Introduction
Ischemic heart disease remains one of the leading
causes of morbidity and mortality across the world (1). Currently, the primary treatment for
ischemic heart disease is reperfusion of the blocked artery.
However, this abrupt reperfusion frequently results in deleterious
secondary damage termed ischemia-reperfusion (I/R) injury (2). I/R triggers a cascade of complex
intracellular events, including the generation of intracellular
reactive oxygen species (ROS), loss of intracellular and
mitochondrial calcium homeostasis and dysfunction of
microcirculation, which subsequently activate multiple pathways
leading to myocardial stunning, cardiomyocyte death, microvascular
obstruction and arrhythmias, all eventually leading to increased
infarct sizes (3,4). Therefore, understanding the
underlying mechanisms of cardiac I/R injury is necessary for the
development of novel therapeutic strategies for the prevention and
treatment of this pathology.
Numerous studies have reported that ROS, including
the superoxide anion (O2•−), hydrogen
peroxide (H2O2) and the hydroxyl radical
(•OH), are critically involved in the pathophysiology of
myocardial I/R (M-I/R) injuries. Induction of oxidative
modification of intracellular molecules by ROS further activates
stress signaling pathways, which leads to cardiomyocyte apoptosis
(5,6). An overload of ROS may trigger the
activation of mitogen-activated protein kinase (MAPK) signaling and
aggravate lethal cellular processes, particularly during I/R
injury. Inhibition of p38, c-Jun NH2-terminal kinases
(JNK) and/or extracellular signal-regulated kinases (ERK) 1 and 2
(all three of which are MAPKs, namely MAPK 8, 3 and 1,
respectively), may protect the heart from I/R injury by reducing
cardiomyocyte death and infarct size, in addition to attenuating
left ventricular function impairment, as demonstrated in M-I/R
models (7–10). Moreover, ROS may also activate
signal transducer and activator of transcription (STAT) 3 during
I/R, which has been demonstrated to protect the myocardium from
ischemia and oxidative stress through the upregulation of
cardioprotective genes and the modulation of mitochondrial
respiration (11,12). In fact, a number of compounds,
including sevoflurane, cilostazol and selenium, have been
demonstrated to have cardioprotective effects and to alleviate
M-I/R injury by activating STAT3 signaling (13–15).
Y-box protein 1 (YB1), a member of the highly
conserved Y-box protein family, is a multifunctional
DNA/RNA-binding protein, which serves important roles during the
process of inflammation, wound healing and the stress response by
regulating gene transcription, RNA splicing and mRNA translation
(16–18). In previous studies, YB1 was also
reported to be an important regulator of metabolic stress by
sequestering mRNA molecules necessary for cell survival during
temporary periods of hypoxia, adenosine triphosphate depletion or
oxidative stress (19–21). However, whether YB1 serves a role
in M-I/R injury remains unknown.
In the present study, the role of YB1 in
cardiomyocyte apoptosis was evaluated using
H2O2-treated H9c2 cells in vitro and
M-I/R injury models in vivo. The results, to the best of our
knowledge, demonstrated for the first time that YB1 expression was
upregulated during H2O2 stimulation, and that
YB1 may protect cardiac myocytes against H2O2
or M-I/R-induced injury by binding to protein inhibitor of
activated STAT 3 (PIAS3) mRNA, resulting in the activation of
STAT3.
Materials and methods
Cell culture and treatment
Rat embryonic cardiomyoblast-derived H9c2 cells
(CLR-1446; American Type Culture Collection, Manassas, VA, USA)
were maintained in Dulbecco's Modified Eagle's medium (DMEM; Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) with 10% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin/streptomycin at 37°C with 5% CO2. H9c2 cells
were seeded in 6-well plates (1×106 cells/well), treated
at 24 h post-seeding with 50 mM H2O2 for 0, 3
or 6 h at 37°C, and harvested for the subsequent experiments.
In vitro cell viability assay
H9c2 cells cultured in 96-well plates
(4×104 cells/well) were treated with 50 mM
H2O2 for 0, 3 or 6 h. Cell viability was
measured using the Cell Counting Kit-8 (Beyotime Institute of
Biotechnology, Haimen, China), according to the manufacturer's
instructions. Absorbance was measured at a wavelength of 450
nm.
Lactate dehydrogenase (LDH) release
assay
H9c2 cells cultured in 96-well plates
(4×104 cells/well) were treated with 50 mM
H2O2 for 0, 3 or 6 h. The activity of LDH was
measured in cell culture supernatants or serum using an LDH Release
Assay kit (Beyotime Institute of Biotechnology), according to the
manufacturer's instructions.
Analysis of cell apoptosis by flow
cytometry
H9c2 cells were cultured and treated with 50 mM
H2O2 for different times as described.
Following treatment, cells were digested with 0.25% trypsin at 37°C
for 5 min and collected by centrifugation at 1,000 × g for 5 min at
4°C. Following two washes with ice-cold PBS, the cells were fixed
in ice-cold 70% ethanol overnight at −20°C and stained with Annexin
V-fluorescein isothiocyanate and propidium iodide (Hangzhou
MultiSciences Biotech Co., Ltd., Hangzhou, China) together for 15
min at room temperature. The apoptotic cells were identified by
EPICS XL flow cytometry (Beckman Coulter, Inc., Brea, CA, USA) and
analyzed by Cell Quest software version FCS2.0 (BD Biosciences, San
Jose, CA, USA). PI and Annexin V-FITC-positive cells were
considered apoptotic cells.
Western blotting
Following treatment, H9c2 cell samples were
collected and lysed with NP40 lysis buffer (Beyotime Institute of
Biotechnology). Following centrifugation at 12,000 × g for 10 min
at 4°C, the supernatant was collected and quantified using a
bicinchoninic acid kit (Beyotime Institute of Biotechnology). The
proteins (15 µg/lane) were separated by 10% SDS-PAGE and
transferred to nitrocellulose membranes. Following blocking with 5%
non-fat milk for 60 min at 37°C, the membranes were incubated with
primary antibodies for 60 min at 37°C. The primary antibodies used
included rabbit-anti-YB1 (cat. no. ab76149; Abcam, Cambridge, UK),
rabbit-anti-phosphorylated P38 (p-P38; cat. no. ab4822; Abcam),
rabbit-anti-P38 (cat. no. ab170099; Abcam),
rabbit-anti-phosphorylated JNK (p-JNK; cat. no. ab4821; Abcam),
rabbit-anti-JNK (cat. no. ab112501; Abcam),
rabbit-anti-phosphorylated ERK1/2 (p-ERK1/2; cat. no. ab215362;
Abcam), rabbit-anti-ERK1/2 (cat. no. ab17942; Abcam),
rabbit-anti-nuclear factor κ B (NF-κB) p65 (cat. no. ab16502;
Abcam), rabbit-anti-phosphorylated NF-κB p65 (p-p65; cat. no.
ab86299; Abcam), rabbit-anti-Janus kinase (JAK)1 (cat. no.
ab133666; Abcam), rabbit-anti-PIAS3 (cat. no. ab22856; Abcam),
mouse-anti-Src homology region 2 domain-containing phosphatase
(SHP)1 (cat. no. ab76202; Abcam), mouse-anti-SHP2 (cat. no.
ab76285; Abcam), mouse-anti-suppressor of cytokine signaling (SOCS)
1 (cat. no. ab211288; Abcam), mouse-anti-SOCS3 (cat. no. ab14939;
Abcam), rabbit-anti-insulin-like growth factor 2 mRNA-binding
protein 1 (IGF2BP1; cat. no. ab82968; Abcam), rabbit-anti-GAPDH
(cat. no. ab181602; Abcam), rabbit-anti-phosphorylated JAK1
(p-JAK1; cat. no. 74129; Cell Signaling Technology, Inc., Danvers,
MA, USA), rabbit-anti-JAK2 (cat. no. 3230; Cell Signaling
Technology, Inc.), rabbit-anti-phosphorylated JAK2 (p-JAK2; cat.
no. 3771; Cell Signaling Technology, Inc.), rabbit-anti-STAT1 (cat.
no. 9172; Cell Signaling Technology, Inc.),
rabbit-anti-phosphorylated STAT1 (p-STAT1; cat. no. 9167; Cell
Signaling Technology, Inc.), rabbit-anti-STAT3 (cat. no. 12640;
Cell Signaling Technology, Inc.), rabbit-anti-phosphorylated STAT3
(p-STAT3; cat. no. 9145, Cell Signaling Technology, Inc.) and
rabbit-anti-SHP2 (cat. no. 3397; Cell Signaling Technology, Inc.).
The primary antibodies were all used at a dilution of 1:1,000 in 5%
bovine serum albumin (BSA; Beyotime Institute of Biotechnology).
Horseradish peroxidase-conjugated secondary antibodies against
rabbit (cat. no. ab6721; Abcam) or mouse (cat. no. ab6789; Abcam)
were used at a dilution of 1:5,000 in 5% BSA. The membranes were
incubated with secondary antibodies for 60 min at 37°C. The protein
levels were first normalized to GAPDH and subsequently normalized
to the experimental controls. Blots were visualized with an
enhanced chemiluminescence kit (Beyotime Institute of
Biotechnology) and quantified with ImageJ version 1.42 software
(National Institutes of Health, Bethesda, MD, USA).
Small interfering RNA (siRNA),
plasmids and transfection
siRNA against YB1 (siYB1) consisted of
5′-UCAUCGCAACGAAGGUUUUTT-3′ and 5′-AAAACCUUCGUUGCGAUGATT-3′. The
scrambled siRNA (siNC) oligonucleotide duplex used as the control
had the following sequences: 5′-UUCUCCGAACGUGUCACGUTT-3′ and
5′-ACGUGACACGUUCGGAGAATT-3′. siRNAs were synthesized by Shanghai
GenePharma Co., Ltd (Shanghai, China). Total RNA was extracted from
H9c2 cells using RNA Isolater Total RNA Extraction Reagent (Vazyme,
Piscataway, NJ, USA) and then reverse-transcribed into cDNA using a
HiScript 1st Strand cDNA Synthesis Kit (Vazyme), according to the
manufacturer's instructions. The YB1 gene was amplified from the
above cDNA by polymerase chain reaction (PCR) using Phanta
Super-Fidelity DNA Polymerase (Vazyme), using forward primer,
5′-CCCAAGCTTATGAGCAGCGAGGCCGAG-3′ and reverse primer,
5′-CCGCTCGAGCTCAGCTGGTGGATC-3′, and then cloned into the
pCMV-flag-N-expression vector (Clontech Laboratories, Inc.,
Mountainview, CA, USA) and sequenced. The PCR thermocycling
conditions were as follows: 95°C for 3 min; 30 cycles of 95°C for
30 sec, 58°C for 30 sec and 72°C for 90 sec; and 72°C for 10 min.
The PIAS3 gene was amplified from cDNA from H9c2 cells by PCR,
using forward primer, 5′-GGAATTCGGATGGCGGAGCTGGGCGAA-3′ and reverse
primer, 5′-GGGGTACCTCAGTCCAAGGAAATGC-3′, cloned into the
pCMV-Myc-N-expression vector (Clontech Laboratories, Inc.) and
sequenced. siRNAs (100 nM) or plasmids (2 µg) were transfected into
H9c2 cells, using Exfect Transfection Reagent (Vazyme), according
to the manufacturer's instructions. At 24 h post-transfection,
cells were processed for further analysis.
Protein co-immunoprecipitation
(COIP)
H9c2 cells were treated with 50 mM
H2O2. After 6 h, the cells were lysed with
NP40 lysis buffer (Beyotime Institute of Biotechnology). Following
centrifugation at 12,000 × g for 10 min at 4°C, the cell lysates
were precleared with protein A/G agarose beads (Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) and an unrelated antibody
from the same species of origin. Subsequently, the samples were
incubated with the anti-YB1 antibody (1:30; cat. no. ab76149;
Abcam) and protein A/G agarose beads overnight at 4°C with
continuous rotation. The beads were washed five times in lysis
buffer, and the immunoprecipitates were eluted from protein A/G
agarose beads by heating at 100°C for 5 min. Following
centrifugation at 12,000 × g for 10 min at 4°C, the samples were
analyzed by western blotting.
RNA preparation and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted from cells using RNA
Isolater Total RNA Extraction Reagent (R401-01; Vazyme). RNA (500
ng) from each sample was reverse-transcribed into cDNA using the
PrimeScript RT reagent kit (Takara Bio, Inc., Otsu, Japan). RT-qPCR
was performed using a 7500 real-time PCR system (Applied
Biosystems; Thermo Fisher Scientific, Inc.) with AceQ qPCR
SYBR® Green Master Mix (Q111-02; Vazyme), under the
following conditions: An initial denaturation at 95°C for 10 min,
and 40 cycles of 95°C for 10 sec, 60°C for 30 sec and 72°C for 10
sec. The uniqueness and sizes of PCR products were confirmed by
generating melting curves. Each sample was evaluated in triplicate
and used for the analysis of the relative transcription data using
the 2−ΔΔCq method (22). RT-qPCR primers were as follows:
YB1, forward 5′-CACCTTACTACATGCGGAGACCT-3′, reverse,
5′-TTGTCAGCACCCTCCATCACT-3′; SHP1, forward
5′-CAGGTCGTCCGACTATTCTGT-3′, reverse, 5′-AGGCTACTGTCTTGGCTAGGA-3′;
SHP2, forward 5′-AGAGGGAAGAGCAAATGTGTCA-3′, reverse,
5′-CTGTGTTTCCTTGTCCGACCT-3′; SOCS1, forward
5′-CTGCGGCTTCTATTGGGGAC-3′, reverse, 5′-AAAAGGCAGTCGAAGGTCTCG-3′;
SOCS3, forward 5′-TGCGCCTCAAGACCTTCAG-3′, reverse,
5′-GCTCCAGTAGAATCCGCTCTC-3′; PIAS3, forward
5′-TTCGCTGGCAGGAACAAGAG-3′, reverse, 5′-GGGCGCAGCTAGACTTGAG-3′; and
GAPDH, forward 5′-TCAACAGCAACTCCCACTCTTCCA-3′ and reverse,
5′-ACCCTGTTGCTGTAGCCGTATTCA-3′. The obtained data were normalized
to the GAPDH expression levels in each sample.
Actinomycin D (ActD) treatment
H9c2 cells cultured in 6-well plates
(1×106 cells/well) were transfected with
p-CMV-flag-vector or p-CMV-flag-YB1. At 24 h post-transfection,
cells were treated with ActD (10 µg/ml) for 0, 1, 2, 4, 6 or 8 h at
37°C. Cellular RNA was extracted and PIAS3 mRNA expression levels
were analyzed via RT-qPCR analysis, as described above.
RNA-binding protein
immunoprecipitation (RIP)
RIP was performed using a Magna RIP kit (Merck KGaA,
Darmstadt. Germany). Briefly, H2O2-treated
H9c2 cells were harvested following two washes with PBS and lysed
with RIP lysis buffer. A total of one-tenth of the supernatant was
retained for the RT-qPCR analysis of the input, and the rest was
incubated with antibodies against YB1 or immunoglobulin G for 24 h.
The 50 µl A/G magnetic beads were added to the supernatant.
Subsequent to immobilizing magnetic bead-bound complexes with a
magnetic separator (Merck KGaA), the supernatant was used to
extract RNAs with phenol:chloroform:isoamyl alcohol reagent at a
ratio of 125:24:1 (all chemicals were purchased from Aladdin
Industrial Corporation, Ontario, CA, USA). A cDNA synthesis kit
(Takara Bio, Inc.) was used to synthesize the first-strand cDNA.
Finally, qPCR was performed for analysis using AceQ qPCR SYBR Green
Master Mix, as described above.
Animal model of M-I/R injury
Adult male Sprague-Dawley (SD) rats (15 rats, 8–9
weeks old, 250–300 g) were obtained from the Model Animal Research
Center of Nanjing University (Nanjing, China). All animal
experiments and procedures were approved by the Institutional
Animal Care and Use Committee of the Medical School of Ningbo
University (Ningbo, China). The animals were housed at 22–24°C
under a 12-h light/dark cycle, with 40–60% humidity and free access
to food and water.
For the M-I/R model (23,24),
rats were anesthetized using thiopental (60 mg/kg,
intraperitoneal). The trachea was cannulated and ventilated using a
rodent ventilator (tidal volume, 2–3 ml; respiratory rate, 65–70
breaths/min; rodent ventilator model 683; Harvard Apparatus,
Holliston, MA, USA). The heart was exposed through left intercostal
thoracotomy (between the fourth and fifth costal spaces) and the
pericardium was cut. Subsequently, ischemia was induced for 30 min
with a 6-0 silk suture, ~1–2 mm distal to the origin of the left
anterior descending coronary artery (LAD), tightening over a
pipette tip to ligate. Successful LAD ligation was characterized by
elevation of the ST segment of the echocardiogram. Reperfusion was
performed for 24 h by removing the tubes and loosening the
suture.
A recombinant lentivirus carrying short hairpin RNA
(shRNA) against YB1 (shYB1) or containing non-specific shRNA (shNC)
as a control were provided by Shanghai GenePharma Co., Ltd. Healthy
adult male SD rats were randomly divided into groups: i) The Sham
group (n=5), sham operation without coronary artery ligation and
received normal saline injection via the tail vein; ii) the
I/R+shNC group (n=5), ischemia induction for 30 min followed by 6 h
reperfusion, and injected with shNC via the tail vein; and iii) the
I/R+shYB1 group (n=5), ischemia induction for 30 min followed by 6
h reperfusion, injected with shYB1 via the tail vein. After 6 h of
reperfusion, echocardiography and cardiac hemodynamic measurements
were performed to assess the heart function. Following the
measurements, the animals were sacrificed and heart tissues and
serum were collected.
Echocardiography analysis
Rats of the Sham group, I/R+shNC group, or I/R+shYB1
group were kept on a heating pad in a left lateral decubitus or
supine position under isoflurane (2%) anesthesia, and
two-dimensional images were recorded using a Vivid 7
echocardiography machine (GE Healthcare, Chicago, IL, USA) equipped
with a 10 MHz phased array transducer. Left ventricle parameters
including interventricular septum thickness, posterior wall
thickness, left ventricle internal diastolic diameter (LVIDd) and
left ventricle internal systolic diameter (LVIDs) were obtained
from M-mode interrogation in a long-axis view. Left ventricle
percentage fractional shortening (LVFS) and ejection fraction
(LVEF) were calculated as follows: LVFS%=(LVIDd - LVIDs)/ LVIDd
×100; LVEF%
=[(LVIDd)3-(LVIDs)3]/(LVIDd)3
×100. All echocardiographic measurements were averaged from at
least three separate cardiac cycles.
Measurement of myocardial infarct
size
Following I/R, the hearts were removed from the
animals (n=5), and snap frozen at −20°C for 24 h. The hearts were
sliced into 2 mm transverse sections, along the ape-base axis,
using a stainless steel heart slicer matrix. The slices were
incubated in 2% triphenyltetrazolium chloride (TTC in 0.1 M
phosphate buffer, pH 7.4) for 20 min at 37°C. The reaction of TTC
with viable parts of tissue produced a red region in the ventricle,
which is distinct from the pale necrotic tissue observed following
fixation in 10% formalin for 24 h at room temperature. The size of
the total left ventricle area and the infarcted area were measured
by planimetry of scanned slices using ImageJ version 1.42.
Statistical analysis
The data are presented as the mean ± standard error
of the mean of at least three independent experiments. All
statistical analyses were performed using with GraphPad Prism 5
(GraphPad Software, Inc., La Jolla, CA, USA). The differences
between two groups were analyzed using a Student's t-test, while
the differences among three or more groups were analyzed using
one-way analysis of variance, followed by Student-Newman-Keuls post
hoc test. P<0.05 was considered to indicate a statistically
significant difference.
Results
YB1 expression is upregulated during
H2O2-induced myocardial injury
To investigate whether YB1 is involved in
cardiomyocyte injury, H9c2 cells grown in vitro were treated
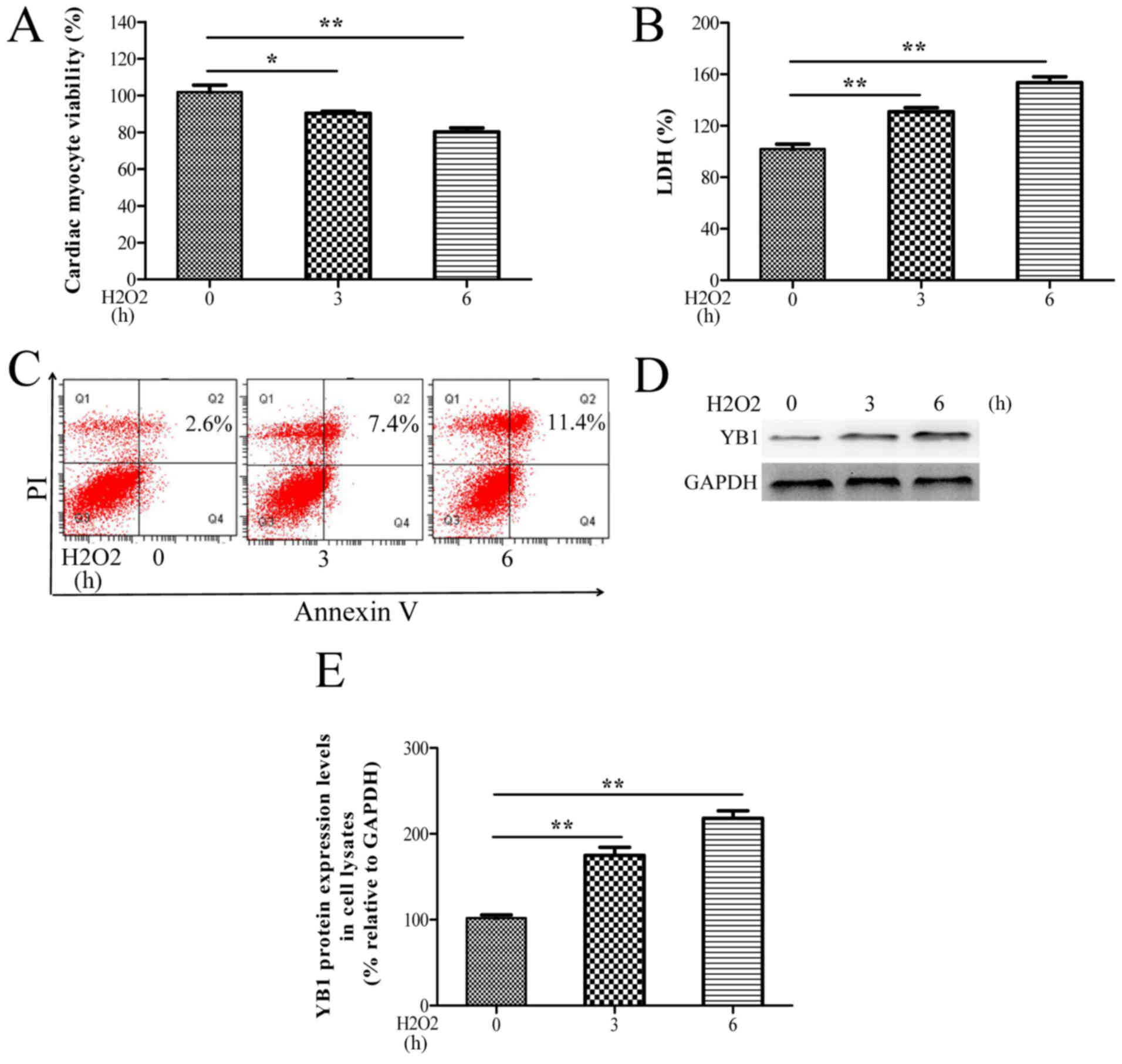
with H2O2. As observed in Fig. 1A-C, H2O2
treatment significantly decreased the cell viability, and increased
the LDH release and apoptosis rates of H9c2 cells in a time
dependent-manner. Moreover, YB1 protein levels progressively
increased during treatment with H2O2
(Fig. 1D and E). These in
vitro data suggested that H2O2-mediated
upregulation of YB1 may be associated with
H2O2-induced cardiomyocyte injury.
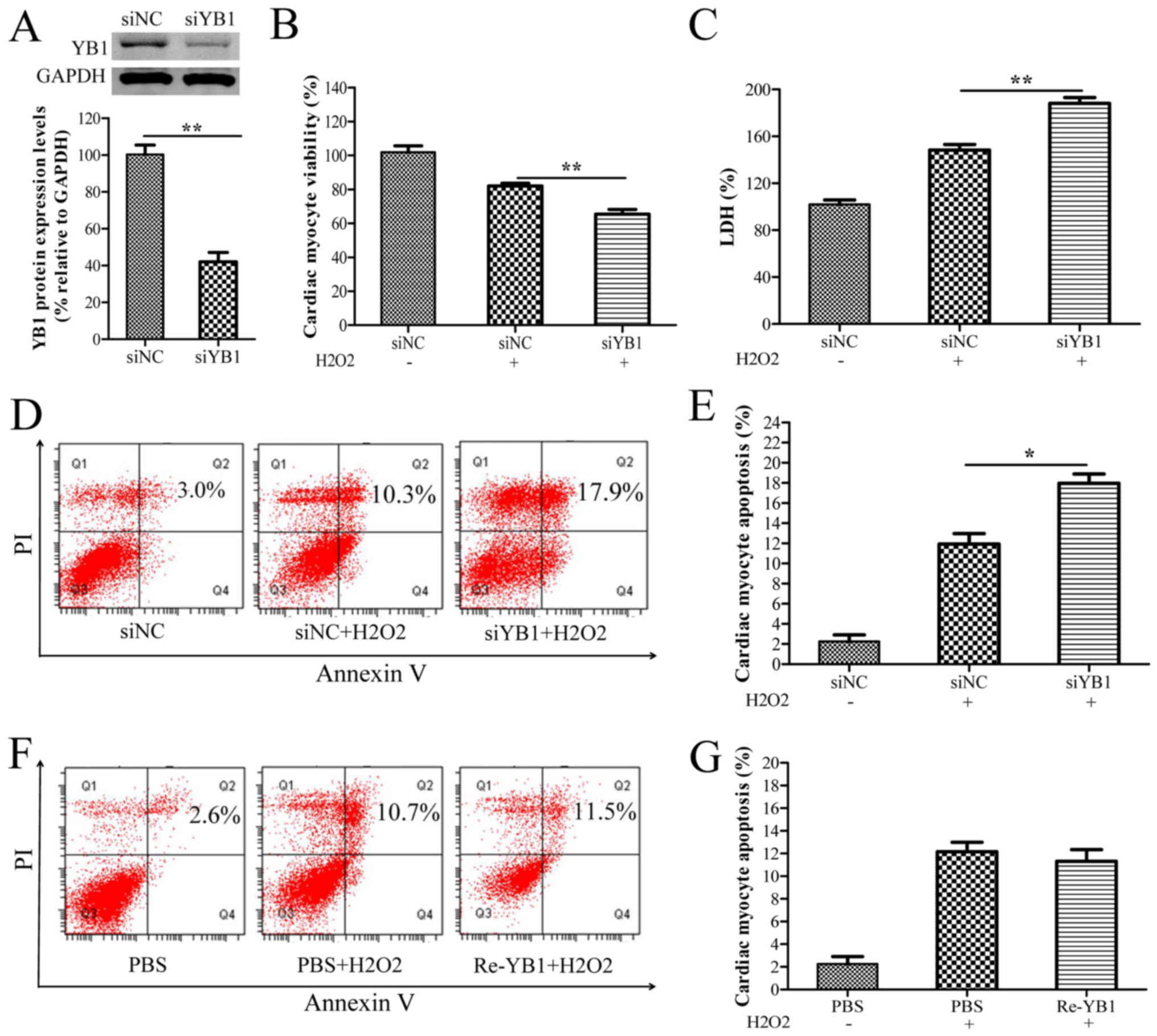
YB1 knockdown enhances
H2O2-induced myocardial injury
To further investigate the roles of YB1 in
H2O2-induced cardiomyocyte injury, YB1 was
knocked-down via siYB1 transfection in H9c2 cells. Western blot
analysis demonstrated that YB1 expression was significantly
decreased in siYB1-transfected cells compared with siNC-transfected
cells (Fig. 2A). Furthermore, YB1
knockdown notably decreased cell viability, while further
increasing the LDH release and apoptosis rates of H9c2 cells
following treatment with H2O2 (Fig. 2B-E), suggesting that YB1 may have
cardioprotective effects in H2O2-induced
cardiomyocyte injury. To further these observations, the effect of
extracellular YB1 treatment on H2O2-induced
cardiomyocyte injury was evaluated. As presented in Fig. 2F and G, the flow cytometry results
demonstrated that stimulation with recombinant YB1 protein did not
affect the apoptosis rates of H2O2-treated
H9c2 cells.
YB1 knockdown inhibits
H2O2-induced STAT3 phosphorylation
Since H2O2 treatment is known
to activate the p38, JNK, ERK and NF-κB signaling pathways, which
induce cell apoptosis (25,26),
it was investigated whether YB1 knockdown enhanced
H2O2-induced myocardial injury. Western blot
analysis demonstrated that the expression levels of phosphorylated
P38 (p-P38), p-JNK, p-ERK1/2 and p-P65 were increased in
H2O2-treated H9c2 cells, compared with those
in nontreated H9c2 cells (Fig.
3A). Moreover, the expression levels of p-P38, p-JNK, p-ERK1/2
and p-P65 in siYB1-transfected H9c2 cells were similar to those in
siNC-transfected cells, suggesting that YB1 knockdown did not
affect the activation of p38, JNK, ERK and NF-κB signaling pathways
following treatment with H2O2 (Fig. 3B and C).
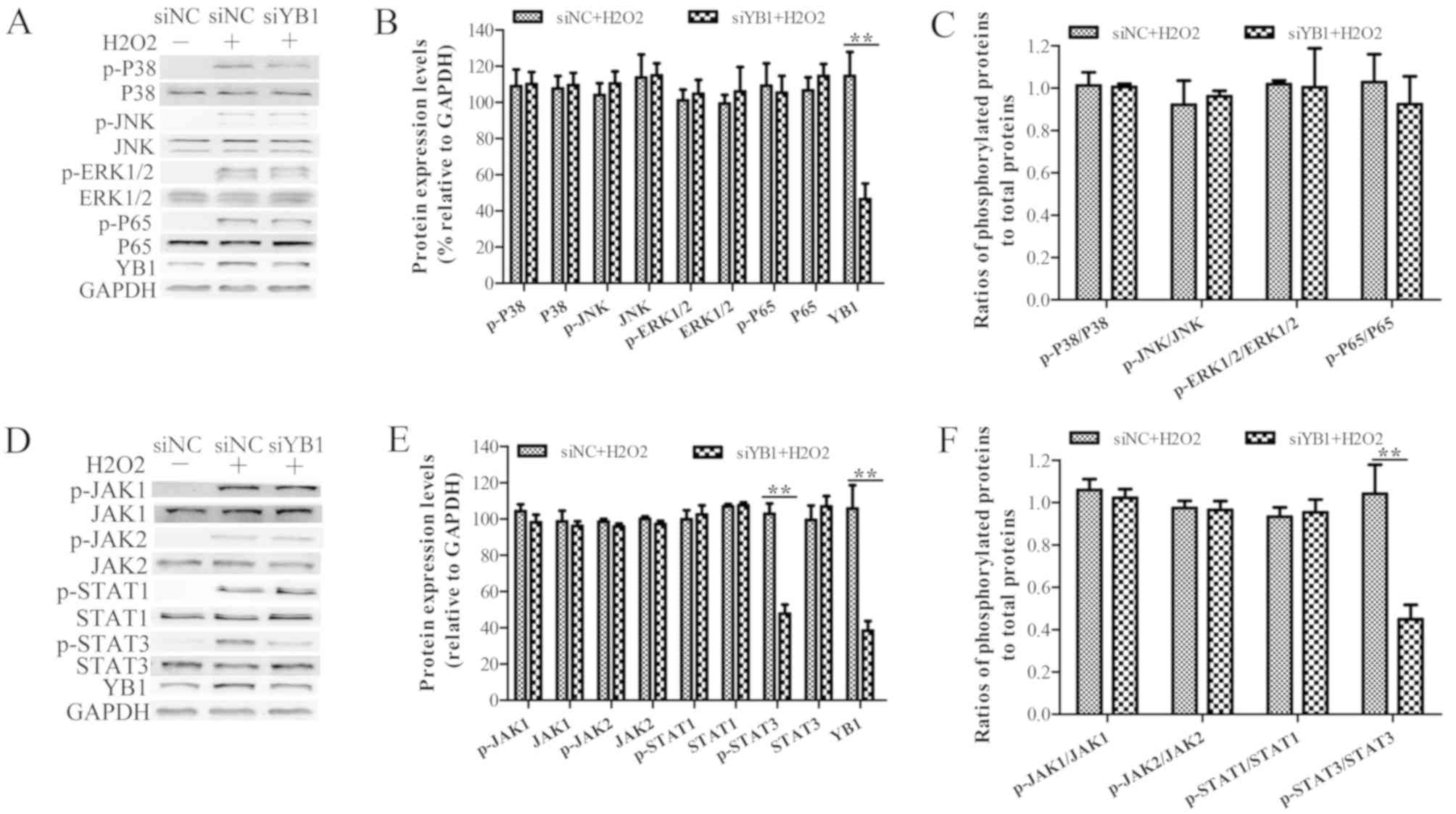 | Figure 3.YB1 knockdown inhibits
H2O2-induced phosphorylation of STAT3. H9c2
cells were transfected with siYB1 or siNC (as a control) for 24 h,
and treated with H2O2 for 6 h. (A) YB1,
p-P38, p-JNK, p-ERK1/2, p-P65, P38, JNK, ERK1/2 and P65 protein
expression levels were detected using western blotting. (B)
Relative expression levels of these proteins were calculated by
normalizing to those of GAPDH, respectively. (C) Ratios of
phosphorylated protein to total proteins after normalization to
GAPDH furthers the observation these proteins are not recruited
following treatment (D) YB1, p-JAK1, p-JAK2, p-STAT1, p-STAT3,
JAK1, JAK2, STAT1 and STAT3 protein expression levels were detected
using western blotting. (E) Relative expression levels of these
proteins were calculated by normalizing to those of GAPDH. (F)
Ratios of phosphorylated protein to total proteins after
normalization to GAPDH demonstrates that STAT3 phosphorylation
levels decrease in the absence of YB1 following treatment. Data are
presented as the mean ± standard error of the mean (n=3).
**P<0.01. YB1, Y-box protein 1; siYB1, small interfering RNA
against YB1; siNC, scrambled small interfering RNA; p-,
phosphorylated; JNK, c-Jun NH2-terminal kinase; ERK,
extracellular signal-regulated kinase; JAK, Janus kinase; STAT,
signal transducer and activator of transcription. |
The JAK-STAT signaling pathway has also been
reported to be involved in H2O2-induced
cardiomyocyte injury. Therefore, to investigate whether YB1
knockdown affected the JAK-STAT pathway and promoted
H2O2-induced myocardial injury, western
blotting was performed. Just as observed in the aforementioned
paragraph, treatment with H2O2 led to an
increase in the phosphorylation levels of JAK1, JAK2, STAT1 and
STAT3 (Fig. 3D). Furthermore, this
assay demonstrated that YB1 knockdown did not significantly affect
the levels of of p-JAK1, p-JAK2 and p-STAT1 following
H2O2 treatment (Fig. 3E and F). However, the protein
levels of p-STAT3 in siYB1-transfected cells were decreased
compared with those observed in siNC-transfected cells, suggesting
that YB1 knockdown may inhibit H2O2-induced
STAT3 phosphorylation.
YB1 interacts with and promotes PIAS3
mRNA decay to facilitate STAT3 phosphorylation
Further to being activated by JAK, STAT3 is also
negatively regulated by phosphatases, including SHP1, SHP2, SOCS1,
SOCS3 and PIAS3. To further investigate how YB1 knockdown inhibited
the H2O2-induced phosphorylation of STAT3,
the interactive association between YB1 and SHP1, SHP2, SOCS1,
SOCS3 and PIAS3 was evaluated using COIP and RIP assays (Fig. 4A and B). The results suggested that
YB1 chiefly interacted with PIAS3 mRNA. Little interaction was
observed between YB1 and SHP1 (protein and mRNA), SHP2 (protein and
mRNA), SOCS1 (protein and mRNA), SOCS3 (protein and mRNA) and PIAS3
protein.
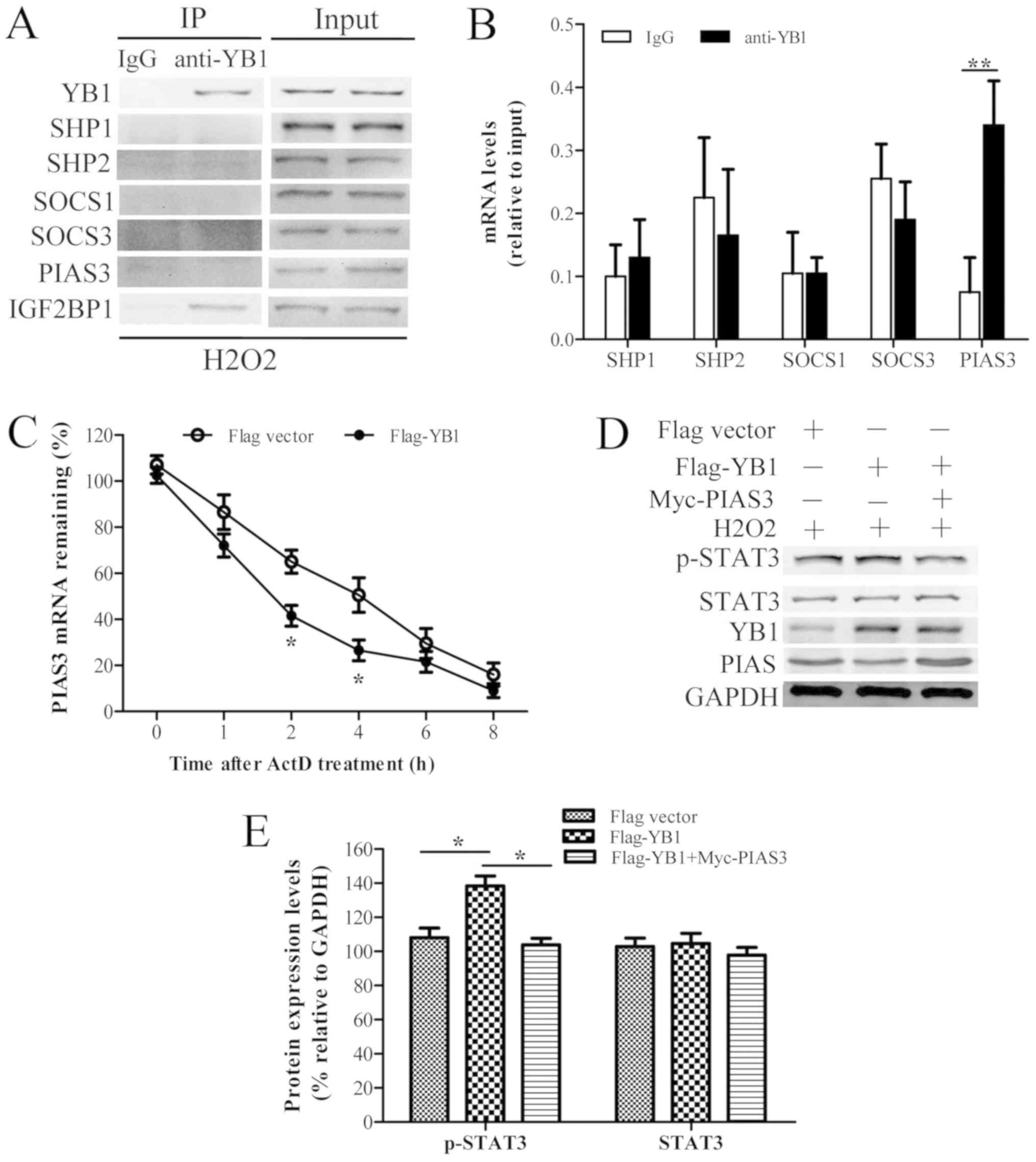 | Figure 4.YB1 interacts with PIAS3 and promotes
its mRNA decay, facilitating STAT3 phosphorylation. (A) H9c2 cells
were treated with H2O2 for 6 h and
immunoprecipitated with anti-YB1 antibody, with IgG as a control.
Following cross-linking with protein A/G agarose beads, the
immunoprecipitated complex was analyzed using western blotting.
IGF2BP1 was used as a positive control. (B) mRNAs of indicated
genes endogenously associated with YB1 in H9c2 cells were detected
by RNA-binding Protein Immunoprecipitation using IgG as a control.
(C) H9c2 cells were transfected with p-CMV-flag-vector (as the
control) or p-CMV-flag-YB1 for 24 h, and treated with ActD (10
µg/ml) for the indicated times. PIAS3 mRNA expression levels were
analyzed with reverse transcription-quantitative polymerase chain
reaction. (D) H9c2 cells were transfected with p-CMV-flag-vector or
p-CMV-flag-YB1 and p-CMV-Myc-PIAS3 for 24 h, and treated with
H2O2 for 6 h. YB1, p-STAT3, STAT3, PIAS3 and
GAPDH protein expression levels were detected using western
blotting. (E) Relative expression levels of these proteins were
calculated by normalizing to those of GAPDH. Data are presented as
the mean ± standard error of the mean (n=3). *P<0.05 and
**P<0.01. YB1, Y-box protein 1; STAT, signal transducer and
activator of transcription; PIAS3, protein inhibitor of activated
STAT 3; IgG, immunoglobulin G; IGF2BP1, insulin-like growth factor
2 mRNA-binding protein 1; ActD, actinomycin D; p-, phosphorylated;
SHP, Src homology region 2 domain-containing phosphatase; SOCS,
suppressor of cytokine signaling. |
Subsequently, the effects of YB1 on PIAS3 mRNA
stability were evaluated by RT-qPCR in YB1-overexpressing H9c2
cells treated with the transcriptional inhibitor ActD. The results
(Fig. 4C) demonstrated that the
amount of PIAS3 mRNA in YB1-overexpressing H9c2 cells was lower
compared with control cells during ActD exposure. Moreover, the
half-life of PIAS3 mRNA in YB1-overexpressing H9c2 cells was also
shorter compared with that of control cells. Therefore, YB1
overexpression may have decreased PIAS3 mRNA stability.
To further these observations, the effects of YB1
overexpression on the protein levels of PIAS3 and STAT3 in H9c2
cells treated with H2O2 were investigated. As
exhibited in Fig. 4D and E, YB1
overexpression in H9c2 cells treated with
H2O2 decreased PIAS3 and increased p-STAT3
protein levels without altering STAT3 total protein levels.
However, PIAS3 overexpression reversed the YB1 overexpression
phenotype by decreasing the phosphorylation levels of STAT3. Taken
together, these results indicated that YB1 may interact with and
promote PIAS3 mRNA decay, facilitating STAT3 phosphorylation in
H9c2 cells.
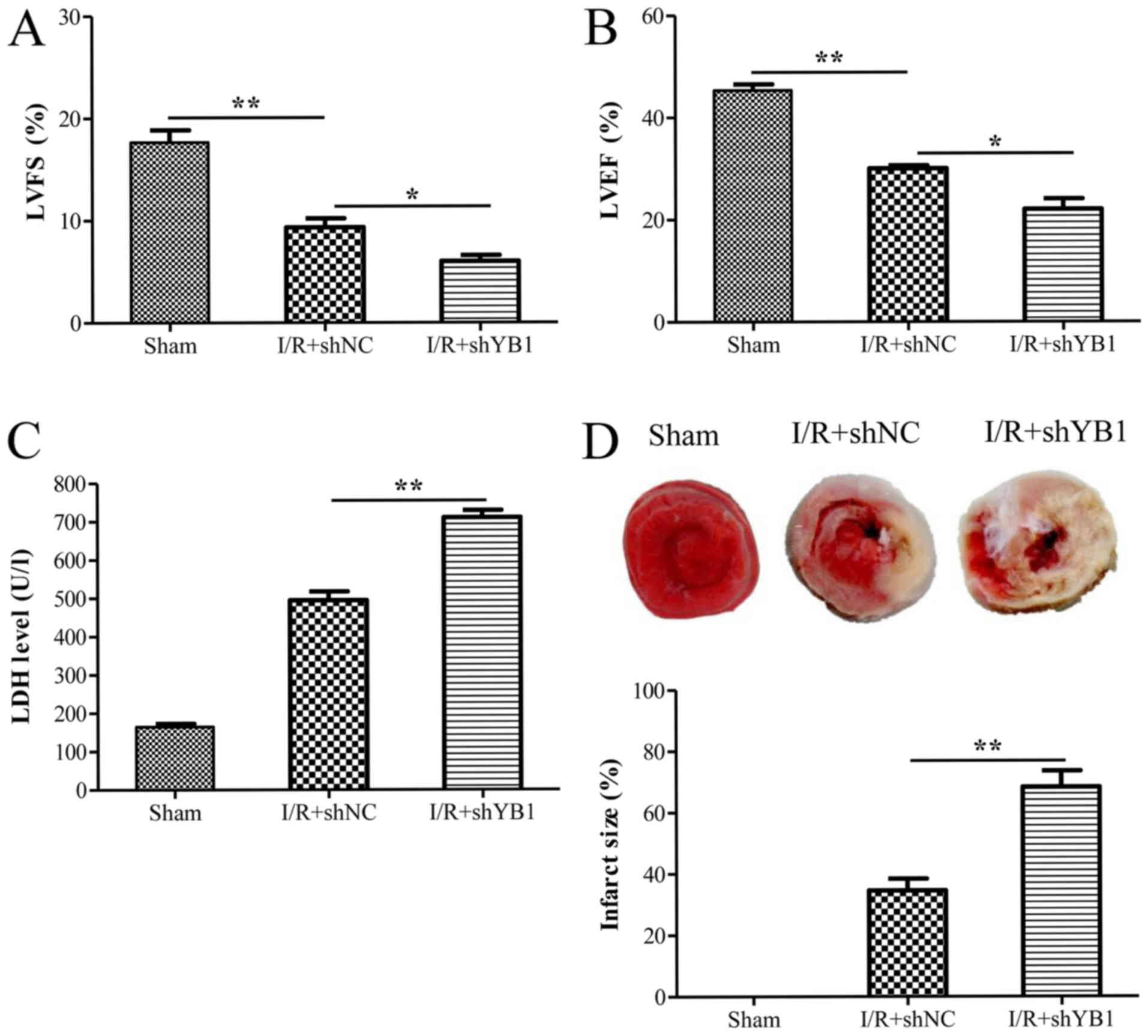
YB1 knockdown aggravates M-I/R injury
in vivo
An in vivo rat M-I/R model was employed to
investigate the effects of YB1 on M-I/R injury. Transthoracic
echocardiography and M-mode tracings were used to evaluate LVFS%
and LVEF% via echocardiographic measurements. As expected, I/R
injury significantly decreased LVFS% and LVEF% (Fig. 5A and B, respectively), compared
with those of the sham group. Moreover, animals injected with shYB1
displayed a significantly decreased LVFS% and LVEF% compared with
I/R-injured rats injected with shNC. Consistently, the LDH levels
in the I/R+shYB1 group were also significantly higher compared with
those of the I/R+shNC group (Fig.
5C). Furthermore, the infarct sizes in I/R+shYB1 group were
significantly increased compared with those in the I/R+shNC group
(Fig. 5D). These results indicated
that YB1 knockdown aggravated myocardial I/R injury in
vivo.
Discussion
Cardiomyocyte apoptosis during the I/R process is
responsible for multiple sequelae of myocardial infarction,
including congestive heart failure, cardiac rupture and ventricular
arrhythmia (27,28). In the present study, the
cardioprotective effect of YB1 in the prevention and control of
I/R-derived cardiac damage was evaluated in vivo and in
vitro. With this approach, it was demonstrated that YB1
inhibited cardiomyocyte apoptosis by promoting STAT3
phosphorylation.
Previous studies reported that YB1 expression is
increased in the regenerating heart following I/R injury, and the
increased expression levels of YB1 have been attributed to
myofibroblast infiltration, which is thought to take place 4–7 days
following I/R (29,30). The present study demonstrated low
expression of YB1 in cardiomyocytes, in contrast with the
previously reported data from myofibroblasts (27). However, in a rat M-I/R model,
pretreatment with lentivirus-shYB1, which caused YB1 knockdown in
heart cells, including cardiomyocytes and myofibroblasts,
aggravated myocardial infarction sizes, which seems inconsistent
with the work of Kamalov et al (29). Due to the positive role of YB1 on
the proliferation and migration of myofibroblasts in the infarct
regions (29), YB1 knockdown in
myofibroblasts may hinder the local scar formation, which
contributes to heart deterioration and heart failure. On the other
hand, the low expression of YB1 in cardiomyocytes may partly
sustain a higher tolerance to I/R induced apoptosis, since YB1
suppression in cardiomyocytes increased apoptosis following
I/R.
To dissect the mechanism of YB1-mediated STAT3
phosphorylation, certain proteins involved in regulating STAT3
phosphorylation were detected, and PIAS3 mRNA was determined to be
regulated by YB1. However, the present study did not evaluate in
detail the YB1 binding region on PIAS3 mRNA, or whether YB1
functions through its cold shock domain or other domains (31,32).
Future experiments should aim to investigate the role of YB1 in
cardiomyocyte proliferation, and whether ectopic expression of YB1
in cardiomyocytes, but not in myofibroblasts, may alleviate
I/R-induced cardiomyocyte apoptosis.
In conclusion, the present study demonstrated that
YB1 expression levels increased in H9c2 cardiomyocytes following
exposure to H2O2. Upregulation of YB1 may
have increased STAT3 phosphorylation by promoting PIAS3 mRNA
degradation. Moreover, lentivirus-mediated YB1 knockdown in a rat
I/R model aggravated infarct size and may have exacerbated the
possibility of heart failure. These results may have implications
in the diagnosis and treatment of a variety of heart diseases
associated with ROS damage, including cardiac hypertrophy, heart
failure, myocardial infarction and M-I/R injury. The potential
physiological roles of YB1 in other cardiac myocyte disease models
should be evaluated in future studies.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SW, FH, ZL, YH and NH performed the experiments. SW,
FH, ZL and XC analyzed the data. SW, FH, ZL, YH, NH and XC designed
the study and drafted the manuscript. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
All animal experiments and procedures were approved
by the Institutional Animal Care and Use Committee of the Medical
School of Ningbo University (Ningbo, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
YB1
|
Y-box protein 1
|
|
I/R
|
ischemia/reperfusion
|
|
M-I/R
|
myocardial ischemia-reperfusion
|
|
ROS
|
reactive oxygen species
|
|
JAK
|
Janus kinase
|
|
STAT
|
signal transducer and activator of
transcription
|
|
COIP
|
co-immunoprecipitation
|
|
RIP
|
RNA-binding protein
immunoprecipitation
|
|
LDH
|
Lactate dehydrogenase
|
|
LVIDd
|
LV internal diastolic diameter
|
|
LVIDs
|
LV internal systolic diameter
|
|
LVFS
|
LV percentage fractional
shortening
|
|
LVEF
|
LV ejection fraction
|
|
SHP1
|
Src homology region 2
domain-containing phosphatase 1
|
|
PIAS3
|
protein inhibitor of activated STAT
3
|
|
ActD
|
actinomycin D
|
|
MAPKs
|
mitogen-activated protein kinases
|
|
ERK1/2
|
extracellular signal-regulated
kinases
|
|
JNK
|
c-Jun NH2-terminal kinases
|
|
NF-κB
|
nuclear factor κB
|
References
|
1
|
Mehta D, Curwin J, Gomes JA and Fuster V:
Sudden death in coronary artery disease: Acute ischemia versus
myocardial substrate. Circulation. 96:3215–3223. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Verma S, Fedak PW, Weisel RD, Butany J,
Rao V, Maitland A, Li RK, Dhillon B and Yau TM: Fundamentals of
reperfusion injury for the clinical cardiologist. Circulation.
105:2332–2336. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Qu S, Zhu H, Wei X, Zhang C, Jiang L, Liu
Y, Luo Q and Xiao X: Oxidative stress-mediated up-regulation of
myocardial ischemic preconditioning up-regulated protein 1 gene
expression in H9c2 cardiomyocytes is regulated by cyclic
AMP-response element binding protein. Free Radic Biol Med.
49:580–586. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Misra MK, Sarwat M, Bhakuni P, Tuteja R
and Tuteja N: Oxidative stress and ischemic myocardial syndromes.
Med Sci Monit. 15:RA209–RA219. 2009.PubMed/NCBI
|
|
5
|
Zweier JL and Talukder MA: The role of
oxidants and free radicals in reperfusion injury. Cardiovasc Res.
70:181–190. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chang H, Sheng JJ, Zhang L, Yue ZJ, Jiao
B, Li JS and Yu ZB: ROS-induced nuclear translocation of calpain-2
facilitates cardiomyocyte apoptosis in tail-suspended rats. J Cell
Biochem. 116:2258–2269. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zheng A, Cao L, Qin S, Chen Y, Li Y and
Zhang D: Exenatide regulates substrate preferences through the p38γ
MAPK pathway after ischaemia/reperfusion injury in a rat heart.
Heart Lung Circ. 26:404–412. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Song ZF, Ji XP, Li XX, Wang SJ, Wang SH
and Zhang Y: Inhibition of the activity of poly (ADP-ribose)
polymerase reduces heart ischaemia/reperfusion injury via
suppressing JNK-mediated AIF translocation. J Cell Mol Med.
12:1220–1228. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Abas L, Bogoyevitch MA and Guppy M:
Mitochondrial ATP production is necessary for activation of the
extracellular-signal-regulated kinases during ischaemia/reperfusion
in rat myocyte-derived H9c2 cells. Biochem J. 349:119–126. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Guo J, Jie W, Kuang D, Ni J, Chen D, Ao Q
and Wang G: Ischaemia/reperfusion induced cardiac stem cell homing
to the injured myocardium by stimulating stem cell factor
expression via NF-kappaB pathway. Int J Exp Pathol. 90:355–364.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kunisada K, Tone E, Fujio Y, Matsui H,
Yamauchi-Takihara K and Kishimoto T: Activation of gp130 transduces
hypertrophic signals via STAT3 in cardiac myocytes. Circulation.
98:346–352. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
O'Sullivan KE, Breen EP, Gallagher HC,
Buggy DJ and Hurley JP: Understanding STAT3 signaling in cardiac
ischemia. Basic Res Cardiol. 111:272016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang J, Zhang J, Yu P, Chen M, Peng Q,
Wang Z and Dong N: Remote ischaemic preconditioning and sevoflurane
postconditioning synergistically protect rats from myocardial
injury induced by ischemia and reperfusion partly via inhibition
TLR4/MyD88/NF-κB signaling pathway. Cell Physiol Biochem. 41:22–32.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li J, Xiang X, Gong X, Shi Y, Yang J and
Xu Z: Cilostazol protects mice against myocardium
ischemic/reperfusion injury by activating a PPARγ/JAK2/STAT3
pathway. Biomed Pharmacother. 94:995–1001. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang C, Deng Y, Lei Y, Zhao J, Wei W and
Li Y: Effects of selenium on myocardial apoptosis by modifying the
activity of mitochondrial STAT3 and regulating potassium channel
expression. Exp Ther Med. 14:2201–2205. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
En-Nia A, Yilmaz E, Klinge U, Lovett DH,
Stefanidis I and Mertens PR: Transcription factor YB-1 mediates DNA
polymerase alpha gene expression. J Biol Chem. 280:7702–7711. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Raffetseder U, Frye B, Rauen T, Jürchott
K, Royer HD, Jansen PL and Mertens PR: Splicing factor SRp30c
interaction with Y-box protein-1 confers nuclear YB-1 shuttling and
alternative splice site selection. J Biol Chem. 278:18241–18248.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen CY, Gherzi R, Andersen JS, Gaietta G,
Jürchott K, Royer HD, Mann M and Karin M: Nucleolin and YB-1 are
required for JNK-mediated interleukin-2 mRNA stabilization during
T-cell activation. Genes Dev. 14:1236–1248. 2000.PubMed/NCBI
|
|
19
|
Coles LS, Lambrusco L, Burrows J, Hunter
J, Diamond P, Bert AG, Vadas MA and Goodall GJ: Phosphorylation of
cold shock domain/Y-box proteins by ERK2 and GSK3beta and
repression of the human VEGF promoter. FEBS Lett. 579:5372–5378.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Roy S, Khanna S, Rink T, Radtke J,
Williams WT, Biswas S, Schnitt R, Strauch AR and Sen CK:
P21waf1/cip1/sdi1 as a central regulator of inducible smooth muscle
actin expression and differentiation of cardiac fibroblasts to
myofibroblasts. Mol Biol Cell. 18:4837–4846. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Eliseeva IA, Kim ER, Guryanov SG,
Ovchinnikov LP and Lyabin DN: Y-box-binding protein 1 (YB-1) and
its functions. Biochemistry (Mosc). 76:1402–1433. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Azizi Y, Faghihi M, Imani A, Roghani M,
Zekri A, Mobasheri MB, Rastgar T and Moghimian M: Post-infarct
treatment with [Pyr(1)]apelin-13 improves myocardial function by
increasing neovascularization and overexpression of angiogenic
growth factors in rats. Eur J Pharmacol. 761:101–108. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Imani A, Faghihi M, Sadr SS, Niaraki SS
and Alizadeh AM: Noradrenaline protects in vivo rat heart against
infarction and ventricular arrhythmias via nitric oxide and
reactive oxygen species. J Surg Res. 169:9–15. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang DK and Kim SJ: Cucurbitacin I
Protects H9c2 Cardiomyoblasts against
H2O2-induced oxidative stress via protection
of mitochondrial dysfunction. Oxid Med Cell Longev.
2018:30163822018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li Y, Liu YJ, Lv G, Zhang DL, Zhang L and
Li D: Propofol protects against hydrogen peroxide-induced apoptosis
in cardiac H9c2 cells is associated with the NF-κB activation and
PUMA expression. Eur Rev Med Pharmacol Sci. 18:1517–1524.
2014.PubMed/NCBI
|
|
27
|
Vaduganathan M, Samman Tahhan A, Greene
SJ, Okafor M, Kumar S and Butler J: Globalization of heart failure
clinical trials: A systematic review of 305 trials conducted over
16 years. Eur J Heart Fail. 20:1068–1071. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Allida SM, Inglis SC, Davidson PM, Lal S,
Hayward CS and Newton PJ: Thirst in chronic heart failure: A
review. J Clin Nurs. 24:916–926. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kamalov G, Varma BR, Lu L, Sun Y, Weber KT
and Guntaka RV: Expression of the multifunctional Y-box protein,
YB-1, in myofibroblasts of the infarcted rat heart. Biochem Biophys
Res Commun. 334:239–244. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kohno K, Izumi H, Uchiumi T, Ashizuka M
and Kuwano M: The pleiotropic functions of the Y-box-binding
protein, YB-1. Bioessays. 25:691–998. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kljashtorny V, Nikonov S, Ovchinnikov L,
Lyabin D, Vodovar N, Curmi P and Manivet P: The cold shock domain
of YB-1 Segregates RNA from DNA by Non-Bonded Interactions. PLoS
One. 10:e01303182015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lu ZH, Books JT and Ley TJ: Cold shock
domain family members YB-1 and MSY4 share essential functions
during murine embryogenesis. Mol Cell Biol. 26:8410–8417. 2006.
View Article : Google Scholar : PubMed/NCBI
|