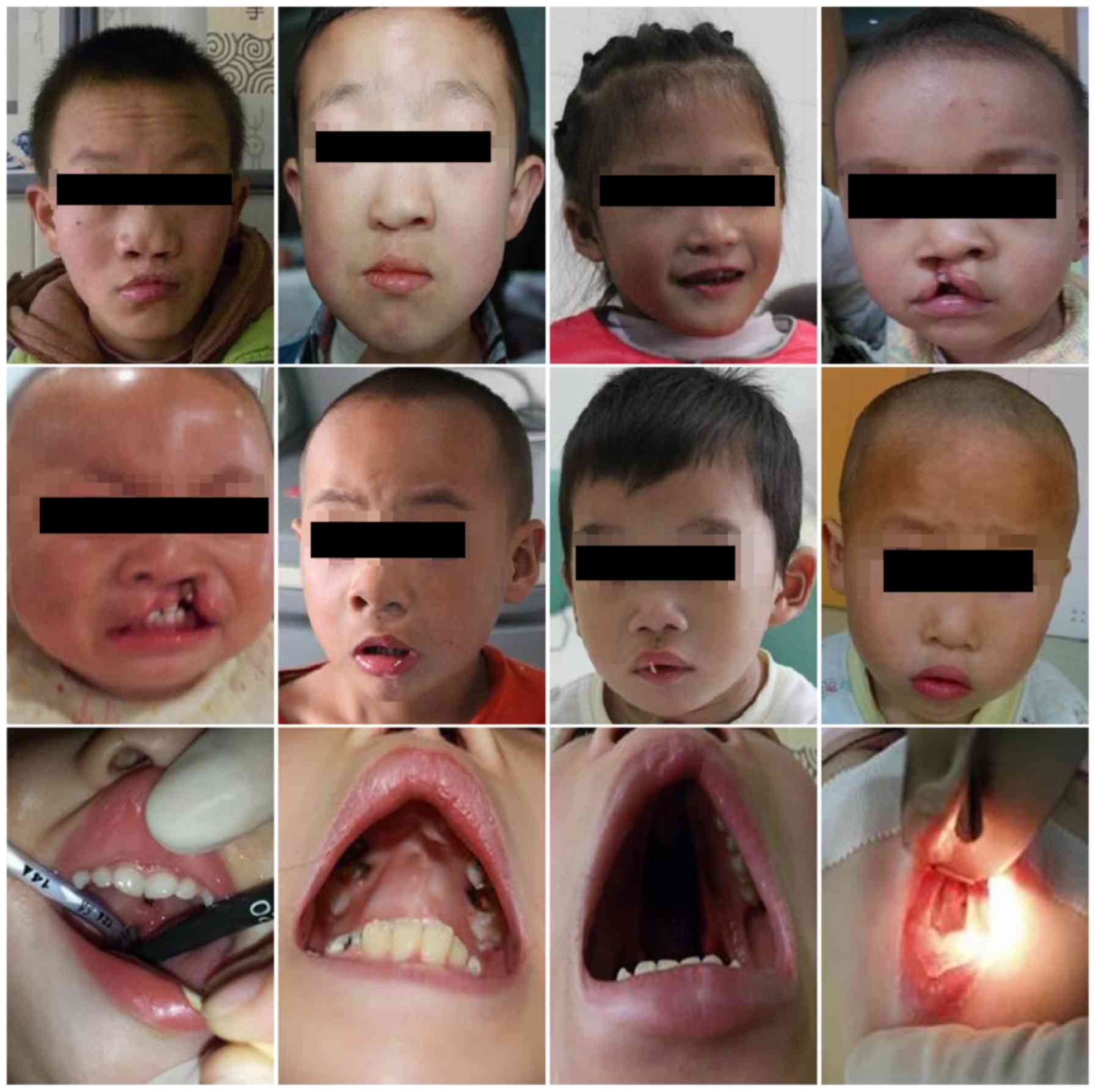
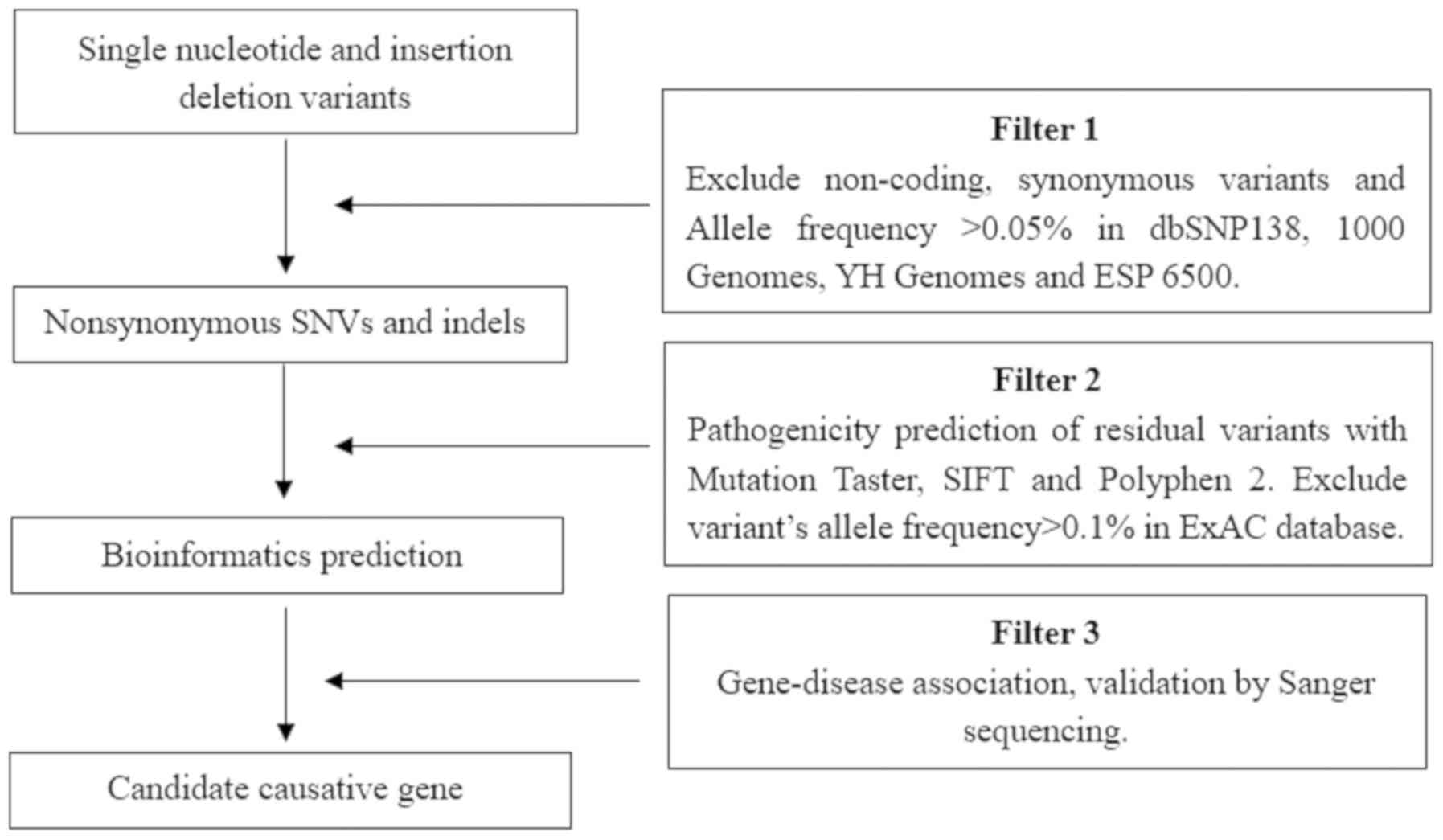
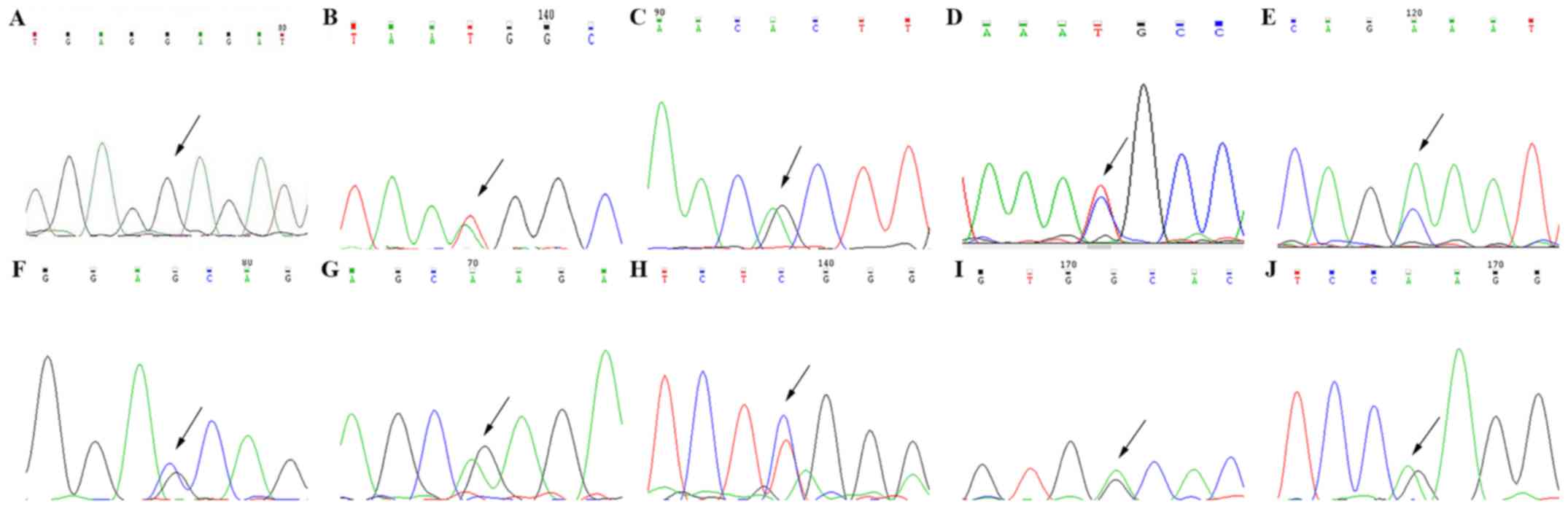
Introduction
Congenital heart disease (CHD), and cleft lip and
palate (CLP) represent birth defects with the highest rates of
incidence worldwide. Furthermore, the incidence rate of CHD in
patients with CLP is 6.5–12.7%, which is notably higher in
comparison with that of the general population (1,2). In
addition to distorting the facial appearance, CLP can negatively
affect normal infant activities, such as suckling and speaking
(3). Clinically, CHD and CLP are
commonly referred to as the main phenotypes, although specific
syndromes are also described in certain patients, such as
velocardiofacial syndrome, solitary median maxillary central
incisor syndrome and Wolf-Hirschhorn syndrome, among others
(4–6). Children with CLP typically require
multiple surgical interventions and numerous sessions of speech
therapy from infancy to early adulthood to achieve near-normal
appearance and function (7).
Genetic studies have suggested that deletion of the chromosome
fragments and single gene mutation are both observed in these
syndromes (4–6,8).
However, the hereditary background of patients with such syndromes
currently remains clear.
With the development of genetic sequencing
technologies, numerous novel methods have been suggested as
important techniques to identify disease-associated genes,
including single nucleotide polymorphism (SNP) array, copy number
variation (CNV) analysis, and targeted and whole exome sequencing
(9–11). Traditionally, genetic testing in
DNA-based diagnostic laboratories involves sequential Sanger
sequencing of known disease genes. However, the diagnostic yield of
next-generation sequencing (NGS) exceeds that of Sanger sequencing
in genetic diseases, since multiple genes can be analyzed in a
single experiment. Thus, the introduction of NGS has provided
revolutionary opportunities for comprehensive genetic testing in
research and diagnostics.
In the present study, the effectiveness and accuracy
of using targeted NGS to determine candidate genes in patients with
CHD concomitant with CLP were assessed.
Patients and methods
Patients
A total of 17 patients with CHD concomitant with CLP
treated at The Second Xiangya Hospital of Central South University
(Changsha, China) between November 2015 and May 2017 were enrolled
into the present study (Fig. 1).
The study group comprised of 14 male and 3 female patients aged
4–108 months (mean age, 42.8±32.9 months) with a mean body weight
of 17.6±6.9 kg (Table I). The
patient selection criteria in terms of CHD were as follows: i)
Exhibiting typical clinical manifestations and symptoms of CHD on
physical examination, including cyanosis and/or cardiac murmur; and
ii) diagnosis of CHD by transthoracic echocardiography (12). In terms of CLP the inclusion
criteria included the following: i) Typical clinical manifestations
and symptoms on physical examination, including cleft lip (CL)
and/or cleft palate (CP) (13,14);
ii) stomatological diagnosis; and iii) amalgamation or non-merger
of other malformations, or growth/mental retardation. The patient
exclusion criteria were as follows: i) Patients without CHD and
CLP; ii) cases diagnosed with trisomy 18 or 21 syndrome; and iii)
refusal of participation by the patient's parents or guardians.
 | Table I.Patient characteristics. |
Table I.
Patient characteristics.
| Characteristic | Value |
|---|
| Total no. | 17 |
| Males/females | 14/3 |
| Mean age
(months) | 42.8±32.9 |
| Mean weight
(kg) | 17.6±6.9 |
| Cardiac
phenotype |
|
|
ASD | 1 |
|
VSD | 7 |
|
DORV | 1 |
| ASD +
VSD | 3 |
| ASD +
PLSVC | 1 |
| TOF +
PLSVC | 1 |
| VSD +
PFO | 1 |
| TOF +
ASD | 1 |
| PDA +
PFO | 1 |
| Maxillofacial
phenotype |
|
| CL | 5 |
| CP | 9 |
|
CLP | 3 |
The study protocol was approved by the Review Board
of The Second Xiangya Hospital of Central South University, and the
relatives of study subjects provided informed consent for
participation. All experiments were performed in accordance with
relevant guidelines and regulations.
Blood sample collection and DNA
extraction
Peripheral blood samples (600 µl) obtained from each
patient were collected into 1.5 ml Eppendorf tubes (Eppendorf,
Hamburg, Germany) containing protein kinase (20 µl) and cell lysate
(200 µl). Tubes were agitated for 1 min, centrifuged for 10 sec at
4°C at 9,295 × g, and subjected to genomic (g)DNA extraction using
a DNeasy Blood and Tissue kit (Qiagen, Inc., Valencia, CA, USA)
according to the manufacturer's protocols using a QIAcube automated
DNA extraction device (Qiagen, Inc.). The gDNA solution generated
was stored at −80°C. Subsequently, a NanoDrop 2000
spectrophotometer (Thermo Fisher Scientific, Inc., Waltham, MA,
USA) was used to determine the quantity and quality of the DNA
samples, and 3 µg DNA from each sample was then used in subsequent
assays (15–17).
SNP array analysis
Genomic DNA samples of the patients were used to
conduct SNP array analysis at a final concentration of 50 ng/ml.
The signal intensities of SNP probes were determined by employing
an Illumina BeadScan genotyping system (Beadstation Scanner 500;
Illumina, Inc., San Diego, CA, USA) with a HumanOmni1-Quad Beadchip
(Illumina, Inc.), according to the manufacturer's protocol.
Targeted NGS
A targeted NGS gene panel for 455 genes that have
been associated with CHD or CLP in previous studies (8,16,18,19)
was employed (Table II). Targeted
NGS, including library construction, capture and sequencing, was
performed by Agilent Technologies, Inc. (Santa Clara, CA, USA).
Enrichment of target regions and library preparation were performed
using a SureSelectXT2 Custom kit (1–499 kb; Agilent Technologies,
Inc.) according to the manufacturer's protocol. Library DNA
concentrations were determined using an Agilent QPCR NGS Library
Quantification kit (G4880A; Agilent Technologies, Inc.), with each
sample at a final concentration of 10 nmol/l. Subsequently, samples
were ordered with a HiSeq2000 sequencing system using TruSeq
chemistry and protocols (version 3; Illumina, Inc.) (20).
| Table II.Target regions. |
Table II.
Target regions.
| Regions
1 | Regions 2 | Regions 3 | Regions 4 | Regions 5 | Regions 6 | Regions 7 | Regions 8 | Regions 9 | Regions
10 |
|---|
| ABCC9 | ACE | ACP6 | ACTA2 | ACTB | ACTC1 | ACTN2 | ACVR1 | ACVR2B | ADRB1 |
| ADRB2 | ADRB3 | AGL | AGT | AGTR1 | AHSA2 | ANKRD1 | APOBEC2 | ARL13B | ASXL2 |
| ATE1 | ATP1A2 | ATP4A | ATP4B | BAT1 | BBIP1 | BBS1 | BBS10 | BBS12 | BBS2 |
| BBS4 | BBS5 | BBS7 | BBS9 | BCL11A | BCL6 | BCL9 | BCOR | BICC1 | BMP7 |
| BMPR1A | BMPR1B | BMPR2 | BUB1B | C1ORF106 | CACNA1C | CACNA2D1 | CALM1 | CALM2 | CALR3 |
| CASQ2 | CAV3 | CCDC39 | CCDC40 | CCT4 | CDH1 | CDH2 | CDKN1C | CER1 | CFC1 |
| CHD1L | CHD7 | LDB3 | FH19 | APPB1 | FIBP | KMT2D | GPD1 | TTC8 | NGF |
| HMGCL | STIL | MKKS | RPSA | ARL6 | NELFA | GRID2 | KCNH2 | TRIM32 | CHRAC1 |
| CHRD | CHRNG | PQBP1 | CITED2 | CLDN7 | CLUL1 | CNTF | COL11A1 | COL11A2 | COL2A1 |
| COL3A1 | CREBBP | CRELD1 | CRHBP | CRX | CRYAB | CSRP1 | CSRP3 | CTLA4 | CTNNA3 |
| CUL3 | CYP11B2 | DAND5 | DAPK3 | DES | DHCR24 | DHCR7 | DHODH | DLL1 | DMRT2 |
| DNAI1 | DNAI2 | DOLK | DOT1L | DPP6 | DPPA4 | DSC2 | DSG2 | DSP | DST |
| DTNA | DVL1 | DVL2 | DZIP1 | EDNRA | EDNRB | EED | EFNB1 | EHMT1 | ELN |
| EMD | EP300 | ESCO2 | EVC | EVC2 | EYA4 | EZH1 | EZH2 | FBN1 | FBN2 |
| FGB | FKTN | FLNA | FLNB | FMO5 | FOXA2 | FOXC1 | FOXC2 | FOXH1 | FOXJ1 |
| FOXL2 | FTO | FXN | GAA | GADL1 | GALNT11 | GATA4 | GATA5 | GATA6 | GATAD1 |
| GDF1 | GJA1 | GJA5 | GJA8 | GJA9 | GLA | GLI2 | GLI3 | GPC3 | GPD1L |
| GPR161 | GPRC6A | GSK3B | HAND1 | HAND2 | HCN4 | HES1 | HES4 | HEY2 | HFE |
| HOXA1 | HUWE1 | HYLS1 | ID2 | IDUA | IER2 | IFNG | IFT122 | IFT172 | IFT20 |
| IFT57 | IFT88 | IGFBP4 | IGFBP5 | IHH | IL10 | IPPK | ISL1 | JAG1 | JARID2 |
| JAZF1 | JPH2 | JUP | KCND2 | KCND3 | KCNE1 | KCNE1L | KCNE2 | KCNE3 | KCNE4 |
| KCNH2 | KCNJ11 | KCNJ2 | KCNJ5 | KCNJ8 | KCNMB1 | KCNQ1 | KDM5A | KDM5B | KDM6A |
|
KIAA0196 | KIAA1841 | KIF3A | KIF3B | KIF3C | KIFAP3 | KLF13 | KRAS | LAMA4 | LAMP2 |
| LBR | LDB3 | LEFTY1 | LEFTY2 | LEMD3 | LIPC | LLPH | LMNA | LPIN1 | LRRC50 |
| LRRC6 | MARK2 | MAX | MED13L | MED20 | MEF2A | MEF2C | METT10D | MGAT1 | MGP |
| MICA | MICB | MID1 | MKKS | MKRN2 | MKS1 | MNDA | MSX2 | MYBPC3 | MYH10 |
| MYH11 | MYH6 | MYH7 | MYL2 | MYL3 | MYLK2 | MYOZ2 | MYPN | NAA15 | NCOR2 |
| NEBL | NEK2 | NEXN | NF1 | NFATC1 | NFATC3 | NFATC4 | NFKBIL1 | NIPBL | NKD1 |
| NKX2-5 | NKX2-6 | NKX3-2 | NODAL | NOS3 | NOTCH1 | NOTCH2 | NOTCH2NL | NOTCH3 | NOTCH4 |
| NOTO | NPHP3 | NPPA | NPPB | NSD1 | NUB1 | NUMBL | NUP188 | OBSCN | OFD1 |
| OSR1 | PAFAH1B1 | PAPOLG | PCMTD2 | PCSK5 | PDLIM3 | PEX1 | PEX13 | PHF8 | PHYHD1 |
| PIFO | PITX2 | PKD1L1 | PKD2 | PKP2 | PLA2G7 | PLAGL1 | PLN | PPM1K | PPP3CA |
| PQBP1 | PRC1 | PRDM1 | PRKAB2 | PRKAG2 | PROX1 | PSEN1 | PSEN2 | PTCH1 | PTCH2 |
| PTPLA | PTPN11 | PTPN22 | PTPRC | RAB10 | RAB23 | RAF1 | RAI1 | RAI2 | RANGRF |
| RAPGEF5 | RBM20 | REL | RFX2 | RFX3 | RIT1 | RNF20 | ROCK2 | ROR2 |
RPGRIP1L |
| RUNX2 | S100Z | SALL1 | SALL2 | SALL4 | SATB2 | SCN1B | SCN3B | SCN4B | SCN5A |
| SDC2 | SDHA | SEL1L3 | SEMA3E | SESN1 | SETBP1 | SGCA | SGCB | SGCD | SGCE |
| SGCG | SHH | SHOC2 | SIX3 | SLC26A2 | SLC2A10 | SLMAP | SMAD2 | SMAD5 | SMARCD3 |
| SMO | SMYD1 | SMYD2 | SNAI1 | SNTA1 | SOD2 | SOS1 | SOX17 | SOX9 | SRF |
| STIL | SUFU | SUPT3H | SUPT5H | SUV420H1 | TAZ | TBX1 | TBX20 | TBX3 | TBX5 |
| TCAP | TCF21 | TCOF1 | TDGF1 | TFAP2A | TFAP2B | TGFB1 | TGFBR1 | TGFBR2 | TGIF1 |
| TLL1 | TMBIM4 | TMEM195 | TMEM43 | TMPO | TNF | TNFRSF21 | TNNC1 | TNNI3 | TNNT2 |
| TP63 | TPM1 | TRDN | TRPM4 | TSC1 | TSEN15 | TTC21B | TTC30A | TTR | TWIST1 |
| TXNDC3 | UBE2B | UBR1 | UMODL1 | USF1 | USP34 | USP44 | VANGL2 | VCL | VEGFA |
| VEGFC | VIT | WDR5 | WHSC1 | WNT3A | XPO1 | ZEB2 | ZFPM1 | ZIC3 | ZNF480 |
| ZNF528 | ZNF534 | ZNF610 | ZNF638 | ZNHIT3 |
Data analysis and filtering
The Ensembl database (release 95; http://www.ensembl.org/) was used for variant
annotation. Filtering was performed with ANNOVAR Documentation
(http://annovar.openbioinformatics.org/), using the
following SNP databases for filtering: dbSNP (build 138; http://www.ncbi.nlm.nih.gov/snp), Exome Variant
Server (release ESP6500SI–V2; http://evs.gs.washington.edu/EVS/), 1000 Genomes
Project (released May 2012; http://www.internationalgenome.org/home) and HapMap
CHB (release 28; http://hapmap.ncbi.nlm.nih.gov/). In order to predict
the possible impact of variants, the following tools were used:
SIFT (version 6.2.1, http://sift.bii.a-star.edu.sg/), Polyphen-2 (version
2.2.2; http://genetics.bwh.harvard.edu/pph2/),
Mutation-Taster (version 2; http://www.mutationtaster.org/) and Human Splicing
Finder (version 3.1, http://www.umd.be/HSF3/). The filtering strategies
used are displayed in Fig. 2.
Variant validation
Variants warranting further investigation included
novel variants, which were predicted to be ‘likely pathogenic’ or
‘pathogenic’ according to PolyPhen-2, Mutation-Taster and SIFT
predictions, or were indicated to be ‘likely pathogenic’ and
possessed minor allele frequencies of <0.1%, as predicted by
ExAC browser (version 0.3.1; http://exac.broadinstitute.org/). Variants and samples
from the parents of certain patients were assessed by Sanger
sequencing. To confirm the disease-associated genes, the relevant
literature was surveyed on PubMed (https://www.ncbi.nlm.nih.gov/pubmed); example
literature searches included: MID1, Opitz G/BBB syndrome,
2007.1.1–2018.10.31, English; TGFBR1, Loeys-Dietz syndrome,
2007.1.1–2018.10.31, English; KIAA0196, Ritscher-Schinzel
syndrome, 2007.1.1–2018.10.31, English; CHD7, CHARGE
syndrome, 2007.1.1–2018.10.31, English.
Polymerase chain reaction (PCR)
Entire exon and exon-intron junctions of genes were
amplified by PCR. Genomic DNA (0.5 µl) obtained from peripheral
blood samples of patients was added to 11 µl double-distilled
water, 0.5 µl forward primer, 0.5 µl reverse primer and 2× PCR
Master Mix (12.5 µl; Nanjing Saihongrui Biotechnology Co., Ltd.,
Nanjing, China) containing 2X Taq DNA Polymerase. qPCR was
conducted as follows: Initial denaturation at 94°C for 5 min; 35
cycles of denaturation at 94°C for 30 sec, annealing at 50°C for 30
sec and extension at 72°C degrees for 30 sec; and final extension
at 72°C degrees for 10 min. Sequences of the PCR products were
determined using an ABI 3100 Genetic Analyzer (Applied Biosystems;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. The primer sequences are listed in Table III.
| Table III.Primer sequences. |
Table III.
Primer sequences.
| Gene | Primers
(5′-3′) |
|---|
| MID1 |
F:CACTTTGTGATAGGAGGCATGA |
|
|
R:ACACATTTCCAGCTACTCCATAG |
| TGFBR1 |
F:CACCAGTACCCTATTGATGGAAA |
|
|
R:AGTTCAGGCAAAGCTGTAGAA |
|
KIAA0196 |
F:CATATTTGGTGCCGCATGTC |
|
|
R:AGAAGCCTGTCTAAGCCATTTA |
| CHD7 |
F:AATGTTTCAGGGTGGAGA |
|
|
R:CAGGACCTTGTTACAGTGAT |
| NPHP3 |
F:CACAATGCAGTATAGCACACAAA |
|
|
R:CTTATCTGTTCCAGCCACACT |
| PCSK5 |
F:GTGCCTTGTTAATCCCTTTACAC |
|
|
R:CAGGCTGCTCTTGCTCTT |
| FBN2 |
F:TAATGAGTGTGTCGCCCTTC |
|
|
R:TGGCACTTAGTATGTTTCCAGAG |
| DLL1 |
F:CTGTCTTTGGTTTGTCTGGTTTC |
|
|
R:AAGTCGTTCACGCCATCC |
| NOTCH3 |
F:TCCTAAACTCACCCTGTCCT |
|
|
R:GCACAGTCGTAAGTGAGGTC |
| COL3A1 |
F:AAGCAGCATCACTGTCATCTAA |
|
|
R:AGAACTGCCCATTTGTGGT |
| CHD1L |
F:GAACTCGCTCTGAGGTTCAA |
|
|
R:AACTTCTAAGGTCACAGGTTAGG |
| FMO5 |
F:GCTGAGTAAAGAGAACACTTGGA |
|
|
R:TGGCTGTTTGGCTAATCTCTAC |
| LLPH |
F:GGATGAACCAAAGGCAAAGAAA |
|
|
R:GAGGAGTTATGCTGGGTTTGA |
|
PPARGC1A |
F:GGATGAACCAAAGGCAAAGAAA |
|
|
R:GAGGAGTTATGCTGGGTTTGA |
| PTPN22 |
F:GGACATAGAGCTGAATTTGCTTC |
|
|
R:CAAAGACAAGCTCTCTATAAGGTAGA |
| TTC30A |
F:GGAACTCTTTATTGTGCCAAAGG |
|
|
R:CTGGACTCATCTGTGACTGTATTC |
| OBSCN |
F:CCACGCTGGACTCCATTAG |
|
|
R:GCACAGATGGGTGGATGAA |
| DAND5 |
F:GTCATTGCTCCTCTCTCTACATC |
|
|
R:ACGTCTTTCTTGGTCCATCTC |
| STIM2 |
F:CTAGAGCTTGTGCATGGGAA |
|
|
R:GACTTTGCTCTGCAGTTTGTAAG |
Results
Screening outcomes
No pathogenic mutations were identified in CNVs
analyzed by high-throughput SNP sequencing. The targeted NGS
results demonstrated that 10 out of the 17 patients (58.8%) carried
gene mutations, including 4 cases (23.5%) that were genetically
diagnosed and 6 cases (35.3%) with unknown etiology; the remaining
7 patients did not carry known mutations. The diseases involved in
the 4 known cases and the associated genes were as follows: Opitz
G/BBB syndrome caused by midline-1 (MID1) gene mutation;
Loeys-Dietz syndrome (LDS) caused by transforming growth factor-β
receptor type I (TGFBR1) gene mutation; Ritscher-Schinzel
syndrome (RSS; also known as 3C syndrome) caused by KIAA0196
gene mutation; and CHARGE syndrome caused by
chromodomain-helicase-DNA-binding protein 7 (CHD7) gene
mutation.
Prediction of gene function
Characteristics of the named gene mutations were
predicted through the PolyPhen-2, SIFT and Mutation Taster programs
(Table IV). Among the genetically
diagnosed cases, the MID1 (c.G1477C, p.A723V) and
TGFBR1 (c.T1400A, p.M467K) gene mutations were predicted to
be ‘pathogenic’, ‘likely pathogenic’ and ‘pathogenic’ by SIFT,
PolyPhen-2 and Mutation Taster, respectively. The CHD7 gene
mutation (c.C4894T, p.R1632C) was predicted to be ‘pathogenic’ by
both SIFT and Mutation Taster, while the PolyPhen-2 program
predicted this mutation to be ‘benign’. The KIAA0196 gene
mutation (c.A2533G, p.T845A) was predicted to be ‘likely
pathogenic’ by PolyPhen-2 and ‘pathogenic’ by Mutation Taster,
while the SIFT program predicted this mutation to have ‘uncertain
significance’. Potential pathogenic genes were identified in the
remaining 6 cases; however, literature searches did not reveal
previously reported associations between the genes and diseases of
interest. Therefore, the cases cannot be diagnosed based upon the
identified mutations.
| Table IV.Results of targeted sequencing. |
Table IV.
Results of targeted sequencing.
|
|
|
|
|
|
|
| Prediction
tool |
|
|
|---|
|
|
|
|
|
|
|
|
|
|
|
|---|
| Patient no. | Age (months) | Sex | CHD | CLP | Gene | Chromosome | SIFT | PolyPhen-2 | Mutation
taster | CCDS | Amino acid |
|---|
| 1 | 108 | Male | VSD | CL | MID1 | Xp22.2 | Pathogenic | Likely
pathogenic | Pathogenic | G1477C | A723V |
| 2 | 48 | Male | TOF, PLSVC | CLP | TGFBR1 | 9q22.3 | Pathogenic | Likely
pathogenic | Pathogenic | T1400A | M467K |
| 3 | 9 | Male | PDA, PFO | CL |
KIAA0196 | 8q24.13 | Uncertain
significance | Likely
pathogenic | Pathogenic | A2533G | T845A |
| 4 | 60 | Male | VSD, ASD | CL | CHD7 | 8q12.2 | Pathogenic | Benign | Pathogenic | C4894T | R1632C |
| 5 | 4 | Female | VSD, ASD | CP | NPHP3 | 3q22.1 | Uncertain
significance | Benign | Pathogenic | C1228A | Q410K |
| 6 | 36 | Male | VSD | CL | PCSK5 | 1q21.1 | Pathogenic | Likely
pathogenic | Pathogenic | C5041G | P1681A |
| 7 | 24 | Male | DORV, VSD | CP | FBN2 | 5q23.3 | Uncertain
significance | Likely
pathogenic | Pathogenic | G6154A | E2052K |
| 8 | 72 | Male | ASD, PLSVC | CP | DLL1 | 6q27 | Pathogenic | Likely
pathogenic | Pathogenic | C1307T | S436L |
| 9 | 84 | Female | VSD | CP | NOTCH3 | 19p13.12 | Pathogenic | Likely
pathogenic | Pathogenic | G515A | G172D |
| 10 | 30 | Male | ASD | CP | COL3A1 | 2q32.2 | Uncertain
significance | Likely
pathogenic | Pathogenic | G1472A | R491Q |
Sanger sequencing
Finally, Sanger sequencing verified each mutation
(Fig. 3), which was followed by
review of the literature in the context of each mutation to obtain
the genetic diagnosis (8,21–23).
The parents of certain patients were also studied through the use
of Sanger sequencing, but the identified gene mutations were not
detected in any of the parents (data not shown).
Discussion
CHD and CLP are characterized by anomalous
anatomical structures, caused by abnormal development of the heart
and large blood vessels in CHD (24), or abnormal fusion of the lip and
palate during embryonic development in CLP (25). These diseases can severely affect
neonatal health, thus representing a burden to families and the
society (16). With the rapid
development of genetic sequencing technology, a number of methods
are considered to be important in identifying disease-associated
genes, including Sanger sequencing, SNP array, CNV analysis, and
targeted and whole exome sequencing.
In the current study, no pathogenic mutations were
identified in CNVs analyzed by high-throughput SNP sequencing.
Certain gene mutations were successfully identified in 10 patients
(58.8%) via targeted NGS. According to the clinical phenotype of
the patient and the mutation site of the candidate pathogenetic
gene, 4 of these patients were diagnosed with a known genetic
syndrome. To the best of our knowledge, it appears that the present
study identified for the first time a mutation (c.G1477C, p.A723V)
in the MID1 gene as a possible cause of ventricular septal
defect and CL in an Opitz G/BBB syndrome patient. The MID1
gene is located on the short arm of the X chromosome, is
approximately 300 kb, and includes 9 coding exons and multiple
non-coding exons. In early embryonic development, the MID1
gene is highly expressed in the heart, facial region and central
nervous system (26,27). In total, >90 different mutations
of the MID1 gene have been reported in the literature, and
point mutations in this gene have been suggested to cause Opitz
G/BBB syndrome (18,21,28–31).
The MID1 protein encoded by the MID1 gene is a ubiquitin
ligase that interacts with the α4 protein, which is linked to the
protein phosphatase PP2A and forms the complex MID1-α4-PP2A
(27,32). This complex is closely associated
with the development of the ventral midline; therefore, this is
also the main reason for the abnormal development of the ventral
midline structure caused by MID1 gene mutation (33).
LDS is characterized by vascular abnormalities
(cerebral, thoracic, and abdominal arterial aneurysms and/or
dissections), skeletal manifestations, craniofacial features (such
as CP) and cutaneous findings (34). Approximately a third of LDS cases
are caused by TGFBR1 mutation, while two thirds are caused
by TGFBR2 mutation; the mutation site is mostly located in
the serine-hydroxybutyrate enzyme activation coding region, located
in the intracellular portion of the TGF-β receptor (35). The TGF-β type I receptor is
necessary for the fusion of the upper lip and soft palate (36,37),
and the TGFBR1 gene serves a major role in the development
of the heart (22). In the present
study, the TGFBR1 gene mutation was predicted to be
‘pathogenic’, ‘likely pathogenic’ and ‘pathogenic’ by the SIFT,
PolyPhen-2 and Mutation Taster programs, respectively. It was also
identified for the first time that mutation (c.T1400A, p.M467K) in
the TGFBR1 gene was a possible cause of tetralogy of Fallot
and CL in the LDS patient.
RSS is a clinically heterogeneous disorder
characterized by distinctive craniofacial features (including CP)
in addition to cerebellar and cardiac anomalies (8). To date, two articles (8,38)
have been reported on cases of RSS caused by KIAA0196 gene
mutation. Mutation (c.A2533G, p.T845A) in KIAA0196 gene was
investigated in the present study, which is a novel candidate gene
involved in heart development. The KIAA0196 gene is situated
at 8q24.13 of chromosome 8, and the encoded protein of this gene is
known as strumpellin, which is comprised of 1,159 amino acids and
is highly conserved (8). This
protein is ubiquitously expressed in multiple systems and is highly
expressed in skeletal muscle. KIAA0196 mutations have been
reported to cause hereditary spastic paraplegia (39), while a complex overlapping
phenotype, particularly with CHD, has been rarely reported. In
2013, Elliott et al (8)
detected KIAA0196 gene mutations in 8 patients with RSS/3C
syndrome. The expression of strumpellin protein was also reduced by
60%, and the patients exhibited abnormal phenotype of heart
development defects. In addition, previous studies have indicated
that there are genes that cause cardiac abnormalities in the 8q24
interval, and suggested that the KIAA0196 gene is
incorporated into the interval (8,40).
These studies also suggested that the KIAA0196 gene may
serve a role in the pathogenesis of cardiac developmental
disorders.
CHARGE syndrome is a congenital condition comprising
of choroid disease, heart disease, atresia choanae, retarded growth
and development, genital hypoplasia, and ear anomalies and/or
deafness; facial palsy, micrognathia, CP and swallowing
difficulties are also common (41). In the present study, the
CHD7 gene mutation (c.C4894T, p.R1632C) was predicted to be
‘pathogenic’ by both SIFT and Mutation Taster, while the PolyPhen-2
program predicted this mutation to be ‘benign’. To date, ~193
mutations of the CHD7 gene have been reported to lead to
CHARGE syndrome (23,42). The CHD7 gene encodes the
CHD7 protein, which serves a role in chromatin remodeling, cell
cycle regulation, apoptosis regulation, transcriptional regulation
and embryonic stem cell diversity (43). Studies have demonstrated that the
CHD7 gene is expressed in a number of fetal tissues,
including the fetal eye, ear, brain cells, olfactory bulb and heart
tube, among others (19,43). The majority of mutations lead to
the production of non-functional CHD7 protein, which may disrupt
chromatin remodeling and gene expression. Regulation changes in
CHD7 gene expression during embryonic development may lead
to symptoms and signs of CHARGE syndrome (19).
The remaining 6 cases were not genetically
diagnosed, although candidate genes were identified, including
nephrocystin-3 (NPHP-3), proprotein convertase
subtilisin/kexin type 5 (PCSK5), fibrillin 2 (FBN2),
delta-like protein 1 (DLL1), Notch 3 (NOTCH3) and
collagen type III α1 chain (COL3A1). The NPHP3 gene
mutation (c.C1228A, p.Q410K) may impact the development of cilia
tissue, while it has been reported that primary ciliary dyskinesia
is associated with the development of CHD (44). In addition, the PCSK5 gene
mutation (c.C5041G, p.P1681A) was predicted to be ‘pathogenic’,
‘likely pathogenic’ and ‘pathogenic’ by the SIFT, PolyPhen-2 and
Mutation Taster tools, respectively. In mice, the PCSK5 gene
causes VACTERL syndrome, which comprises of deformity in vertebral,
anorectal, cardiac, tracheoesophageal, renal, limb and other
systems (45). The FBN2
gene mutation (c.G6154A, p.E2052K) may cause congenital
contractural arachnodactyly. The cardiovascular phenotype of this
syndrome is milder and less common in comparison with that of
Marfan syndrome, and the ascending aorta is also slightly dilated,
which may be combined with other intracardiac malformations
(46). Furthermore, the SIFT,
PolyPhen-2 and Mutation Taster programs respectively predicted the
DLL1 gene mutation (c.C1307T, p.S436L) to be ‘pathogenic’,
‘likely pathogenic’ and ‘pathogenic’. The protein encoded by the
DLL1 gene is a ligand in the Notch signaling pathway, which
mainly regulates the apoptosis of hematopoietic cells and signals
between cells. The Notch signaling pathway serves an important role
in embryonic differentiation and in homeostasis in adults, as well
as in the development of various systems (47). Additionally, the protein encoded by
the NOTCH3 gene, which was found to be affected by mutation
(c. G515A, p. G172D) in the present study, is among the key
proteins in the Notch signaling pathway. The disease caused by the
mutation is an autosomal dominant arteriopathy associated with
subcortical infarcts and leukoencephalopathy (48). Although CHD or CLP caused by
mutations in the NOTCH3 gene has not been reported to date,
the role of the Notch signaling pathway in the growth and
development of various systems is widely recognized (47). The COL3A1 gene mutation
(c.G1472A, p.R491Q) was predicted to be ‘likely pathogenic’ and
‘pathogenic’ by PolyPhen-2 and Mutation Taster, respectively,
whereas the SIFT program predicted this mutation to be of
‘uncertain significance’. This gene mutation may cause
Ehlers-Danlos syndrome and aortic aneurysm. According to review of
the literature, it was noted that all of the mutation sites
determined in the current study are novel mutation sites that have
not been previously reported to the best of our knowledge. Samples
obtained from the parents of certain patients were also examined by
Sanger sequencing, however, these parents did not carry the
identified genes mutations. The results should be further verified
in an animal model, such as zebra fish or mouse.
When considering all forms of genetic sequencing
technology, the advantages of targeted NGS are evident. Firstly,
the amount of tedious and repetitive work for researchers is
reduced, owing to the fact that this method can rapidly analyze
large quantities of genetic information. NGS enables thousands of
genes to be analyzed simultaneously, or a smaller subset of genes
(a ‘mini-genome’ or disease-specific panel) to be examined in a
single assay. However, the limitation of sample size and the
possibility of leak detection of base point mutation exist in SNP
array technology. Secondly, targeted NGS not only allows focusing
on specific genes associated with pathological expression, but can
also improve the coverage and expressive quality of exons due to
its efficient enrichment. Large-scale parallel sequencing of a
specifically selected part of the genome (for example, the exome or
a specific set of genes relevant to a disease phenotype) leads to a
higher sequencing coverage as compared with that of whole-genome
sequencing (49). Furthermore, for
a specific phenotype or disease, targeted sequencing has lower cost
and is more rapid than whole exome sequencing, since this
technology reduces the genetic discovery that may be irrelevant to
the disease (49). In addition,
targeted NGS may be more clinically useful in comparison with other
sequencing techniques, owing to faster turnaround time (reduced
sequencing volume and associated data analysis), higher and more
reliable coverage, and the ability to avoid incidental findings.
However, this method is associated with certain disadvantages
including the limitation of requiring known virulence genes and its
lack of suitability for a single sample.
Notably, the implementation of NGS in clinical
practice has altered the way genetic counsellors and other
clinicians approach genetic testing. Molecular diagnostics may now
be performed at an early stage of disease, often enabling a broader
set of therapeutic options and a lengthened window of opportunity
to ameliorate disease progression (50). The identification of underlying
genetic defects can also improve diagnosis of the disease prior to
genetic counselling and enable prenatal testing.
In conclusion, using targeted NGS technology, the
present study determined 10 individual mutations (58.8%) in
candidate disease genes, which are possible causes of CHD and CLP
in patients. The targeted NGS was demonstrated to be an effective
and accurate method for providing a specific diagnosis of CHD and
CLP, despite the presence of diverse phenotypes.
Acknowledgements
The authors would like to thank the State Key
Laboratory of Medical Genetics of China (Changsha, China) for the
technical assistance provided.
Funding
This study was supported by the Hunan Provincial
Natural Science Foundation of China (grant no. 2015JJ4085) and the
Scientific Research Foundation for the Returned Overseas Chinese
Scholars, State Education Ministry (2014; grant no. 1685).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HB and TZ conceived and designed the study, and
drafted the manuscript. HB and LL collected the data. HB, TZ and SH
were involved in data cleaning and verification. HB, TZ and ZT
analyzed the data. All authors were involved in the final draft of
the manuscript.
Ethics approval and consent to
participate
The study protocol was approved by the Review Board
at The Second Xiangya Hospital of Central South University (China),
and the relatives of study subjects provided informed consent. All
experiments were performed in accordance with relevant guidelines
and regulations.
Patient consent for publication
Written informed consent was obtained from the their
parent, guardian or next of kin for the publication of any
associated data and accompanying images.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CHD
|
congenital heart disease
|
|
CLP
|
cleft lip and palate
|
|
SNP
|
single nucleotide polymorphisms
|
|
CNVs
|
copy number variations
|
|
CL
|
cleft lip
|
|
CP
|
cleft palate
|
|
NGS
|
next-generation sequencing
|
|
PCR
|
polymerase chain reaction
|
|
LDS
|
Loeys-Dietz syndrome
|
|
RSS
|
Ritscher-Schinzel syndrome
|
References
|
1
|
Liang CD, Huang SC and Lai JP: A survey of
congenital heart disease in patients with oral clefts. Acta
Paediatr Taiwan. 40:414–417. 1999.PubMed/NCBI
|
|
2
|
Barbosa MM, Rocha CM, Katina T, Caldas M,
Codorniz A and Medeiros C: Prevalence of congenital heart diseases
in oral cleft patients. Pediatr Cardiol. 24:369–374. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bill J, Proff P, Bayerlein T, Weingaertner
J, Fanghänel J and Reuther J: Treatment of patients with cleft lip,
alveolus and palate-a short outline of history and current
interdisciplinary treatment approaches. J Craniomaxillofac Surg. 34
(Suppl 2):S17–S21. 2006. View Article : Google Scholar
|
|
4
|
Ho KS, South ST, Lortz A, Hensel CH, Sdano
MR, Vanzo RJ, Martin MM, Peiffer A, Lambert CG, Calhoun A, et al:
Chromosomal microarray testing identifies a 4p terminal region
associated with seizures in Wolf-Hirschhorn syndrome. J Med Genet.
53:256–263. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Biswas AB and Furniss F: Cognitive
phenotype and psychiatric disorder in 22q11.2 deletion syndrome: A
review. Res Dev Disabil. 53-54:242–257. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Poelmans S, Kawamoto T, Cristofoli F,
Politis C, Vermeesch J, Bailleul-Forestier I, Hens G, Devriendt K,
Verdonck A and Carels C: Genotypic and phenotypic variation in six
patients with solitary median maxillary central incisor syndrome.
Am J Med Genet A. 167A:2451–2458. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Abulezz TA: Cleft lip and palate: An
experience of a developing center in egypt. J Craniofac Surg.
28:e731–e734. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Elliott AM, Simard LR, Coghlan G, Chudley
AE, Chodirker BN, Greenberg CR, Burch T, Ly V, Hatch GM and
Zelinski T: A novel mutation in KIAA0196: Identification of a gene
involved in Ritscher-Schinzel/3C syndrome in a First Nations
cohort. J Med Genet. 50:819–822. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tang M, Yang YF, Xie L, Chen JL, Zhang WZ,
Wang J, Zhao TL, Yang JF and Tan ZP: Duplication of 10q22.3-q23.3
encompassing BMPR1A and NGR3 associated with congenital heart
disease, microcephaly, and mild intellectual disability. Am J Med
Genet A. 167A:3174–3179. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zeng H, Tang JG, Yang YF, Tan ZP and Tan
JQ: A novel homozygous SACS mutation identified by whole-exome
sequencing in a consanguineous family with autosomal recessive
spastic ataxia of charlevoix-saguenay. Cytogenet Genome Res.
152:16–21. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Guo T, Tan ZP, Chen HM, Zheng DY, Liu L,
Huang XG, Chen P, Luo H and Yang YF: An effective combination of
whole-exome sequencing and runs of homozygosity for the diagnosis
of primary ciliary dyskinesia in consanguineous families. Sci Rep.
7:79052017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Koestenberger M: Transthoracic
echocardiography in children and young adults with congenital heart
disease. ISRN Pediatr. 2012:7534812012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Allori AC, Mulliken JB, Meara JG,
Shusterman S and Marcus JR: Classification of Cleft Lip/Palate:
Then and now. Cleft Palate Craniofac J. 54:175–188. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gatti GL, Freda N, Giacomina A, Montemagni
M and Sisti A: Cleft lip and palate repair. J Craniofac Surg.
28:1918–1924. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hu S, Yang Y, Liu L, Tan Z and Zhao T:
High-resolution single nucleotide polymorphism arrays identified an
atypical microdeletion of the Williams-Beuren syndrome interval in
a patient presenting with a different phenotype. Mol Med Rep.
15:2709–2712. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu L, Bu H, Yang Y, Tan Z, Zhang F, Hu S
and Zhao T: A targeted, next-generation genetic sequencing study on
tetralogy of fallot, combined with cleft lip and palate. J
Craniofac Surg. 28:e351–e355. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen JL, Zhu X, Zhao TL, Wang J, Yang YF
and Tan ZP: Rare copy number variations containing genes involved
in RASopathies: Deletion of SHOC2 and duplication of PTPN11. Mol
Cytogenet. 7:282014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu CH, Liu YF, Yu JS, Ng YY, Chen SJ, Su
PH and Chen JY: A MID1 gene mutation in a patient with Opitz G/BBB
syndrome that altered the 3D structure of SPRY domain. Am J Med
Genet A. 158A:726–731. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jongmans MC, Admiraal RJ, van der Donk KP,
Vissers LE, Baas AF, Kapusta L, van Hagen JM, Donnai D, de Ravel
TJ, Veltman JA, et al: CHARGE syndrome: The phenotypic spectrum of
mutations in the CHD7 gene. J Med Genet. 43:306–314. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tan ZP, Xie L, Deng Y, Chen JL, Zhang WZ,
Wang J, Yang JF and Yang YF: Whole-exome sequencing identifies
Y1495X of SCN5A to be associated with familial conduction disease
and sudden death. Sci Rep. 4:56162014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang X, Chen Y, Zhao S, Markljung E and
Nordenskjöld A: Hypospadias associated with hypertelorism, the
mildest phenotype of Opitz syndrome. J Hum Genet. 56:348–351. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Muramatsu Y, Kosho T, Magota M, Yokotsuka
T, Ito M, Yasuda A, Kito O, Suzuki C, Nagata Y, Kawai S, et al:
Progressive aortic root and pulmonary artery aneurysms in a neonate
with Loeys-Dietz syndrome type 1B. Am J Med Genet A. 152A:417–421.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Husu E, Hove HD, Farholt S, Bille M,
Tranebjærg L, Vogel I and Kreiborg S: Phenotype in 18 Danish
subjects with genetically verified CHARGE syndrome. Clin Genet.
83:125–134. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Srivastava D: HAND proteins: Molecular
mediators of cardiac development and congenital heart disease.
Trends Cardiovasc Med. 9:11–18. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sisti A and Oranges CM: Evidence-based
medicine: Unilateral cleft lip and nose repair. Plast Reconstr
Surg. 136:118e–119e. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pinson L, Augé J, Audollent S, Mattéi G,
Etchevers H, Gigarel N, Razavi F, Lacombe D, Odent S, Le Merrer M,
et al: Embryonic expression of the human MID1 gene and its
mutations in Opitz syndrome. J Med Genet. 41:381–386. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Short KM, Hopwood B, Yi Z and Cox TC: MID1
and MID2 homo- and heterodimerise to tether the rapamycin-sensitive
PP2A regulatory subunit, alpha 4, to microtubules: Implications for
the clinical variability of X-linked Opitz GBBB syndrome and other
developmental disorders. BMC Cell Biol. 3:12002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hüning I, Kutsche K, Rajaei S, Erlandsson
A, Lovmar L, Rundberg J and Stefanova M: Exon 2 duplication of the
MID1 gene in a patient with a mild phenotype of Opitz G/BBB
syndrome. Eur J Med Genet. 56:188–191. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Migliore C, Athanasakis E, Dahoun S,
Wonkam A, Lees M, Calabrese O, Connell F, Lynch SA, Izzi C,
Pompilii E, et al: Complex rearrangement of the exon 6 genomic
region among Opitz G/BBB syndrome MID1 alterations. Eur J Med
Genet. 56:404–410. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Preiksaitiene E, Krasovskaja N, Utkus A,
Kasnauskiene J, Meskiene R, Paulauskiene I, Valeviciene NR and
Kucinskas V: R368X mutation in MID1 among recurrent mutations in
patients with X-linked Opitz G/BBB syndrome. Clin Dysmorphol.
24:7–12. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ji X, Xing Y, Xu Y, Liu Y, Chen Y, Tao J
and Xiao B: A novel mutation in MID1 in a patient with X-linked
Opitz G/BBB syndrome. Gene. 537:140–142. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Van den Veyver IB, Cormier TA, Jurecic V,
Baldini A and Zoghbi HY: Characterization and physical mapping in
human and mouse of a novel RING finger gene in Xp22. Genomics.
51:251–261. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schweiger S and Schneider R: The MID1/PP2A
complex: A key to the pathogenesis of Opitz BBB/G syndrome.
Bioessays. 25:356–366. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Van Laer L, Dietz H and Loeys B:
Loeys-Dietz syndrome. Adv Exp Med Biol. 802:95–105. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Loeys BL, Schwarze U, Holm T, Callewaert
BL, Thomas GH, Pannu H, De Backer JF, Oswald GL, Symoens S,
Manouvrier S, et al: Aneurysm syndromes caused by mutations in the
TGF-beta receptor. N Engl J Med. 355:788–798. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dudas M, Kim J, Li WY, Nagy A, Larsson J,
Karlsson S, Chai Y and Kaartinen V: Epithelial and ectomesenchymal
role of the type I TGF-beta receptor ALK5 during facial
morphogenesis and palatal fusion. Dev Biol. 296:298–314. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li WY, Dudas M and Kaartinen V: Signaling
through Tgf-beta type I receptor Alk5 is required for upper lip
fusion. Mech Dev. 125:874–882. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Valdmanis PN, Meijer IA, Reynolds A, Lei
A, MacLeod P, Schlesinger D, Zatz M, Reid E, Dion PA, Drapeau P and
Rouleau GA: Mutations in the KIAA0196 gene at the SPG8 locus cause
hereditary spastic paraplegia. Am J Hum Genet. 80:152–161. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jahic A, Kreuz F, Zacher P, Fiedler J,
Bier A, Reif S, Rieger M, Krüger S, Beetz C and Plaschke J: A novel
strumpellin mutation and potential pitfalls in the molecular
diagnosis of hereditary spastic paraplegia type SPG8. J Neurol Sci.
347:372–374. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dauber A, Golzio C, Guenot C, Jodelka FM,
Kibaek M, Kjaergaard S, Leheup B, Martinet D, Nowaczyk MJ,
Rosenfeld JA, et al: SCRIB and PUF60 are primary drivers of the
multisystemic phenotypes of the 8q24.3 copy-number variant. Am J
Hum Genet. 93:798–811. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Pagon RA, Graham JM Jr, Zonana J and Yong
SL: Coloboma, congenital heart disease, and choanal atresia with
multiple anomalies: CHARGE association. J Pediatr. 99:223–227.
1981. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cho HJ, Song MH, Choi SY, Kim J, Lee J,
Kim UK, Bok J and Choi JY: Genetic analysis of the CHD7 gene in
Korean patients with CHARGE syndrome. Gene. 517:164–168. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zentner GE, Layman WS, Martin DM and
Scacheri PC: Molecular and phenotypic aspects of CHD7 mutation in
CHARGE syndrome. Am J Med Genet A. 152A:674–686. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Picard C, McCarl CA, Papolos A, Khalil S,
Lüthy K, Hivroz C, LeDeist F, Rieux-Laucat F, Rechavi G, Rao A, et
al: STIM1 mutation associated with a syndrome of immunodeficiency
and autoimmunity. N Engl J Med. 360:1971–1980. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Szumska D, Pieles G, Essalmani R, Bilski
M, Mesnard D, Kaur K, Franklyn A, El Omari K, Jefferis J, Bentham
J, et al: VACTERL/caudal regression/Currarino syndrome-like
malformations in mice with mutation in the proprotein convertase
Pcsk5. Genes Dev. 22:1465–1477. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ramos Arroyo MA, Weaver DD and Beals RK:
Congenital contractural arachnodactyly. Report of four additional
families and review of literature. Clin Genet. 27:570–581. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chiba S: Notch signaling in stem cell
systems. Stem Cells. 24:2437–2447. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Di Donato I, Bianchi S, De Stefano N,
Dichgans M, Dotti MT, Duering M, Jouvent E, Korczyn AD,
Lesnik-Oberstein SA, Malandrini A, et al: Cerebral autosomal
dominant arteriopathy with subcortical infarcts and
leukoencephalopathy (CADASIL) as a model of small vessel disease:
Update on clinical, diagnostic, and management aspects. BMC Med.
15:412017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Nijman IJ, Mokry M, van Boxtel R, Toonen
P, de Bruijn E and Cuppen E: Mutation discovery by targeted genomic
enrichment of multiplexed barcoded samples. Nat Methods. 7:913–915.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Pei Y and Watnick T: Diagnosis and
screening of autosomal dominant polycystic kidney disease. Adv
Chronic Kidney Dis. 17:140–152. 2010. View Article : Google Scholar : PubMed/NCBI
|