the molecular control of reactive oxygen species toxicology.
Toxicology. 153:83–104. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rice ME, Forman RE, Chen BT, Avshalumov
MV, Cragg SJ and Drew KL: Brain antioxidant regulation in mammals
and anoxia-tolerant reptiles: Balanced for neuroprotection and
neuromodulation. Comp Biochem Physiol C Toxicol Phamacol.
133:515–525. 2002. View Article : Google Scholar
|
|
4
|
Poljsak B, Šuput D and Milisav I:
Achieving the balance between ROS and antioxidants: When to use the
synthetic antioxidants. Oxid Med Cell Longev. 2013:9567922013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Son Y, Cheong YK, Kim NH, Chung HT, Kang
DG and Pae HO: Mitogen-activated protein kinases and reactive
oxygen species: How can ROS activate MAPK pathways? J Signal
Transduct. 2011:7926392011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Saeed U, Durgadoss L, Valli RK, Joshi DC,
Joshi PG and Ravindranath V: Knockdown of cytosolic Glutaredoxin 1
leads to loss of mitochondrial membrane potential: Implication in
neurodegenerative diseases. PLoS One. 3:e24592008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sanderson TH, Reynolds CA, Kumar R,
Przyklenk K and Hüttemann M: Molecular mechanisms of
ischemia-reperfusion injury in brain: Pivotal role of the
mitochondrial membrane potential in reactive oxygen species
generation. Nol Neurobiol. 47:9–23. 2013. View Article : Google Scholar
|
|
8
|
Jao SC, Ospina English SM, Berdis AJ,
Starke DW, Post CB and Mieyal JJ: Computational and mutational
analysis of human glutaredoxin (thioltransferase): Probing the
molecular basis of the low pKa of cysteine 22 and its role in
catalysis. Biochemistry. 45:4785–4796. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Okuda M, Inoue N, Azumi H, Seno T, Sumi Y,
Hirata KI, Kawashima S, Hayashi Y, Itoh H, Yodoi J and Yokoyama M:
Expression of glutaredoxin in human coronary arteries: Its
potential role in antioxidant protection against atherosclerosis.
Arterioscler Thromb Vasc Biol. 21:1483–1487. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pai HV, Starke DW, Lesnefsky EJ, Hoppel CL
and Mieyal JJ: What is the functional significance of the unique
location of glutaredoxin 1 (GRx1) in the intermembrane space of
mitochondria? Antioxid Redox Signal. 9:2027–2033. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Peltoniemi M, Kaarteenaho-Wiik R, Säily M,
Sormunen R, Pääkkö P, Holmgren A, Soini Y and Kinnula VL:
Expression of glutaredoxin is highly cell specific in human lung
and is decreased by transforming growth factor-beta in vitro and in
interstitial lung diseases in vivo. Hum Pathol. 35:1000–1007. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cater MA, Materia S, Xiao Z, Wolyniec K,
Ackland SM, Yap YW, Cheung NS and La Fontaine S: Glutaredoxin1
protects neuronal cells from copper-induced toxicity. Biometals.
27:661–672. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
El-Andaloussi S, Holm T and Langel U:
Cell-penetrating peptides: Mechanisms and applications. Curr Pharm
Des. 11:3597–3611. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Beerens AM, Al Hadithy AF, Rots MG and
Haisma HJ: Protein transduction domains and their utility in gene
therapy. Curr Gene Ther. 3:486–494. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Morris MC, Depollier J, Mery J, Heitz F
and Divita G: A peptide carrier for the delivery of biologically
active proteins into mammalian cells. Nat Biotechonol.
19:1173–1176. 2001. View Article : Google Scholar
|
|
16
|
Ahn EH, Kim DW, Shin MJ, Kim HR, Kim SM,
Woo SJ, Eom SA, Jo HS, Kim DS, Cho SW, et al: PEP-1-PEA-15 protects
against toxin-induced neuronal damage in a mouse model of
Parkinson's disease. Biochim Biophys Acta. 1840:1686–1700. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim DW, Lee SH, Shin MJ, Kim K, Ku SK,
Youn JK, Cho SB, Park JH, Lee CH, Son O, et al: PEP-1-FK506BP
inhibits alkali burn-induced corneal inflammation on the rat model
of corneal alkali injury. BMB Rep. 48:618–623. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim HR, Kim DW, Jo HS, Cho SB, Park JH,
Lee CH, Choi YJ, Yeo EJ, Park SY, Kim ST, et al: Tat-biliverdin
reductase A inhibits inflammatory response by regulation of MAPK
and NF-κB pathways in Raw 264.7 cells and edema mouse model. Mol
Immunol. 63:355–366. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim MJ, Park M, Kim DW, Shin MJ, Son O, Jo
HS, Yeo HJ, Cho SB, Park JH, Lee CH, et al: Transduced PEP-1-PON1
proteins regulate microglial activation and dopaminergic neuronal
death in a Parkinson's disease model. Biomaterials. 64:45–56. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim YN, Jung HY, Eum WS, Kim DW, Shin MJ,
Ahn EH, Kim SJ, Lee CH, Yong JI, Ryu EJ, et al: Neuroprotective
effects of PEP-1-carbonyl reductase 1 against
oxidative-stress-induced ischemic neuronal cell damage. Free Radic
Biol Med. 69:181–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jo HS, Yeo HJ, Cha HJ, Kim SJ, Cho SB,
Park JH, Lee CH, Yeo EJ, Choi YJ, Eum WS and Choi SY: Transduced
Tat-DJ-1 protein inhibits cytokines-induced pancreatic RINm5F cell
death. BMB Rep. 49:297–302. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shin MJ, Kim DW, Jo HS, Cho SB, Park JH,
Lee CH, Yeo EJ, Choi YJ, Kim JA, Hwang JS, et al: Tat-PRAS40
prevent hippocampal HT-22 cell death and oxidative stress induced
animal brain ischemic insults. Free Radic Biol Med. 97:250–262.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bradford MM: A rapid and sensitive method
for the quantitation of microgram quantities of protein utilizing
the principle of protein-dye binding. Anal Biochem. 72:248–254.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
An SY, Youn GS, Kim H, Choi SY and Park J:
Celastrol suppresses expression of adhesion molecules and
chemokines by inhibiting JNK-STAT1/NF-κB activation in
poly(I:C)-stimulated astrocytes. BMB Rep. 50:25–30. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jo HS, Kim DS, Ahn EH, Kim DW, Shim MJ,
Cho SB, Park JH, Lee CH, Yeo EJ, Choi YJ, et al: Protective effects
of Tat-NQO1 against oxidative stress-induced HT-22 cell damage, and
ischemic injury in animals. BMB Rep. 49:617–622. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sohn EJ, Shin MJ, Kim DW, Son O, Jo HS,
Cho SB, Park JH, Lee CH, Yeo EJ, Choi YJ, et al: PEP-1-GSTpi
protein enhanced hippocampal neuronal cell survival after oxidative
damage. BMB Rep. 49:382–387. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Niizuma K, Yoshioka H, Chen H, Kim GS,
Jung JE, Katsu M, Okami N and Chan PH: Mitochondrial and apoptotic
neuronal death signaling pathways in cerebral ischemia. Biochim
Biophys Acta. 1802:92–99. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pradeep H, Diya JB, Shashikumar S and
Rajanikant GK: Oxidative stress-assassin behind the ischemic
stroke. Folia Neuropathol. 50:219–230. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yu JT, Lee CH, Yoo KY, Choi JH, Li H, Park
OK, Yan B, Hwang IK, Kwon YG, Kim YM, et al: Maintenance of
anti-inflammatory cytokines and reduction of glial activation in
the ischemic hippocampal CA1 region preconditioned with
lipopolysaccharide. J Neurol Sci. 296:69–78. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bachschmid MM, Xu S, Maitland-Toolan KA,
Ho YS, Cohen RA and Matsui R: Attenuated cardiovascular hypertrophy
and oxidant generation in response to angiotensin II infusion in
glutaredoxin-1 knockout mice. Free Radic Biol Med. 49:1221–1229.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Murata H, Ihara Y, Nakamura H, Yodoi J,
Sumikawa K and Kondo T: Glutaredoxin exerts an antiapoptotic effect
by regulating the redox state of Akt. J Biol Chem. 278:50226–50233.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Akterin S, Cowburn RF, Miranda-Vizuete A,
Jiménez A, Bogdanovic N, Winblad B and Cedazo-Minguez A:
Involvement of glutaredoxin-1 and thioredoxin-1 in beta-amyloid
toxicity and Alzheimer's disease. Cell Death Differ. 13:1454–1465.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Joliot A and Prochiantz A: Transduction
peptides: From technology to physiology. Nat Cell Biol. 6:189–196.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dietz GP: Cell-penetrating peptide
technology to deliver chaperones and associated factors in diseases
and basic research. Curr Pharm Biotechnol. 11:167–174. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Popiel HA, Nagai Y, Fujikake N and Toda T:
Protein transduction domain-mediated delivery of QBP1 suppresses
polyglutamine-induced neurodegeneration in vivo. Mol Ther.
15:303–309. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Barrera G: Oxidative stress and lipid
peroxidation products in cancer progression and therapy. ISRN
Oncol. 2012:1372892012.PubMed/NCBI
|
|
37
|
Emerit J, Edeas M and Bricaire F:
Neurodegenerative diseases and oxidative stress. Biomed
Pharmacother. 58:39–46. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ho YS, Xiong Y, Ho DS, Gao J, Chua BH, Pai
H and Mieyal JJ: Targeted disruption of the glutaredoxin 1 gene
does not sensitize adult mice to tissue injury induced by
ischemia/reperfusion and hyperoxia. Free Radic Biol Med.
43:1299–1312. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Valavanidis A, Vlachogianni T and Fiotakis
C: 8-hydroxy-2′-deoxyguanosine (8-OHdG): A critical biomarker of
oxidative stress and carcinogenesis. J Environ Sci Health C Environ
Carcinog Ecotoxicol Rev. 27:120–139. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lahair MM, Howe CJ, Rodriguez-Mora O,
McCubrey JA and Franklin RA: Molecular pathways leading to
oxidative stress-induced phosphorylation of Akt. Antioxid Redox
signal. 8:1749–1756. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kwon SH, Hong SI, Kim JA, Jung YH, Kim SY,
Kim HC, Lee SY and Jang CG: The neuroprotective effects of
Lonicera japonica THUNB. Against hydrogen peroxide-induced
apoptosis via phosphorylation of MAPKs and PI3K/Akt in SH-SY5Y
cells. Food Chem Toxicol. 49:1011–1019. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hwang SL and Yen GC: Modulation of Akt,
JNK, and p38 activation is involved in citrus flavonoid-mediated
cytoprotection of PC12 cells challenged by hydrogen peroxide. J
Agric Food Chem. 57:2576–2582. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yang B, Oo TN and Rizzo V: Lipid rafts
mediate H2O2 prosurvival effects in cultured endothelial cells.
FASEB J. 20:1501–1503. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ruffels J, Griffin M and Dickenson JM:
Activation of ERK1/2, JNK and PKB by hydrogen peroxide in human
SH-SY5Y neuroblastoma cells: Role of ERK1/2 in H2O2-induced cell
death. Eur J Pharmacol. 483:163–173. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu X, Jann J, Xavier C and Wu H:
Glutaredoxin 1 (Grx1) protects human retinal pigment epithelial
cells from oxidative damage by preventing AKT glutathionylation.
Invest Ophtalmol Vis Sci. 56:2821–2832. 2015. View Article : Google Scholar
|
|
46
|
Fulda S, Gorman AM, Hori O and Samali A:
Cellular stress responses: Cell survival and cell death. Int J Cell
Biol. 2010:2140742010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Villegas SN, Njaine B, Linden R and Carri
NG: Glial-derived neurotrophic factor (GDNF) prevents ethanol
(EtOH) induced B92 glial cell death by both PI3K/AKT and MEK/ERK
signaling pathways. Brain Res Bull. 71:116–126. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Reddy PH: Role of mitochondria in
neurodegenerative diseases: Mitochondria as a therapeutic target in
Alzheimer's disease. CNS Spectr. 14 8 Suppl 7:8–13; discussion
16-8. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Gupta S, Kass GE, Szeqezdi E and Joseph B:
The mitochondrial death pathway: A promising therapeutic target in
diseases. J Cell Mol Med. 13:1004–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Gu ZT, Wang H, Li L, Liu YS, Deng XB, Huo
SF, Yuan FF, Liu ZF, Tong HS and Su L: Heat stress induces
apoptosis through transcription-independent p53-mediated
mitochondrial pathways in human umbilical vein endothelial cell.
Sci Rep. 4:44692014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Niizuma K, Endo H and Chan PH: Oxidative
stress and mitochondrial dysfunction as determinants of ischemic
neuronal death and survival. J Neurochem. 1 109 Suppl:133–138.
2009. View Article : Google Scholar
|
|
52
|
Zhang S, Ong CN and Shen HM: Involvement
of proapoptotic Bcl-2 family members in parthenolide-induced
mitochondrial dysfunction and apoptosis. Cancer Lett. 211:175–188.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Würstle ML, Laussmann MA and Rehm M: The
central role of initiator caspase-9 in apoptosis signal
transduction and the regulation of its activation and activity on
the apoptosome. Exp Cell Res. 318:1213–1220. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kalogeris T, Baines CP, Krenz M and
Korthuis RJ: Cell biology of ischemia/reperfusion injury. Int Rev
Cell Mol Biol. 298:229–317. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ito D, Tanaka K, Suzuki S, Dembo T and
Fukuuchi Y: Enhanced expression of Iba1, ionized calcium-binding
adapter molecule 1, after transient focal cerebral ischemia in rat
brain. Stroke. 32:1208–1215. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chen Y and Swanson RA: Astrocytes and
brain Injury. J Cereb Blood Flow Metab. 23:137–149. 2003.
View Article : Google Scholar : PubMed/NCBI
|





















