Introduction
In mice, the pronuclear stage is a vital period in
which the activation of zygotic genes begins (1–3).
During the pronuclear stage, the zygote undergoes meiosis to form
the female pronucleus, and serial reactions in the sperm nucleus
occur to form the male pronucleus. In a previous study, it was
reported that female and male pronuclear migration in zygotes
depends on microtubules and organelles (4,5), and
that simulated microgravity, generated by a rotary cell culture
system (RCCS), disturbs spindle organization to inhibit mouse
oocyte maturation (6), as the
microtubules and chromosomes cannot form a complete spindle.
Long non-coding RNAs (lncRNAs), which evolve rapidly
and demonstrate little if any sequence conservation (7), are spliced transcripts that do not
encode proteins; lncRNAs range in length from 200 to several
thousand nucleotides. Previous studies have reported that there are
>1,000 promoter-associated non-coding RNA/gene pairs at the time
of zygotic gene activation (ZGA) (8), ~5,563 novel lncRNAs in mouse
cleavage-stage embryos (9), and
2,733 novel lncRNAs in human preimplantation embryos, as determined
via single-cell RNA sequencing (RNA-Seq) (10). Numerous lncRNAs have been revealed
to serve key roles in post-transcriptional, translational and
epigenetic regulation, and in embryogenesis without having any
apparent function (11–15). Some lncRNAs associate with
promoters to activate partner gene expression (8). Previous investigations have mainly
focused on early oocytes or later embryonic development, and the
global gene expression profiles of lncRNAs have been revealed using
single-cell RNA-Seq (10,16–22).
However, the biological functions of lncRNAs in mouse pronuclear
migration are not well understood.
Although previous studies have investigated whether
the disruption of microtubules inhibits pronuclear migration in
mouse zygotes, it remains unclear as to whether these microtubular
abnormalities are induced by lncRNAs. Therefore, a RCCS was used to
generate a model of pronuclear migration defects, and the relative
lncRNA expression patterns in the mouse zygote were investigated to
understand the biological roles of lncRNAs during pronuclear
migration.
Materials and methods
Mouse zygote collection and
culture
All animal procedures were carried out according to
the guidelines developed by the China Council on Animal Care, and
the protocols were approved by the Animal Care and Use Committee of
South China Agricultural University (Guangzhou, China). Mice were
maintained under the following conditions: Temperature, 18–22°C;
humidity, 50–60%; 10–14 h light/dark cycle; ad libitum
access to food and water. Superovulation of 20 adult female mice
(C57BL/6; age, 6–12-weeks; weight, 18–25 g; Guangdong Medical
Laboratory Animal Center, Foshan, China) was induced via
intraperitoneal injection of 5 IU pregnant mare serum gonadotropin
(Ningbo Second Hormone Factory, Ningbo, China), followed by
injection of 5 IU human chorionic gonadotropin (hCG; Ningbo Second
Hormone Factory) after 48 h. The mice were then placed in
individual cages with an adult male mouse (C57BL/6; age, 8–24
weeks; weight, 20–50 g; Guangdong Medical Laboratory Animal
Center). The female mice were screened for vaginal plugs the
following morning (15 h post-hCG), and mice with vaginal plugs were
subsequently dissected for collect ion of their zygotes. A total of
16 h following hCG administration, the zygotes were collected from
the ampullae of the oviducts, and the cumulus cells were removed
with 300 IU/ml hyaluronidase. The zygotes were subsequently
cultured in potassium simplex optimization medium supplemented with
a solution of 1% Eagle's Basal Medium and Minimum Essential Media
non-essential amino acids, 1% penicillin-streptomycin (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, UK), 95 mM NaCl, 2.5
mM KCl, 0.35 mM KH2PO4, 1.71 mM
CaCl2, 25 mM NaHCO3, 1 mM bovine serum
albumin, 1 mM glutamine, 0.2 mM pyruvate, 10 mM sodium DL-lactate,
0.2 mM D-glucose, 0.2 mM MgSO4 and 0.01 mM EDTA, and
were overlaid with embryo-tested mineral oil in a humidified
atmosphere containing 5% CO2 at 37°C. Unless otherwise
specified, all reagents were obtained from Sigma-Aldrich (Merck
KGaA, Darmstadt, Germany).
A total of 20 h post-hCG administration, when the
pronucleus was observable, the zygotes were randomly divided into
two groups (50 zygotes/group). One group was cultured at 37°C in an
atmosphere containing 5% CO2 under normal gravity for 9
h (three repetitions, groups C1, C2 and C3), after which, the
zygotes (in which the pronuclei were adjacent to each other) were
collected immediately. The other group was cultured at 37°C in an
atmosphere containing 5% CO2 in the RCCS (Synthecon
Inc., Houston, TX, USA), under simulated microgravity for 9 h, as
described previously (three repetitions, groups R1, R2 and R3)
(6). The zygotes exhibiting
disrupted pronuclear migration were collected immediately. Zygotes
collected from the two groups were transferred to 80 µl RNA
extraction buffer (Qiagen RNeasy Mini kit; Qiagen, Inc., Valencia,
CA, USA) for RNA isolation and were stored at −80°C until
sequencing.
Immunofluorescence and laser-scanning
confocal microscopy
The immunohistochemical assay was performed as
previously described (6). Zygotes
were fixed with 4% paraformaldehyde for 30 min at room temperature,
blocked in 1% bovine serum albumin (BSA; Sigma-Aldrich; Merck KGaA)
for 30 min at room temperature, and incubated with a mouse
monoclonal anti-α-tubulin antibody (1:200; cat. no. T8328;
Sigma-Aldrich; Merck KGaA) at 4°C overnight. Zygotes were washed
three times in washing buffer (6),
including 0.02% NaN3 (20 mg/ml), 0.01% Triton-X, 0.2%
non-fat dry milk, 2% normal goat serum (cat. no. AR0009; Wuhan
Boster Biological Technology, Ltd., Wuhan, China), 0.1 M glycine,
2% BSA and 95.77% PBS, prior to each step. The zygotes were
sequentially incubated with an Alexa Fluor® 568-labelled
goat anti-mouse immunoglobulin G secondary antibody (1:100; cat.
no. A11031; Invitrogen; Thermo Fisher Scientific, Inc.) for 1 h at
37°C in the dark. Finally, the zygotes were washed, stained with 10
µg/ml Hoechst 33342 (Molecular Probes; Thermo Fisher Scientific,
Inc.) for 10 min at 37°C in the dark to detect DNA, mounted in PBS
containing 50% glycerol (anti-fading reagent) and 25 mg/ml
NaN3, and then examined with a Zeiss laser-scanning
confocal microscope (Carl Zeiss AG, Oberkochen, Germany).
Single-cell cDNA amplification and
RNA-Seq library preparation
To prepare single-cell cDNA, all the samples were
amplified using the Smart-Seq2 method (23), and a cDNA product of 1–2 kb in
length was obtained. Subsequently, single-cell cDNA was purified
with the Ampure XP kit (Beckman Coulter, Inc., Brea, CA, USA). The
concentration and fragment distribution of the single-cell cDNA
obtained from normal gravity and simulated microgravity zygote
samples were subsequently assessed in a Qubit® 3.0
fluorometer (Thermo Fisher Scientific, Inc.), and the High
Sensitivity DNA Assay kit in the Bioanalyzer 2100 system (Agilent
Technologies, Inc., Santa Clara, CA, USA).
Single-cell cDNA (20 ng) was used as the initial
material for library construction. Initially, a
Bioruptor® Sonication system (20–60 kHz; 4°C; 30 sec;
Diagenode, Inc., Denville, NJ, USA) was employed to generate small
fragments ~300 bp in length. Subsequently, the library fragments
were subjected to end repair via the addition of a poly(A) tail and
an adapter sequence. The Beckman Ampure XP kit was used to purify
fragments after each reaction. Polymerase chain reaction (PCR) was
subsequently performed, and different index tags, which were used
for distinguishing samples from each other during sequencing, were
added to each sample. The PCR products were retrieved via 2%
agarose gel electrophoresis in order to select for 4,000-bp DNA
fragments, from which the library was constructed. The Agilent
Bioanalyzer 2100 system (Agilent Technologies, Inc.) was then used
to assess the quality of the libraries.
Deep sequencing and expression
analysis of lncRNAs
The libraries were sequenced on the Illumina HiSeq
2500 platform (Illumina, Inc., San Diego, CA, USA) using 150-bp
paired-end sequencing. Sequencing reads were assessed with the
FASTX tool kit (http://hannonlab.cshl.edu/fastx_toolkit/) and were
then subjected to standard quality control criteria to remove short
(<30 bp) and low-quality (quality score <20) reads. After
trimming the adaptor sequences, high-quality clean reads were
obtained for the following analysis. Firstly, the reads were mapped
to the mouse reference genome (mm9; genome.ucsc.edu/) with TopHat v1.4.0 software,
assembled with Cufflinks v2.2.0 and annotated with RefSeq
(www.ncbi.nlm.nih.gov/refseq) to
remove the annotated genes known to encode proteins or small RNAs
(24). Genes/transcripts with
fragments per kilobase transcriptome per million reads (FPKM)
>10 were retained, meaning that if there was only one exon, the
length could be >2,000 bp, whereas if more than two exons were
present, the length could be >200 bp. The coding potential of
the transcripts was then identified by CPC 2.0 software;
subsequently, transcripts with coding potential were excluded, and
the final lncRNAs were obtained (25). Finally, the read counts for each
novel transcript were converted into FPKM, values which were used
to calculate gene expression.
Screening and cluster analysis of
differentially expressed lncRNAs
Differences in the expression of the lncRNAs were
calculated based on the Q-value, and genes with similar
expression patterns were directly reflected in the cluster analysis
conducted using the hierarchical complete linkage clustering method
in R software (R version 3.4.3; www.r-project.org/). Significant differences in lncRNA
expression levels were determined based on a Q-value cut-off
0.05 and a minimum fold change (FC) of 1.5 using DEseq software
package version: 1.20.0 (26). The
differentially expressed lncRNAs were replaced by log10 values
(data values), and the Euclidean distance was calculated. The
results were further analysed using R packages; specifically, the
‘heatmap 2’ function of the ‘gplots’ package, which was used to
draw heat maps of the differentially expressed lncRNAs.
Reverse transcription-quantitative PCR
(RT-qPCR) and statistical analysis
RT-qPCR was performed to validate the RNA-Seq
results for lncRNA expression. Total RNA was extracted from 100
fertilized embryos using the Qiagen RNeasy Mini kit (Qiagen, Inc.),
according to the manufacturer's protocol. RNA then underwent RT
using the PrimeScript™ RT Reagent kit with gDNA Eraser (perfect
real time; Takara Bio, Inc., Otsu, Japan). The relative expression
levels of the target lncRNAs were measured by RT-qPCR using the
Power SYBR-Green RT-PCR kit (Toyobo Life Science, Osaka, Japan) in
a Bio-Rad CFX96 PCR system (Bio-Rad Laboratories, Inc., Hercules,
CA, USA). H2A histone family member Z was employed as a
housekeeping gene, and each sample was analysed three times. The
lncRNA-specific PCR conditions were as follows: Initial
denaturation at 95°C for 5 min, followed by 40 cycles of
denaturation at 95°C for 10 sec, annealing at 60°C for 10 sec and
extension at 72°C for 30 sec. RT-qPCR primers used in these
analyses are presented in Table
I.
 | Table I.Primer sequences of the lncRNAs and
housekeeping gene for reverse transcription-quantitative polymerase
chain reaction. |
Table I.
Primer sequences of the lncRNAs and
housekeeping gene for reverse transcription-quantitative polymerase
chain reaction.
| Gene lncRNA | Primer sequence
(5′-3′) | Product length
(bp) |
|---|
| H2afz | F:
ACAGCGCAGCCATCCTGGAGTA | 202 |
|
| R:
TTCCCGATCAGCGATTTGTGGA |
|
| lnc013878 | F:
ACCTGTGCCAAATGAGGCTT | 181 |
|
| R:
CTGCCAGTGTCTAAGGTGCT |
|
| lnc019773 | F:
GTCAGCTCTACAACCGCAGA | 182 |
|
| R:
TCCCGGTATTTAGGAGGGGG |
|
| lnc025630 | F:
TCAATGTCTGAATCGCCAACC | 160 |
|
| R:
GCATGGTGACAGCTTTTCATAATAC |
|
| lnc023277 | F:
GCGAGCCTTCCCGTTATCAT | 116 |
|
| R:
TACTGCGGCGTTTCCTTCTC |
|
| lnc013100 | F:
GTGGCTTGCTCATACCAGGA | 149 |
|
| R:
GTTTTGTGCAGAGCCATCCC |
|
| lnc019410 | F:
CCGGTTTATCCACGTCTGCT | 115 |
|
| R:
GAACATCACTTGTGGCAGCG |
|
| lnc032797 | F:
TGTTGTAACGGAGCACCTGAT | 104 |
|
| R:
AATCCCAGACGACTCCGGT |
|
| lnc006988 | F:
ACGGGCTCATCATTATCACTCTG | 109 |
|
| R:
TTCATGGGAGGTTGGCAGTAA |
|
| lnc001078 | F:
ACCAGTTTGTTTCTCTGTTGATGC | 124 |
|
| R:
CCTTCAGTGTCCCTGTTCCCT |
|
| lnc007956 | F:
TCTTCCTCTCGCCCCTAGTC | 100 |
|
| R:
ATCTGAGCTTCTCAACCCTGG |
|
The comparative quantification cycle (Cq) method was
employed to quantify the relative expression of lncRNAs. The
expression levels of the lncRNAs were analysed as FC values using
the ΔCq method (27). All data
were log transformed, and the results were comparable to the
RNA-Seq data.
Analysis of functional enrichment and
the lncRNA-gene network
BLAST v2.2.24 (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and RNAplex
software (https://omictools.com/rnaplex-tool) were used to
predict and identify the target genes (mRNAs) of the lncRNAs,
whereas Cytoscape V3.6.1 software was used to construct the
lncRNA-gene network (28). The
stage-specific lncRNAs, showing differential expression between the
two groups, and their target genes were chosen to construct the
network. For further analysis, tubulin β-4B class IVb
(Tubb4b) and lnc007956 were chosen to validate the
association between a target gene and lncRNA. However, it is
difficult to induce gene transfer in early embryos. The 293FT cell
line has the advantage of easier culture, higher transfer
efficiency and easier experimental operation; it is usually used to
verify the target association of microRNA/lncRNAs in mammalian
cells (29). In the present study,
293FT cells (cat. no. R70007; Thermo Fisher Scientific), which were
grown to 70–80% confluence in 96-well plates, were co-transfected
with a dual-luciferase reporter (cat. no. E1330; Promega
Corporation, Madison, WI, USA) carrying wild-type or mutant
Tubb4b (sequence was mutated from
5′-TGGTGCCCTTCCCTCGCCTGCACTTCTTCATGCC-3′ to
5′-TGGTGCACTTCACTCGCCTGCACGTCTGCATATC-3′; 100 ng) and a plasmid
carrying the lncRNA [100 ng; pcDNA3.1(−), cat. no. V795-20;
Invitrogen; Thermo Fisher Scientific, Inc.] using
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.) at 37°C in an atmosphere containing 5%
CO2 for 24 h. Luciferase activity was measured using the
Dual-Luciferase Reporter Assay system (Promega Corporation) and
compared with Renilla luciferase activity, according to the
manufacturer's protocol 36 h post-transfection. This experiment was
repeated more than three times and results were analysed using a
Student's t-test (Microsoft Excel 2010; Microsoft Corporation,
Redmond, WA, USA). P<0.05 was considered to indicate a
statistically significant difference.
LncRNA functions were predicted based on the
corresponding target genes through Gene Ontology (GO) analysis. GO
enrichment in terms of molecular function, cellular component and
biological process categories was assessed using the MGI Goslim
Database (www.informatics.jax.org/gotools/MGI_GO_Slim.html) and
the R package GSEABase. The most enriched terms may reflect lncRNA
functions. Hypergeometric tests with the Benjamini and Hochberg
false discovery rate were performed using the default parameters in
order to adjust the Q-value.
Results
Simulated microgravity negatively
affects mouse pronuclear migration
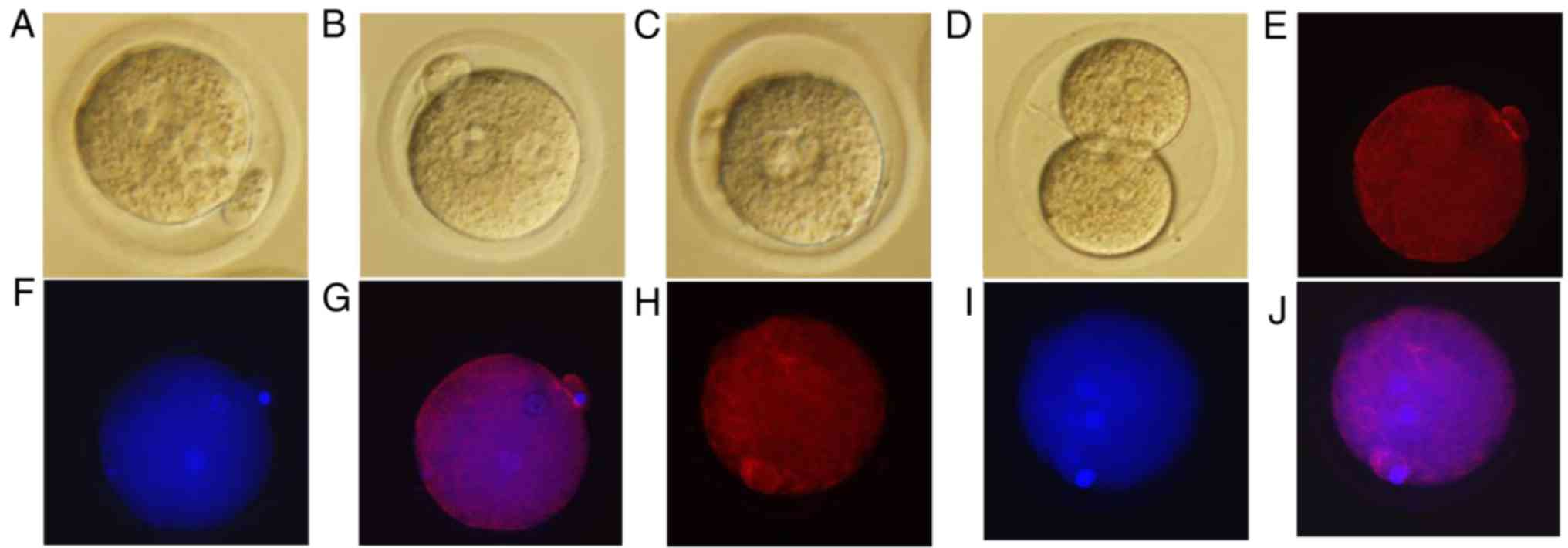
To investigate the effects of simulated microgravity
on mouse pronuclear migration, zygotes in which the male and female
pronuclei were forming were cultured for spontaneous maturation
under RCCS (Fig. 1A) or normal
gravity (Fig. 1B) conditions for 9
h. Subsequently, the zygotes cultured under simulated microgravity
were cultured at 37°C in an atmosphere containing 5% CO2
under normal gravity for 10 h (Fig.
1C) and 40 h, whereas the culture conditions for the control
group were unchanged. Subsequently, for each group, the proportions
of complete pronuclear migration, and of embryos at the 2-cell and
4-cell stages, were recorded following the onset of embryonic
development (Table II). During
further culture, the control zygotes exhibited orderly embryonic
development (Fig. 1D), whereas
those in the simulated microgravity group were stalled at the
pronuclear merge stage (Fig. 1C).
The zygotes cultured in the RCCS completed pronuclear migration
after 10 h under normal gravity (Fig.
1C) but could not progress to the 2-cell stage (Table II). The present study further
investigated whether tubulin protein expression was altered in the
two groups using immunofluorescence; tubulin protein was disturbed
in the RCCS group (Fig. 1E-G)
compared with in the control group (Fig. 1H-J), which tubulin protein had
assembled nearby the male and female pronuclei. Pronuclear
migration was markedly compromised when zygotes were cultured under
simulated microgravity conditions, and alterations in the
configuration of microtubules were observed after staining for
tubulin protein (Fig. 1E-J).
| Table II.In vitro development of the
embryos under different culture conditions. |
Table II.
In vitro development of the
embryos under different culture conditions.
|
|
| Rate of development
at the indicated stage (%) |
|---|
|
|
|
|
|---|
| Group | Number of cultured
zygotes | Completed
migration | 2-cell stage | 4-cell stage |
|---|
| Control | 292 | 100.00 | 90.75 | 52.74 |
| RCCS | 257 | 78.21a | 0.79b | 0.00b |
RNA-Seq and lncRNA data for mouse
zygotes during the pronuclear stage
The Illumina HiSeq 2500 platform was used to perform
RNA-Seq on the six cDNA libraries (groups C1, C2, C3, R1, R2 and
R3), and 125-bp paired-end reads were generated. The number of raw
reads was >40 million (Table
III). Low-quality reads were filtered, and the clean reads
still included >81.58% of the raw data. Among the clean reads,
92.29% exhibited perfect BLAST hits against the mouse reference
genome (https://blast.ncbi.nlm.nih.gov/Blast.cgi), and
>85.19% of the total mapped reads were uniquely mapped (Table III), indicating that the data
were credible and could be used for further analyses.
| Table III.Summary of the draft reads of the six
libraries obtained via RNA sequencing. |
Table III.
Summary of the draft reads of the six
libraries obtained via RNA sequencing.
| Sample | Raw reads | Clean reads
(%) | Mapped reads
(%) | Unique mapped reads
(%) |
|---|
| Control 1 | 47,771,190 | 43,350,036
(90.75) | 41,613,034
(95.99) | 37,050,242
(89.04) |
| Control 2 | 54,131,806 | 45,000,374
(83.13) | 43,786,923
(97.30) | 39,498,918
(90.21) |
| Control 3 | 64,009,546 | 52,978,050
(82.77) | 51,500,762
(97.21) | 46,592,730
(90.47) |
| RCCS 1 | 49,911,144 | 42,055,314
(84.26) | 40,902,021
(97.26) | 36,680,028
(89.68) |
| RCCS 2 | 61,916,186 | 51,834,282
(83.72) | 47,837,489
(92.29) | 40,753,247
(85.19) |
| RCCS 3 | 57,604,508 | 46,994,506
(81.58) | 45,471,826
(96.76) | 41,184,968
(90.57) |
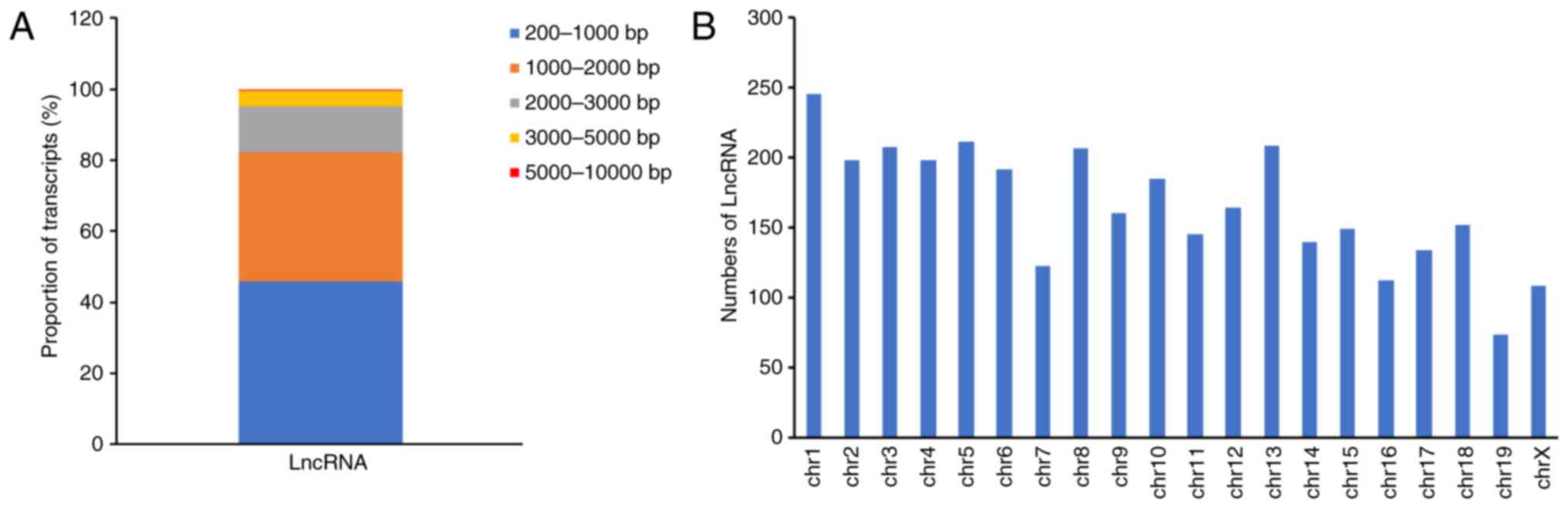
After the removal of protein-coding genes and
transcripts that were <200 bp, a total of 6,254 novel lncRNA
transcripts were obtained from the 3,307 expressed loci in the
RNA-Seq analysis. The lncRNAs varied between 200 and 7,804 bp in
length, with 45.8% falling between 200 and 1,000 bp, 36.3% between
1,000 and 2,000 bp, 13.1% between 2,000 and 3,000 bp, 4.4% between
3,000 and 5,000 bp, and 0.4% between 5,000 and 10,000 bp (Fig. 2A). The lncRNA genes had an average
length of 1,305 bp, and the lncRNA transcripts were mainly located
on mouse chromosomes 1–19 and X (Fig.
2B). Therefore, the existence of a large number of lncRNA
transcripts at the mouse pronuclear stage was confirmed.
Differential expression analysis of
lncRNAs
To analyse the differential expression of lncRNAs in
mouse zygotes between the normal gravity and simulated microgravity
culture conditions, lncRNA sequencing was performed to explore the
key lncRNAs during mouse pronuclear migration. A total of 52
lncRNAs were significantly differentially expressed between the
control and RCCS groups in the mouse zygotes, and unsupervised
hierarchical clustering and scatterplot analysis (FC>1.5 and
P<0.05) based on the FPKM values (log2 transformed) were
conducted to select the key lncRNAs that affect mouse pronuclear
migration (Fig. 3A and B).
Compared with in the control group, the RCCS group included 19
lncRNAs that were upregulated and 33 lncRNAs that were
downregulated (data not shown).
Verification through RT-qPCR
To validate the RNA-Seq results for lncRNA
expression levels, 10 differentially expressed lncRNAs (lnc013878,
lnc019773, lnc025630, lnc023277, lnc013100, lnc019410, lnc032797,
lnc006988, lnc001078 and lnc007956) were selected for testing in
mouse zygotes using RT-qPCR. The RT-qPCR results confirmed that the
expression patterns were similar to those obtained via RNA-Seq
(Fig. 3C). These results indicated
that the RNA-Seq data were credible and could be used to study
pronuclear migration in the mouse zygote.
LncRNA-gene interaction network
analysis
In the present study, the target genes (mRNAs) of
the differentially expressed lncRNAs were predicted and identified
using BLAST and RNAplex software, in order to investigate the
function of the lncRNAs regulating mouse pronuclear migration
(30). Subsequently, a lncRNA-gene
interaction network between differentially expressed lncRNAs and
their target genes was constructed with Cytoscape software. A total
of 668 network nodes (40 lncRNAs and 628 protein-coding genes) and
2,289 lncRNA-gene connections were identified in the network (data
not shown). Furthermore, 12 lncRNA-gene pairs [lnc006745-actinin-α4
(Actn4), lnc007956-Actn4, lnc013100-Actn4,
lnc013782-Actn4, lnc017097-Actn4,
lnc019869-Actn4, lnc025838-Actn4,
lnc027046-Actn4, lnc005454-Tubb4b,
lnc007956-Tubb4b, lnc019410-Tubb4b and
lnc019607-Tubb4b] that may be associated with pronuclear
migration were identified (Fig.
4A).
To further determine whether lnc007956 binds
directly to Tubb4b, two dual-luciferase reporter vectors
were constructed, with the wild-type or mutant target sequence of
Tubb4, inserted at the 3′ end of the firefly luciferase
gene. Subsequently, the effects of lnc007956 on these reporters in
293FT cells were tested. The results demonstrated that lnc007956
transfection significantly inhibited reporter activity (P<0.01),
whereas there was no effect on mutant reporter activity (Fig. 4B), suggesting that the predicted
binding site in Tubb4b is a bona fide target of
lnc007956.
GO analysis
To investigate the functions of the lncRNAs in mouse
early embryonic development, the target genes of the differentially
expressed lncRNAs were enriched through GO analysis. Based on this
analysis, 39 enriched GO terms potentially associated with
biological processes, 18 enriched GO terms potentially associated
with molecular functions and 67 enriched GO terms potentially
associated with cellular components were identified. Additionally,
15 GO terms, including cellular process, protein transport,
binding, catalytic activity, membrane-bounded organelle, protein
complex and cortical cytoskeleton, were significantly associated
with mouse pronuclear migration (Fig.
4C).
Discussion
In mice, the completion of pronuclear migration is a
crucial step in ZGA and serves an important role in development of
the early embryo. To generate a model of pronuclear migration
defects, in order to evaluate the role of pronuclear progression
during early embryonic development, an RCCS was used to simulate
microgravity conditions in a previous study and was revealed to
inhibit polar body extrusion by disrupting microtubule organization
during mouse oocyte maturation (6). To investigate the progress of
pronuclear migration, simulated microgravity was used to alter the
organization of the microtubules in mouse zygotes and it was
demonstrated that zygotes cultured under simulated microgravity
exhibit a delay in the assembly of male and female pronuclei, and
that progression to the 2-cell stage is also affected. As ZGA
begins approximately at the pronuclear stage and transcription of a
wide variety of transcripts occurs at the 2-cell stage in mice
(1,2), zygotes with defects in pronuclear
migration may possess altered activation of specific genes at the
pronuclear stage, which are required for progression to the 2-cell
stage. The present results suggested that simulated microgravity
altered gene expression in mouse zygotes to prevent progression to
the 2-cell stage; therefore, zygotes cultured in the RCCS represent
a good model for studying the impact of pronuclear migration in
mice.
Increasing number of mRNA and lncRNA expression
profiles during early embryonic development have been obtained via
single-cell RNA-Seq (10), and the
molecular mechanisms by which many of these RNAs modulate early
embryonic development have been elucidated (20,31,32).
However, whether alterations in genes associated with pronuclear
migration are regulated by mRNA/lncRNAs remains unknown. Based on
the high resolution of RNA-Seq, single-cell RNA-Seq can be used to
examine rare cell types, identify molecules of low abundance,
capture brief events and detect weak associations masked in bulk
experiments (33,34). Therefore, single-cell RNA-Seq
represents a promising tool for exploring the expression levels of
lncRNAs in mouse pronuclear migration; this approach was used in
the present study to elucidate the lncRNA profile during mouse
pronuclear migration and identified 6,254 novel lncRNA transcripts
from the 3,307 expressed loci. In addition, previous studies
identified 5,563 novel lncRNAs in mouse cleavage-stage embryos
(9), and 2,733 novel lncRNAs in
human preimplantation embryos (10), thus confirming that there are
numerous lncRNAs present during early mammalian embryonic
development and that lncRNAs may serve a vital role in the
pronuclear stage. Through further investigation, 52 differentially
expressed lncRNAs were identified and 10 of these lncRNAs were
selected to validate the accuracy of the RNA-Seq results via
RT-qPCR. The RT-qPCR results were concordant with the RNA-Seq
results, demonstrating that the results of single-cell RNA-Seq were
reliable.
It remains unclear as to how many lncRNAs are
involved in mouse pronuclear migration. Therefore, the functions of
lncRNAs were predicted based on their association with known
protein-coding genes and a lncRNA-target gene co-expression network
was constructed. Subsequently, based on the identification of
Tubb4b and Actn4 as target genes, 12 lncRNAs linked
to microfilaments and microtubules were identified, which may
affect mouse pronuclear migration. It has been reported that
microfilaments and microtubules are essential proteins for male and
female pronuclei (3,35–37).
During mouse pronuclear migration, specific tools and associated
proteins (such as microfilaments and microtubules) become
activated, and the sperm centrosome forms a sperm aster, to bring
the male and female pronuclei together to complete migration
(3,37).
Tubulins, which include eight α and nine β isotypes,
are the proteins that form microtubules, which are cytoskeletal
elements of all eukaryotic cells that participate in various
essential cellular functions (38). Tubb4b, also referred to as
the tubulin β-2C chain, is tightly associated with active
spermatogenesis in mice (39).
Actn4 belongs to the α-actinin family of cytoskeletal
proteins that display unique characteristics associated with
cytoskeletal organization, signal transduction, regulation of gene
expression, protecting cells from mechanical stress and controlling
cell movement (40–42). Knockdown of Actn4 expression
in keratinocytes and murine lung fibroblasts not only impairs the
directionality of cell migration but also reduces cell
proliferation (43,44). In the dual-luciferase reporter
assay, lnc007956 was revealed to bind to the predicted binding site
in Tubb4b mRNA, thus indicating that lnc007956 may regulate
mouse pronuclear migration by binding to Tubb4b. Therefore,
lncRNAs may be considered to regulate mouse pronuclear migration,
and lncRNA defects could result in abnormalities of microtubules
and microfilaments.
According to GO analysis, lncRNA target genes were
independently enriched in cellular process-associated terms,
including protein transport, binding, catalytic activity,
membrane-bounded organelle, protein complex and cortical
cytoskeleton. In Caenorhabditis elegans embryos, migration
of the female pronucleus is associated with organelles, which
promote the movement of the female pronucleus along the
microtubules to the sperm centrosome (35,36).
LncRNAs may control the molecules involved in this process in mice,
thereby affecting mouse pronuclear migration. Notably, these data
greatly improve the understanding of early embryonic development
and may lead to the development of highly efficient markers for
analysing the molecular mechanisms of zygote pronuclear migration.
The present study provided basic data, which may improve the
treatment of physiological reproductive disorders.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Key
R&D Program of China (grant no. 2017YFD0501902), the National
Natural Science Foundation of China (grant nos. 31402072 and
31572397), the Guangdong Province Science and Technology Plan
Project (grant no. 2015A020208015) and the Guangdong Provincial
Education Department Talent Project (grant no. 2017KQNCX013).
Availability of data and materials
The datasets used and/or analysed during the current
study are available from the corresponding author on reasonable
request. The datasets generated and/or analysed during the current
study are also available in the Gene Expression Omnibus repository,
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE118001
Authors' contributions
MF designed the study, performed the experiments,
analyzed the data, and wrote the manuscript. ND collected zygotes
from the two groups and recorded the number of embryos at the
2-cell and 4-cell stages. YB participated in the cell culture
experiments. KW and ZZ collected and analyzed the data. HW and LM
provided guidance with regards to the experiments and analytical
methods, analyzed the data, revised the manuscript, and provided
administrative and financial support; YC, ZC, FG and LL were
responsible for collection and assembly of data. SZ conceived the
idea, designed the experiments, provided administrative and
financial support, and gave final approval of the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
All animal procedures were carried out according to
the guidelines developed by the China Council on Animal Care, and
the protocols were approved by the Animal Care and Use Committee of
Guangdong Province, China.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
Actn4
|
actinin-α4
|
|
CQ
|
quantification cycle
|
|
FC
|
fold change
|
|
GO
|
Gene Ontology
|
|
hCG
|
human chorionic gonadotropin
|
|
lncRNAs
|
long non-coding RNAs
|
|
RCCS
|
rotary cell culture system
|
|
RNA-Seq
|
RNA sequencing
|
|
Tubb4b
|
tubulin β-4B class IVb
|
|
ZGA
|
zygotic gene activation
|
References
|
1
|
Aoki F, Worrad DM and Schultz RM:
Regulation of transcriptional activity during the first and second
cell cycles in the preimplantation mouse embryo. Dev Biol.
181:296–307. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Latham KE, Garrels JI, Chang C and Solter
D: Quantitative analysis of protein synthesis in mouse embryos. I.
Extensive reprogramming at the one- and two-cell stages.
Development. 112:921–932. 1991.PubMed/NCBI
|
|
3
|
Gundersen GG and Worman HJ: Nuclear
positioning. Cell. 152:1376–1389. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Clift D and Schuh M: Restarting life:
Fertilization and the transition from meiosis to mitosis. Nat Rev
Mol Cell Biol. 14:549–562. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schatten G, Simerly C and Schatten H:
Microtubule configurations during fertilization, mitosis, and early
development in the mouse and the requirement for egg
microtubule-mediated motility during mammalian fertilization. Proc
Natl Acad Sci USA. 82:4152–4156. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu C, Guo X, Wang F, Li X, Tian XC, Li L,
Wu Z and Zhang S: Simulated microgravity compromises mouse oocyte
maturation by disrupting meiotic spindle organization and inducing
cytoplasmic blebbing. PLoS One. 6:e222142011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kutter C, Watt S, Stefflova K, Wilson MD,
Goncalves A, Ponting CP, Odom DT and Marques AC: Rapid turnover of
long noncoding RNAs and the evolution of gene expression. PLoS
Genet. 8:e10028412012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hamazaki N, Uesaka M, Nakashima K, Agata K
and Imamura T: Gene activation-associated long noncoding RNAs
function in mouse preimplantation development. Development.
142:910–920. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang K, Huang K, Luo Y and Li S:
Identification and functional analysis of long non-coding RNAs in
mouse cleavage stage embryonic development based on single cell
transcriptome data. BMC Genomics. 15:8452014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yan L, Yang M, Guo H, Yang L, Wu J, Li R,
Liu P, Lian Y, Zheng X, Yan J, et al: Single-cell RNA-Seq profiling
of human preimplantation embryos and embryonic stem cells. Nat
Struct Mol Biol. 20:1131–1139. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang KC and Chang HY: Molecular mechanisms
of long noncoding RNAs. Mol Cell. 43:904–914. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mercer TR, Dinger ME and Mattick JS: Long
non-coding RNAs: Insights into functions. Nat Rev Genet.
10:155–159. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mercer TR and Mattick JS: Structure and
function of long noncoding RNAs in epigenetic regulation. Nat
Struct Mol Biol. 20:300–307. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ulitsky I and Bartel DP: lincRNAs:
Genomics, evolution, and mechanisms. Cell. 154:26–46. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pauli A, Rinn JL and Schier AF: Non-coding
RNAs as regulators of embryogenesis. Nat Rev Genet. 12:136–149.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hamatani T, Carter MG, Sharov AA and Ko
MS: Dynamics of global gene expression changes during mouse
preimplantation development. Dev Cell. 6:117–131. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang QT, Piotrowska K, Ciemerych MA,
Milenkovic L, Scott MP, Davis RW and Zernicka-Goetz M: A
genome-wide study of gene activity reveals developmental signaling
pathways in the preimplantation mouse embryo. Dev Cell. 6:133–144.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xie D, Chen CC, Ptaszek LM, Xiao S, Cao X,
Fang F, Ng HH, Lewin HA, Cowan C and Zhong S: Rewirable gene
regulatory networks in the preimplantation embryonic development of
three mammalian species. Genome Res. 20:804–815. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tang F, Barbacioru C, Nordman E, Bao S,
Lee C, Wang X, Tuch BB, Heard E, Lao K and Surani MA: Deterministic
and stochastic allele specific gene expression in single mouse
blastomeres. PLoS One. 6:e212082011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li XY, Cui XS and Kim NH: Transcription
profile during maternal to zygotic transition in the mouse embryo.
Reprod Fertil Dev. 18:635–645. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tang F, Barbacioru C, Wang Y, Nordman E,
Lee C, Xu N, Wang X, Bodeau J, Tuch BB, Siddiqui A, et al: mRNA-Seq
whole-transcriptome analysis of a single cell. Nat Methods.
6:377–382. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fan X, Zhang X, Wu X, Guo H, Hu Y, Tang F
and Huang Y: Single-cell RNA-seq transcriptome analysis of linear
and circular RNAs in mouse preimplantation embryos. Genome Biol.
16:1482015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Serra L, Chang DZ, Macchietto M, Williams
K, Murad R, Lu D, Dillman AR and Mortazavi A: Adapting the
smart-seq2 protocol for robust single worm RNA-seq. Bio Protoc.
8:e27292018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Trapnell C, Roberts A, Goff L, Pertea G,
Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL and Pachter L:
Differential gene and transcript expression analysis of RNA-seq
experiments with TopHat and Cufflinks. Nat Protoc. 7:562–578. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kong L, Zhang Y, Ye ZQ, Liu XQ, Zhao SQ,
Wei L and Gao G: CPC: Assess the protein-coding potential of
transcripts using sequence features and support vector machine.
Nucleic Acids Res. 35:W345–W349. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Love MI, Huber W and Anders S: Moderated
estimation of fold change and dispersion for RNA-seq data with
DESeq2. Genome Biol. 15:5502014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Scmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wu GQ, Wang X, Zhou HY, Chai KQ, Xue Q,
Zheng AH, Zhu XM, Xiao JY, Ying XH, Wang FW, et al: Evidence for
transcriptional interference in a dual-luciferase reporter system.
Sci Rep. 5:176752015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tafer H and Hofacker IL: RNAplex: A fast
tool for RNA-RNA interaction search. Bioinformatics. 24:2657–2663.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cote I, Vigneault C, Laflamme I, Laquerre
J, Fournier E, Gilbert I, Scantland S, Gagne D, Blondin P and
Robert C: Comprehensive cross production system assessment of the
impact of in vitro microenvironment on the expression of messengers
and long non-coding RNAs in the bovine blastocyst. Reproduction.
142:99–112. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Plourde D, Vigneault C, Lemay A, Breton L,
Gagne D, Laflamme I, Blondin P and Robert C: Contribution of oocyte
source and culture conditions to phenotypic and transcriptomic
variation in commercially produced bovine blastocysts.
Theriogenology. 78:116–131. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shalek AK, Satija R, Adiconis X, Gertner
RS, Gaublomme JT, Raychowdhury R, Schwartz S, Yosef N, Malboeuf C,
Lu D, et al: Single-cell transcriptomics reveals bimodality in
expression and splicing in immune cells. Nature. 498:236–240. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wills QF, Livak KJ, Tipping AJ, Enver T,
Goldson AJ, Sexton DW and Holmes C: Single-cell gene expression
analysis reveals genetic associations masked in whole-tissue
experiments. Nat Biotechnol. 31:748–752. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wuhr M, Dumont S, Groen AC, Needleman DJ
and Mitchison TJ: How does a millimeter-sized cell find its center?
Cell Cycle. 8:1115–1121. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chew TG, Lorthongpanich C, Ang WX, Knowles
BB and Solter D: Symmetric cell division of the mouse zygote
requires an actin network. Cytoskeleton (Hoboken). 69:1040–1046.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Reinsch S and Gonczy P: Mechanisms of
nuclear positioning. J Cell Sci. 111:2283–2295. 1998.PubMed/NCBI
|
|
38
|
Gadadhar S, Bodakuntla S, Natarajan K and
Janke C: The tubulin code at a glance. J Cell Sci. 130:1347–1353.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sarkar H, Arya S, Rai U and Majumdar SS: A
study of differential expression of testicular genes in various
reproductive phases of hemidactylus flaviviridis (Wall Lizard) to
derive their association with onset of spermatogenesis and its
relevance to mammals. PLoS One. 11:e1511502016. View Article : Google Scholar
|
|
40
|
Thomas DG and Robinson DN: The fifth
sense: Mechanosensory regulation of alpha-actinin-4 and its
relevance for cancer metastasis. Semin Cell Dev Biol. 71:68–74.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhao X, Khurana S, Charkraborty S, Tian Y,
Sedor JR, Bruggman LA and Kao HY: α Actinin 4 (ACTN4) regulates
glucocorticoid receptor-mediated transactivation and
transrepression in podocytes. J Biol Chem. 292:1637–1647. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hsu KS and Kao HY: Alpha-actinin 4 and
tumorigenesis of breast cancer. Vitam Horm. 93:323–351. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shao H, Wang JH, Pollak MR and Wells A:
α-actinin-4 is essential for maintaining the spreading, motility
and contractility of fibroblasts. PLoS One. 5:e139212010.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hamill KJ, Hopkinson SB, Skalli O and
Jones JC: Actinin-4 in keratinocytes regulates motility via an
effect on lamellipodia stability and matrix adhesions. FASEB J.
27:546–556. 2013. View Article : Google Scholar : PubMed/NCBI
|