Introduction
Pulmonary hypertension (PH) is a physiological
pathological disorder with several clinical manifestations, which
finally leads to cardiovascular and respiratory diseases (1). According to several data, the current
worldwide prevalence of PH is ~97/1,000,000 individuals, and the
mortality rate 5–10/100,000 (2,3). PH
is a pan-vasculopathy involving several cell types, including
endothelial cells, smooth muscle cells (SMCs) and fibroblasts, all
of which constitute vascular cells (4). A wide spectrum of genetic factors and
environmental stimuli, including hypoxia, are the pathological
factors of PH. Pulmonary artery (PA) SMCs (PASMCs), the major
component of the vasculature, serve a key role in the response to
hypoxia, and the dysregulation of their functions is closely
associated with the occurrence and development of PH (5). Recently, several studies demonstrated
that the inhibited apoptosis and enhanced proliferation of PASMCs,
induced by chronic exposure to hypoxia, were major contributors to
the development of hypoxic PH (HPH) (6). However, the molecular mechanisms of
the hypoxia-induced dysfunction of PASMCs remain unclear.
MicroRNAs (miRNAs) are small endogenous noncoding
RNAs and they serve an important role in regulating gene expression
by targeting the 3′-untranslated region (3′-UTR) of mRNA to repress
its translation or accelerate its degradation (7,8).
miRNAs have been identified to serve important roles in diverse
biological processes, including cellular development,
differentiation and proliferation by several previous studies
(8,9). Several cardiovascular diseases,
including PH, exhibit a decreased miRNA expression, resulting in
disease progression (10–12). However, the molecular role of
miRNAs in these pathologies has not yet been fully elucidated.
Transforming growth factor-β1 (TGF-β1), which is a
multifunctional cytokine, serves an important role in regulating
cell differentiation, proliferation and extracellular matrix
deposition, which is directly associated with the occurrence and
development of pulmonary hypertension (13–15).
The TGF-β1 pathway signals are mediated by specific type I and II
serine/threonine kinase receptors. Activin receptor-like kinase-1
(ALK1) is a TGF-β-type I receptor that transmits signals via the
ALK1/Smad1/5 pathways (16).
Although several studies have suggested that ALK1 is expressed
primarily in endothelial cells in the vasculature (13,17–19),
ALK1 mRNA and protein expression in PASMCs in vitro and in
mouse pup lungs in vivo were also detected in other studies
(20,21). miRNA expression has been
demonstrated to be associated with the activation of the TGF-β1
pathway (22); however, the
association between miRNA and TGF-β1 in the pathogenesis of HPH has
not yet been fully elucidated.
In the present study, the role of miR-98 in HPH was
examined. It was identified that the levels of miR-98 expression
were decreased in the lung tissues of HPH rat models and rat PASMCs
under hypoxia, as compared with those of the controls. Furthermore,
the downregulation of miR-98 was involved in the hypoxia-induced
proliferation and apoptosis of PASMCs. In addition, it was
demonstrated that ALK1 was the direct target of miR-98. The
conclusions of the present study provided novel insights into the
pathogenesis of HPH and potential targets for its clinical
diagnosis and treatment.
Materials and methods
HPH rat model and lung tissue
preparation
Adult male Wistar rats (weighing 200±10 g; age, ~8
weeks) were obtained from the Animal Experimental Centre of Nanjing
Medical University (Nanjing, China). A total of ~60 rats were used
in the experimental model, and were fed with standard rat chow and
allowed water ad libitum. The animals were maintained under
a 12:12 light: dark cycle. The 60 rats were distributed into five
groups: The normoxia group (10 rats); hypoxia group (10 rats);
normoxia control (N-control, 10 rats); hypoxia control (H-control,
15 rats); and hypoxia + miR-98 agomir, (H + miR-98 agomir, 15
rats). miR-98 agomir (Guangzhou RiboBio Co., Ltd.; sequence,
5′-UGAGGUAGUAAGUUGUAUUGUU-3′) was used to mimic the overexpression
of miR-98. miR-98 agomir (250 µg/kg at the volume of 100 µl) was
injected into the tail vein. For hypoxic exposure, rats were placed
into a normobaric hypoxic chamber (percentage of inspired
O2=10%) for 8 h/day. For the control groups, the rats
were maintained in a similar normoxic chamber for 3 weeks, as
described previously (23). Right
ventricular systolic pressure (RVSP) was measured to confirm the
successful establishment of the HPH rat model. At the end of the 3
week exposure period, each rat was treated with an intraperitoneal
(i.p.) injection of pentobarbital (120 mg/kg). The thorax was then
opened and the heart and lungs removed and placed onto a flat
plate. The study was approved by the Ethical Committee of Xuzhou
Medical University (Xuzhou, China).
Cell culture
PA segments were collected from a group of 5 rats
not included in the HPH rat model immediately following sacrifice.
The samples were then minced into small fragments using ophthalmic
scissors, followed by digesting with 0.25% trypsin (Thermo Fisher
Scientific, Inc.) at 37°C for 40 min. Next, 0.2% collagenase (Merck
KGaA) was added to treat the fragments at 37°C for an additional 4
h. Cell suspension was filtered and dissociated with a 40 µm cell
strainer (BD Biosciences), followed by centrifugation at 850 × g
for 10 min at 4°C. Rat PASMCs were incubated in Dulbecco's modified
Eagle's medium (DMEM; Thermo Fisher Scientific, Inc.) supplied with
10% fetal bovine serum (Thermo Fisher Scientific, Inc.), 100 U/ml
penicillin, and 100 µg/ml streptomycin at 37°C in a humidified
incubator. The cell line 293 was obtained from the Cell Bank of
Type Culture Collection of Chinese Academy of Science and
maintained in DMEM with 10% FBS at 37°C in a humidified
incubator.
Prediction of miR-98 target genes
Genes targeted by miR-98 were predicted using PicTar
(version 2; http://pictar.bio.nyu.edu) (24), TargetScan (version 5.1; http://www.targetscan.org/) combined with miRanda
(version 3.3a; http://www.microrna.org/) as previously described
(25).
Hypoxia treatment of PASMCs
PAMSCs at ~60% confluence were exposed to difference
oxygen concentrations (0, 3 and 5%) by connecting the incubator
with a chamber that was equilibrated with a water-saturated gas
mixture of (0, 3 and 5%) O2, 5% CO2 and (95,
92 and 90%) N2 at 37°C for 24–48 h, as described
previously (26).
Transfection
A total of 24 h prior to transfection, suspended rat
PASMCs at 50% confluence were seeded onto 12-well plates. The
miR-98 mimics (5′-UGAGGUAGUAAGUUGUAUUGUU-3′), inhibitors
(5′-AACAAUACAACUUACUACCUC-3′) or negative control (NC) sequence,
(5′-GUGUAACACGUCUAUACGCCCA-3) were purchased from Guangzhou RiboBio
Co., Ltd. Mimics or inhibitors were transfected into PASMCs with
Lipofectamine® 3000 reagent (Thermo Fisher Scientific,
Inc.). The final concentration of miRNAs was 60 nM. To overexpress
ALK1 in rat primary PASMCs, coding sequences of ALK1 were cloned
into a lentiviral vector (GeneCopoeia, Inc.) (lenti-ALK1) and
packaged into lentiviruses particles by Obio Technology (Shanghai)
Co., Ltd. The transfected cells (60% confluence, 6 h after
transfection) were incubated with lentiviruses for ~6 h at 37°C,
then the media was replaced with normal media. After 24, 48 or 72
h, the cells were harvested for subsequent experiments.
RNA extraction and reverse
transcription quantitative polymerase chain reaction (RT-qPCR)
TRIzol® reagent (Thermo Fisher
Scientific, Inc.) was used to extract total RNA from lung tissues
or PASMCs according to the manufacturer's protocol. RNAs (1 µg) was
used for cDNA reverse transcription using an M-MLV kit (Takara
Biotechnology Co., Ltd.). RT-qPCR was performed for the detection
of ALK1 and ALK4 gene expression. The forward and reverse primers
were as follows: Rat ALK1 forward, 5′-ACCCAAACTCCTTCGGAGGAG-3′; rat
ALK1 reverse, 5′-CGCTGCTTCTCCTGCCTTC-3′; rat ALK4 forward,
5′-TGACCTGAGGGTGCCCAGTG-3′; rat ALK4 reverse,
5′-TGAGGGGTCCTCCATGTCCAG-3′; β-actin forward,
5′-GAGTACGATGAGTCCGGCCCC-3′; and β-actin reverse,
5′-GCAGCTCAGTAACAGTCCGCCT-3′. β-actin was used as an internal
control. The relative expression levels of ALK1 and ALK4 were
quantified using the Applied Biosystems 7500 Real-Time PCR System
(Thermo Fisher Scientific, Inc.) with Power SYBR1 Green PCR Master
Mix (Thermo Fisher Scientific, Inc.). The thermocycling conditions
were as follows: Initial denaturation at 95°C for 10 min, followed
by 40 cycles at 95°C for 15 sec, 60°C for 30 sec and 72°C for 30
sec. The relative mRNA expression levels were analyzed through
using the expressed relative to the threshold cycle values (ΔCq),
and then converted to fold changes using the 2−ΔΔCq
method (27).
The expression levels of miR-98 were analyzed and
quantified separately using a stem-loop RT-PCR assay, as described
previously (28,29). RNA was extracted from tissues or
cells using the TRIzol® reagent (Thermo Fisher
Scientific, Inc.). Total RNA (1 µg) was used for cDNA reverse
transcription using a Mir-X™ miRNA First Strand Synthesis Kit (cat.
no. 638315; Takara Biotechnology Co., Ltd.). For the RT reaction,
the samples were incubated for 1 h at 37°C, and the reaction was
terminated at 85°C for 5 min. qPCR was performed for the detection
of the expression levels of miR-98. The primers used for qPCR
analysis were the following: miR-98 forward,
5′-TGAGGTAGTAAGTTGTATTGTT-3′; qPCR miR-98 reverse,
5′-GCTGTCAACGATACGCTACGTAACG-3′; qPCR 5S forward,
5′-GTCTACGGCCATACCACCCIGAAC; and qPCR 5S reverse,
5′-CTGTCAACGATACGCTACGTAACG. RNA input was normalized to the level
of rat 5S rRNA. RT-qPCR was performed using SYBR1 Green PCR Master
Mix (Thermo Fisher Scientific, Inc.) on a 7500 system (Thermo
Fisher Scientific, Inc.). The thermocycling conditions were as
follows: Initial denaturation at 95°C for 10 min and 40 cycles at
95°C for 15 sec, 60°C for 30 sec and 72°C for 30 sec. The
expression level of miR-98 were normalized to the expression levels
of 5S rRNA, and fold changes were calculated by relative
quantification (2−ΔΔCq) (27).
Cell proliferation assay
A Cell Counting Kit-8 (CCK-8; Nanjing KeyGen
Biotech, Co., Ltd.) assay was used to analyze cell proliferation,
as described previously (26).
PASMCs were seeded in 96-well plates at a density of
3×103 cells per well and cultured at 37°C in 5%
CO2 for 24 h. Following treatment with 0, 24 and 48 h,
CCK-8 solution (10% combined with DMEM) was added to each well and
incubated for an additional 2 h. Cell proliferation was detected
and analyzed by scanning with a microplate reader (Bio-Rad
Laboratories, Inc.) at 450 nm. Each experiment was performed in
triplicate.
Cell apoptosis assay
Apoptosis was analyzed using a flow cytometer (BD
Biosciences). Briefly, an annexin-V fluorescein isothiocyanate and
propidium iodide double-stain assay was performed in accordance
with the manufacturer's protocol (FITC Annexin V Apoptosis
Detection Kit I; BD Biosciences). The results were analyzed using
FlowJo software (version 7.5.5; Tree Star, Inc.). Each experiment
was performed in triplicate.
Measurement of RVSP
A total of 10 rats from each group was used to
measure RVSP by right heart catheterization, as previously
described (30). Briefly, rats
were anesthetized by an i.p. injection of 35 mg/kg pentobarbital
sodium. An additional dose of pentobarbital sodium (17.5 mg/kg) was
administered when further anesthesia was necessary. A 1.2 French
Pressure Catheter (Transonic Systems, Inc.) was connected to the
Scisense FA-404 recorder (Transonic Systems, Inc.). Following
exposure of the right jugular vein, the catheter was inserted into
the vein, advanced into the superior vena cava, and finally into
the RV. RVSP was continuously recorded for 45 min.
Luciferase reporter assay
The 3′-UTR-Luc reporter of ALK1 was constructed by
the ligation of ALK1 3′-UTR PCR product into the Xhol and
BamH I sites of the pGL3 basic vector (Promega Corporation).
The mutant reporter was generated from pGL3-wild type (WT)-ALK1
3′-UTR-Luc by replacing the binding site of miR-98 with restriction
enzyme cutting site of BamH I: CGGATCCG. For the luciferase
reporter assay, cells were cultured in 96-well plates and then
transfected with WT or mutant luciferase reporter plasmids, and
then miR-98 mimics with Lipofectamine® 3000 reagent
(Thermo Fisher Scientific, Inc.). After incubation for 24 h,
luciferase activity of each well was measured using a
Dual-Luciferase Reporter Assay System (Promega Corporation)
according to the protocol of the manufacturer (Promega
Corporation). Renilla luciferase activity was utilized as an
internal control.
Western blot analysis
For western blot analysis, total protein was
extracted using radioimmunoprecipitation assay lysis buffer
(Beyotime Institute of Biotechnology) and quantified using the
Bradford method. A total of ~30 µg total protein from each sample
was loaded and separated by 10% SDS-PAGE and then transferred onto
polyvinylidene fluoride membranes (EMD Millipore). Following
blocking with 5% non-fat milk in PBST buffer at 37°C for 1 h,
membranes were incubated with the primary antibody at 4°C
overnight. Subsequently, the membranes were incubated with
horseradish peroxidase (HRP)-conjugated secondary antibodies (Santa
Cruz Biotechnology, Inc.; cat. no. sc-2004; 1:2,000 dilution) for 1
h at 37°C and finally detected using an ECL substrate kit (Tanon
Science and Technology Co., Ltd.). Chemiluminescence was detected
using the ChemiDoc XRS+ (Bio-Rad Laboratories, Inc.).
The secondary antibodies conjugated with HRP (cat. no. sc-2004;
1:2,000 dilution) and primary antibodies against β-actin (cat. no.
sc-70319; 1:1,000 dilution) were purchased from Santa Cruz
Biotechnology, Inc., ALK1 (cat. no. ab108207; 1:1,000 dilution),
cleaved caspase 3 (cat. no. ab13847; 1:1,000 dilution),
proliferating cell nuclear antigen (PCNA; cat. no. ab92552; 1:1,000
dilution), smad1 (cat. no. ab66737; 1:1,000 dilution) and P-smad1
(cat. no. ab73211; 1:1,000 dilution) primary antibodies were
purchased from Abcam. The densitometric was analyzed using ImageJ
(version 1.51J8; National Institutes of Health).
Histology
After the rats were anesthetized, the lung tissues
were obtained immediately, then the tissues were sliced into tissue
blocks, and immersed in 4% paraformaldehyde for overnight fixation
at 37°C. Fixed tissues were then dehydrated in ascending alcohol
series, cleared, and embedded in paraffin wax. The tissues were cut
into 4-µm thick sections using a Leica slicer (Leica Microsystems,
Inc.) and stained with hematoxylin and eosin (H&E) using a
standard method as described previously (31). Additionally, Masson staining and
PCNA immunohistochemistry were used to study the effect of the
miR-98 agomir on the development of HPH, as described previously
(32). For immunohistochemistry,
the slides were boiled (~100°C) in citrate antigen retrieval
solution (Beyotime Institute of Biotechnology) for 1.5 min and
cooled at room temperature for 30 min. After a 15-min incubation in
3% hydrogen peroxide, sections were blocked with 10% goat serum
(Beyotime Institute of Biotechnology; cat. no. C0265) for 60 min at
37°C and then incubated with primary antibody against PCNA (cat.
no. ab92552; Abcam; 1:100 dilution) overnight at 4°C. After washing
with PBST buffer for three times, sections were incubated with
HRP-conjugated anti-rabbit (cat. no. ZDR-5306; ZSBio; OriGene
Technologies, Inc.; 1:500 dilution) for 60 min at 37°C.
Localization of peroxidase conjugates was determined using a
3′-diaminobenzidine kit (cat. no. ZLI-9018; ZSBio; OriGene
Technologies, Inc.) as a chromogen and H&E for counterstaining.
Samples were photographed using a light microscope (magnification,
×400).
Statistical analysis
All data are presented as the mean ± standard
deviation. GraphPad Prism 6.0 software (GraphPad Software, Inc.)
was used for statistical analysis. Unpaired two-tailed Student's
t-test and one-way ANOVA followed by Tukey-Kramer post-hoc test
were used to determine statistical significance. P<0.05 was
considered to indicate a statistically significant difference.
Results
miR98 is downregulated by hypoxia in
an HPH rat model and PASMCs
The HPH rat model was established by subjecting the
rats to a hypoxic assault with 10% O2 for 3 weeks, as
described previously (23), and PH
indicators, including the morphology of pulmonary vessels and the
RVSP, were detected. H&E staining indicated that the pulmonary
vascular smooth muscle layer was significantly thickened in the
hypoxic-induced group compared with the normoxic group (Fig. 1A), and the vascular lumen was
significantly narrowed. Furthermore, the RVSP was increased
significantly in the hypoxic-induced rats (Fig. 1B). These results demonstrated that
the HPH rat model had been successfully established. In order to
examine the expression levels of miR-98 in the HPH rat model, the
total lung tissue RNA was extracted and examined by RT-qPCR. As
indicated in Fig. 1C, the level of
miR-98 was significantly decreased in the HPH model compared with
the control. To investigate the effect of hypoxia on the expression
of miR-98 in PASMCs, the optimal hypoxic stimulation concentration
was first selected by determining the O2 concentration
gradient for cell induction (data not shown). As demonstrated in
Fig. 1D, the expression of miR-98
in rat primary PASMCs was decreased as the O2
concentration gradually decreased. The expression of miR-98 was
significantly downregulated after 24 h of hypoxia stimulation, and
further downregulated after 48 h (Fig.
1D). These results revealed that the downregulation of miR-98
may be associated with the hypoxia-induced development of pulmonary
hypertension.
Effect of miR-98 on PASMC
proliferation and apoptosis
PASMC proliferation is a well-known mechanism of
vascular remodeling in HPH. In the present study, a CCK-8 assay was
used to assess the effect of miR-98 on PASMC proliferation. As
demonstrated in Fig. 2A and B,
PASMC proliferation was significantly enhanced following the
transfection of cells exposed to hypoxia for 24 and 48 h with
anti-miR-98 inhibitors. Conversely, transfection with miR-98 mimics
significantly inhibited the hypoxia-induced enhancement of rat
primary PASMC proliferation. Furthermore, a cell proliferation
marker, PCNA, was analyzed. The protein level of PCNA was markedly
downregulated in cultured miR-98 mimic-transfected cells and
upregulated in anti-miR-98 inhibitor-transfected cells (Fig. 2C). The RT-qPCR results suggested a
high efficiency of miR-98 inhibitors and mimics in PASMCs (Fig. 2H). These data suggested that the
downregulation of miR-98, which resulted in PASMC proliferation,
was induced by hypoxia and accelerated vascular remodeling in
HPH.
Furthermore, to explore the molecular mechanism
underlying the modulation of miR-98 in PASMC proliferation, PASMC
apoptosis was analyzed by flow cytometry, and the results indicated
that the overexpression of miR-98 significantly induced cell
apoptosis, whereas transfection with anti-miR-98 inhibitors
prevented hypoxia-induced apoptosis (Fig. 2D and E). As caspase 3 serves a key
role in the process of apoptosis, its cleaved form is often used as
an indicator of apoptosis levels. In the present study, the protein
level of cleaved caspase 3 in PASMCs transfected with miR-98 mimics
or inhibitors and then exposed to hypoxia was examined. It was
identified that the protein level of cleaved caspase 3 was
consistently decreased in cells transfected with miR-98 inhibitors
(Fig. 2F and G). By contrast,
transfection of cells with miR-98 mimics increased cleaved caspase
3 activity. These results suggested that miR-98 upregulation leads
to PASMC apoptosis.
ALK1 is a direct target of miR-98
In order to elucidate the molecular mechanism
through which miR-98 affects HPH, TargetScan and MiRanda were used
to identify putative binding sites for miR-98. A number of mRNAs
may be targets of miR-98, including the ALK gene family, which is
associated with the occurrence and progression of HPH (13). The bioinformatics analysis of the
potential targets of miR-98 indicated that the ALK family genes,
including ALK1 and ALK4, may be targets of miR-98 (Fig. 3A). Next, the expression of these
two genes, ALK1 and ALK4, was analyzed in HPH lung tissue. As
demonstrated in Fig. 3B, the
expression of ALK1 in hypoxia-induced HP was significantly
increased, while that of ALK4 was increased, but not significantly.
Therefore, ALK1 was selected as the potential target gene of miR-98
in HPH. To additionally confirm whether ALK1 is the target of
miR-98, luciferase reporter plasmids carrying the 3′-UTR miR-98
wild type or miR-98 mutant-binding sites of ALK1 was constructed,
and 293 cells were co-transfected with either miR-98 mimics or NC.
Compared with the control, transfection with miR-98 mimics
decreased the luciferase activities significantly (Fig. 3C). However, the miR-98 mimics did
not affect the luciferase activity in the mutant construct, and
their luciferase activities did not exhibit any significant
differences when compared with the control (Fig. 3C). In addition, transfection with
miR-98 mimics significantly decreased the mRNA level of ALK1 in
PASMCs, while transfection with miR-98 inhibitors increased it
(Fig. 3D). Western blot analysis
data indicated that the protein level of ALK1 in PASMCs was
decreased when transfected with miR-98 mimics. Conversely, the
protein level of ALK1 was increased in the cells expressing
anti-miR-98 (Fig. 3E and F). These
data indicated that miR-98 directly modulated the ALK1 expression
by binding to the 3′-UTR of ALK1.
 | Figure 3.ALK1 is a direct target of miR-98 in
PASMCs. (A) Diagram of miR-98 seed sequence, indicating that it
matched the 3′-UTR of the ALK1 and ALK4 genes. (B) RT-qPCR analysis
of the ALK1 and ALK4 expression in lung pulmonary arteries from H
and N rats. **P<0.01 (n=10). (C) Luciferase reporter assays in
293 cells, following co-transfection of cells with WT or mut 3′-UTR
ALK1 and miR-98 mimics. (D) RT-qPCR analysis of the ALK1 expression
24 h following transfection with miR-98 mimics, inhibitors or NC.
(E) Western blot analysis results of the ALK1 expression in PASMCs
transfected with miR-98 mimics, inhibitors or NC. (F) Densitometric
analysis of the western blot analysis of ALK1. All experiments were
repeated 3 times. *P<0.05, **P<0.01. ALK, activin
receptor-like kinase; miR, microRNA; UTR, untranslated region;
PASMCs, pulmonary artery smooth muscle cells; RT-qPCR; reverse
transcription quantitative polymerase chain reaction; N, normoxic;
H, hypoxic; NS, not significant; WT, wild type; mut, mutant; NC,
negative control. |
Confirmation of miR-98 functions by
targeting ALK1
To additionally examine the role of ALK1 in
mediating the function of miR-98, rescue experiments were
conducted. PASMCs were transiently transfected with miR-98 mimics
and a ALK1 overexpression vector carried by lentiviral particles
(lenti-ALK1), and subsequently exposed to hypoxia for 24 h or 72 h.
Cell survival, proliferation and apoptosis assays indicated that
the re-introduction of ALK1 into the miR-98-overexpressing cells
inhibited PASMC apoptosis (Fig. 4A and
B), and enhanced cell survival and proliferation (Fig. 4C). It must be noted that the time
of PASMCs exposure to hypoxia in these 2 experiments was different;
the data in Fig. 4B was obtained
24 h after transfection, and the data from Fig. 4C was measured 72h after
transfection. Western blot analysis results suggested that ALK1
overexpression inhibited the protein level of cleaved caspase 3.
Concomitantly, the protein levels of phosphorylated Smad1, one of
the target signaling pathways of ALK1, and the
proliferation-associated protein PCNA were significantly increased
(Fig. 4D-F). These results
suggested that miR-98 was significantly downregulated in HPH,
leading to an increase in the levels of ALK1 protein, which
promoted the activation of Smad signaling pathways, thereby
inhibiting SMC apoptosis, promoting cell proliferation and leading
to the development of HPH.
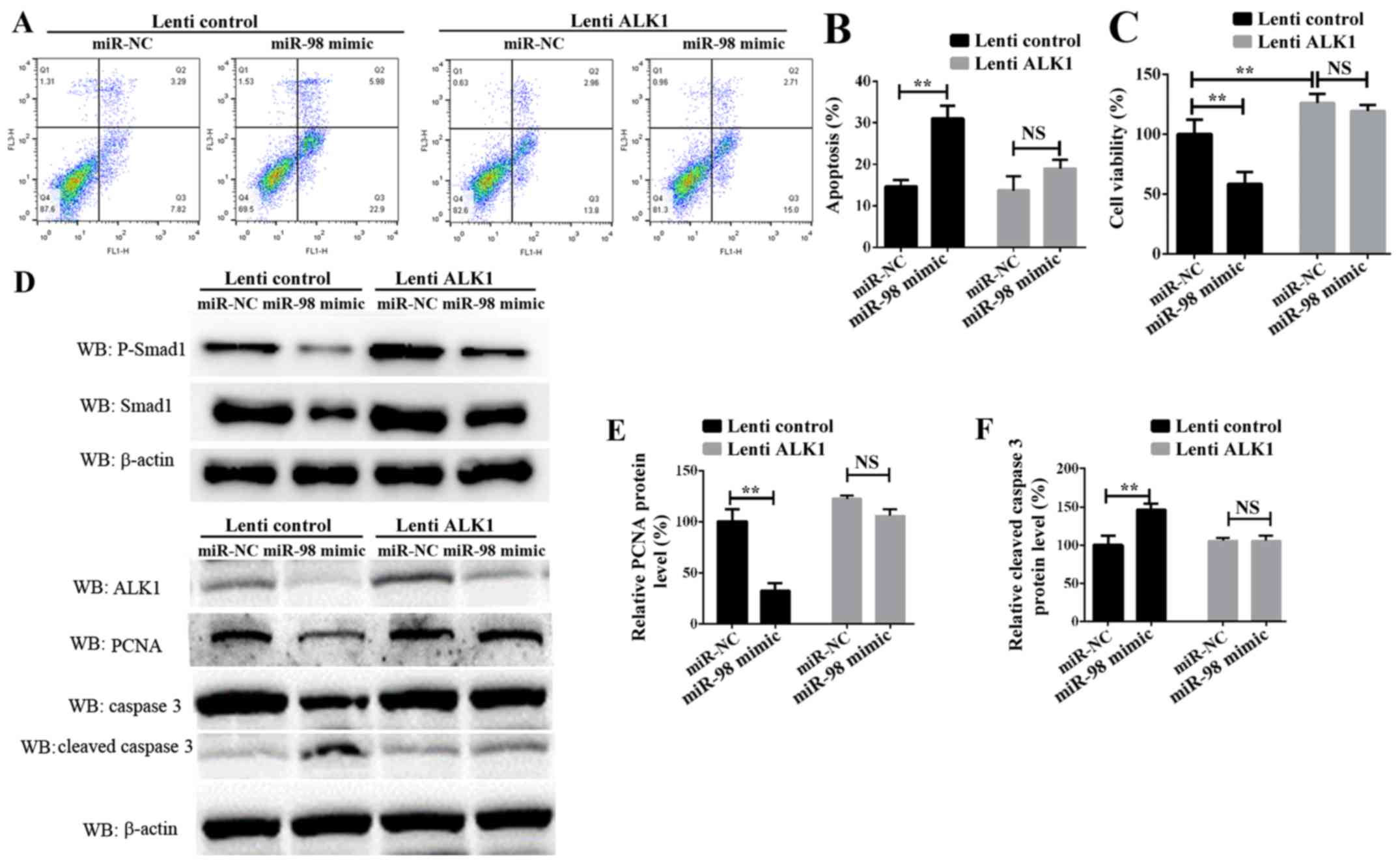 | Figure 4.Confirmation of miR-98 functions by
targeting ALK1 in PASMCs. (A) Representative FACS analysis of
Annexin V and PI staining of PASMCs following transfection with
miR-98 mimics and lenti-ALK1 particles, and subsequent exposure to
hypoxia for 24 h. (B) Percentage of apoptotic cells analyzed by
FACS. (C) Cell Counting Kit-8 assay was used to analyze PASMC
proliferation following transfection of PASMCs and exposure to
hypoxia for 72 h. (D) Upper panel, western blot analysis of
phosphorylated Smad1 and total Smad1; lower panel, western blot
analysis of ALK1, cleaved caspase 3 and PCNA levels in PASMCs
transfected with miR-98 mimics and ALK1 overexpression vector and
subsequent exposure to hypoxia for 24 h. Densitometric analysis of
(E) PCNA and (F) cleaved caspase 3 levels. All experiments were
repeated 3 times. **P<0.01. miR, microRNA; ALK1, activin
receptor-like kinase-1; PASMCs, pulmonary artery smooth muscle
cells; PI, propidium iodide; NS, not significant; CCK-8, Cell
Counting Kit-8; lenti-ALK1, ALK1 overexpression vectors carrying
lentiviral particles; FACS, fluorescence-activated cell sorting;
PCNA, proliferating cell nuclear antigen. |
In vivo effect of miR-98 agomir
treatment on the expression of ALK1 and hypoxia-induced pulmonary
vasoconstriction
To examine the effect of miR-98 agomir, a
chemically-modified miR-98 mimic, on the expression of ALK1 and the
development of HPH, an animal model of HPH was established by
exposing rats to hypoxia and a tail vein injection of miR-98 agomir
or its control. The rats were divided into three groups, including
normoxia control (N-control), hypoxia control (H-control) and
hypoxia + miR-98 agomir (H + miR-98 agomir). The results
demonstrated that the miR-98 agomir injection significantly
increased miR-98 in the lung tissue, as compared with the untreated
group. In addition, the mRNA level of ALK1 was significantly
decreased following the tail injection of miR-98 mimics (Fig. 5B). Furthermore, western blot
analysis results indicated that the miR-98 agomir significantly
decreased the levels of ALK1 protein expression, and inhibited the
phosphorylation of Smad1 (Fig.
5A). To explore the biological effect of miR-98, the RVSP was
measured. It was identified that the RVSP was markedly decreased in
miR-98 agomir-treated rats under hypoxia (Fig. 5C). Furthermore, H&E staining
demonstrated that the thickening of the smooth muscle layer of the
pulmonary small blood vessels was improved by miR-98 agomir
treatment (Fig. 5D). The results
of the Masson staining protocol suggested that the collagen
deposition in the pulmonary muscle arterioles induced by hypoxia
was also improved by miR-98 agomir treatment (Fig. 5E). Hyperproliferation of SMCs
induced by hypoxia was also inhibited following miR-98 agomir
treatment, as the protein level of PCNA was decreased in the H +
miR-98 agomir group compared with the H-control group (Fig. 5F and G). All of these results
indicated that treatment with miR-98 agomir decreased the
expression of ALK1 and attenuated hypoxia-induced vascular
remodeling.
 | Figure 5.Effect of miR-98 agomir treatment on
hypoxia-induced pulmonary vasoconstriction in the HPH rat model.
(A) Western blot analysis of ALK1 and phosphorylated Smad1 levels
in lung pulmonary arteries from normoxic and hypoxic rats with or
without miR-98 agomir treatment. (B) Reverse transcription
quantitative polymerase chain reaction analysis of ALK1 and miR-98
expression in lung pulmonary arteries from N and H rats with or
without miR-98 agomir treatment. (C) Measurement of RVSP. (D)
Representative images of hematoxylin and eosin staining of lung
pulmonary arteries (magnification, ×200). (E) Representative images
of Masson staining of lung pulmonary arteries (magnification,
×200). (F) Representative immunohistochemical staining of PCNA in
N-control, H-control and H + miR-98 agomir treatment groups
(magnification, ×400). (G) Semi-quantitative analysis of
immunohistochemical staining of PCNA (analyzed by ImageJ).
*P<0.05, **P<0.01 and ***P<0.001. All experiments were
repeated 3 times. miR, microRNA; HPH, hypoxic pulmonary
hypertension; ALK1, activin receptor-like kinase-1; N, normoxia; H,
hypoxia; RVSP, right ventricular systolic pressure; PCNA,
proliferating cell nuclear antigen; N-control, normoxic control
group; H-control, hypoxia induced group; H + miR-98 agomir, hypoxia
induced with miR-98 agomir. |
Discussion
Although the mechanisms of PH have been studied for
several decades, they remain unclear. Chronic hypoxia involving
multiple molecular signaling pathways has been reported to be an
important reason for the occurrence and development of PH. In
previous decades, several studies have demonstrated that the
association between miRNAs and vascular remodeling, the development
of hypertrophy, failure in the heart muscle function and
vasculature, all are relevant (33,34).
In addition, several miRNAs have been suggested to be aberrantly
expressed and involved in the development of PH (10,35,36).
Although great progress has been made with regards to the role of
miRNAs in PH, these studies have not fully clarified their
mechanisms in HPH, which require additional study.
The present study revealed that the expression of
miR-98 was significantly decreased in the lung tissues of the HPH
rat model and PASMCs under hypoxia compared with the controls.
However, the association between miR-98 and the pathological
process of HPH was not examined. Based on all of these results, it
was reasonable to hypothesize that miR-98 may serve an important
role in the development of HPH. In order to examine our hypothesis,
an HPH animal model and primary rat PASMCs were used to examine the
function of miR-98 in the development of HPH. As expected, miR-98
overexpression inhibited hypoxia-induced PASMC proliferation. Rat
PASMC apoptosis was also markedly increased following miR-98
transfection. These results suggested that miR-98 may serve an
important role in hypoxia-induced PASMC hyperproliferation. miRNAs
exert their functions via targeting different target genes; for
example miR-328 regulates HPH by targeting insulin growth factor 1
receptor and l-Type calcium channel-α1C (37). Therefore, the present study
hypothesized that miR-98 regulated several target genes to modify
the regulation network and inhibit PASMC proliferation in the
present study. Using online bioinformatics software, including
TargetScan, MiRanda and PicTar, several candidate targets for
miR-98 were predicted.
Though gain-of-function approaches, it was confirmed
that ALK1 is a direct target gene of miR-98. Firstly, the mRNA
expression of ALK1 was identified to be increased in the lung
tissues of HPH rats, while that of ALK4, which was revealed to be a
target of miR-98 in breast cancer cells, was not (38). Secondly, data from the present
study demonstrated that the overexpression of miR-98 notably
decreased the relative luciferase activity of the WT but not the
mutant vector containing ALK1 3′-UTR. Thirdly, the overexpression
of miR-98 inhibited the mRNA and protein expression of ALK1.
Finally, miR-98 overexpression inhibited cell proliferation and
promoted apoptosis in PASMCs; however, the levels of proliferation
were restored and apoptosis was inhibited when ALK1 was
overexpressed.
However, it must be noted that when the PASMCs
transfected with miR-98 mimics and ALK1 overexpression lenti-ALK1
particles were exposed to hypoxia for 24 h, and then analyzed ~30 h
after transfection, it is possible that the cells had not
completely recovered from the toxicity of transfection, and that
the levels of overexpression of ALK1 were not high. This may
explain why the overexpression of ALK1 did not change the levels of
cell apoptosis at first. However, the results from the present
study indicated that, after transfection for 72 h, the
overexpression of ALK1 significantly inhibited the levels of
apoptosis.
In addition, it was indicated that the forced
expression of ALK1 was also degraded by overexpression of miR-98;
therefore, it may be better to use resistant mutants of ALK1 in
future studies. It was concluded that ALK1 is a direct and pivotal
target gene of miR-98 in the development of HPH.
ALK1 is a specific type I receptor that transmits
signals via the ALK1/Smad1/5 pathways. Several studies have
demonstrated that the TGF-β/ALK1 signaling pathway serves a key
role in idiopathic pulmonary arterial hypertension and experimental
hypoxic PH via a direct effect on pulmonary endothelial cells,
leading to the overproduction of growth factors and inflammatory
cytokines that are involved in the pathogenesis of PAH (13,18,39–41).
In addition, several previous data have indicated that ALK1 is
essential for hypoxia-induced PASMC proliferation (13,42).
The present study demonstrated that ALK1 overexpression restored
the proliferation of PASMC, which was inhibited by miR-98
mimics.
In conclusion, to the best of our knowledge, the
present study was the first to provide evidence that miR-98 serves
an important role in vasoconstriction and remodeling in the
pathogenesis of HPH. The inhibitory effect of miR-98 on
vasoconstriction may be attributable to the hypoxia-induced
inhibition of the ALK1 expression. These results provide novel
insight into the molecular mechanisms of HPH and suggest a
potential target for treatment of HPH. However, these results
require verification through additional clinical studies.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from The
Science and Technology Bureau of Xuzhou (grant no. BK2016109).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
XiaZ conceived and designed the experiments. QL and
XinZ performed the experiments, and XiaZ and QL analyzed the data
and wrote the manuscript.
Ethics approval and consent to
participate
All aspects of the present study were approved by
the Ethics Committee of the Municipal Hospital Affiliated to Xuzhou
Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Simonneau G, Gatzoulis MA, Adatia I,
Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, Kumar RK,
Landzberg M, Machado RF, et al: Updated clinical classification of
pulmonary hypertension. Turk Kardiyol Dern Arsivi (Turkish). 42
(Suppl):S45–S54. 2014.
|
|
2
|
D'Alonzo GE, Barst RJ, Ayres SM, Bergofsky
EH, Brundage BH, Detre KM, Fishman AP, Goldring RM, Groves BM,
Kernis JT, et al: Survival in patients with primary pulmonary
hypertension. Results from a national prospective registry. Ann
Intern Med. 115:343–349. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
McLaughlin VV, Sitbon O, Badesch DB, Barst
RJ, Black C, Galiè N, Rainisio M, Simonneau G and Rubin LJ:
Survival with first-line bosentan in patients with primary
pulmonary hypertension. Eur Respir J. 25:244–249. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chan SY and Loscalzo J: Pathogenic
mechanisms of pulmonary arterial hypertension. J Mol Cell Cardiol.
44:14–30. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tang H, Desai AA and Yuan JX: Genetic
insights into pulmonary arterial hypertension. Application of
whole-exome sequencing to the study of pathogenic mechanisms. Am J
Respir Crit Care Med. 194:393–397. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhou W, Negash S, Liu J and Raj JU:
Modulation of pulmonary vascular smooth muscle cell phenotype in
hypoxia: Role of cGMP-dependent protein kinase and myocardin. Am J
Physiol Lung Cell Mol Physiol. 296:L780–L789. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bushati N and Cohen SM: microRNA
functions. Annu Rev Cell Dev Biol. 23:175–205. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Krol J, Loedige I and Filipowicz W: The
widespread regulation of microRNA biogenesis, function and decay.
Nat Rev Genet. 11:597–610. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bertero T, Lu Y, Annis S, Hale A, Bhat B,
Saggar R, Saggar R, Wallace WD, Ross DJ, Vargas SO, et al:
Systems-level regulation of microRNA networks by miR-130/301
promotes pulmonary hypertension. J Clin Invest. 124:3514–3528.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Caruso P, Dempsie Y, Stevens HC, McDonald
RA, Long L, Lu R, White K, Mair KM, McClure JD, Southwood M, et al:
A role for miR-145 in pulmonary arterial hypertension: Evidence
from mouse models and patient samples. Circ Res. 111:290–300. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Potus F, Graydon C, Provencher S and
Bonnet S: Vascular remodeling process in pulmonary arterial
hypertension, with focus on miR-204 and miR-126 (2013 Grover
Conference series). Pulm Circ. 4:175–184. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gore B, Izikki M, Mercier O, Dewachter L,
Fadel E, Humbert M, Dartevelle P, Simonneau G, Naeije R, Lebrin F
and Eddahibi S: Key role of the endothelial TGF-beta/ALK1/endoglin
signaling pathway in humans and rodents pulmonary hypertension.
PLoS One. 9:e1003102014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Savage-Dunn C: TGF-beta signaling.
WormBook. 9:1–12. 2005.
|
|
15
|
Yan Y, Wang XJ, Li SQ, Yang SH, Lv ZC,
Wang LT, He YY, Jiang X, Wang Y and Jing ZC: Elevated levels of
plasma transforming growth factor-β1 in idiopathic and heritable
pulmonary arterial hypertension. Int J Cardiol. 222:368–374. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Massague J: TGF-beta signal transduction.
Ann Rev Biochem. 67:753–791. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Oh SP, Seki T, Goss KA, Imamura T, Yi Y,
Donahoe PK, Li L, Miyazono K, ten Dijke P, Kim S and Li E: Activin
receptor-like kinase 1 modulates transforming growth factor-beta 1
signaling in the regulation of angiogenesis. Proc Natl Acad Sci U S
A. 97:2626–2631. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Uznanska-Loch B, Wiklo K,
Kulczycka-Wojdala D, Szymańska B, Chrzanowski Ł, Wierzbowska-Drabik
K, Trzos E, Kasprzak JD and Kurpesa M: Genetic variants in a Polish
population of patients with pulmonary arterial hypertension:
Sequencing of BMPR2, ALK1, and ENG genes. Kardiol Pol. 76:852–859.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chida A, Shintani M, Yagi H, Fujiwara M,
Kojima Y, Sato H, Imamura S, Yokozawa M, Onodera N, Horigome H, et
al: Outcomes of childhood pulmonary arterial hypertension in BMPR2
and ALK1 mutation carriers. Am J Cardiol. 110:586–593. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang H, Du L, Zhong Y, Flanders KC and
Roberts JD Jr: Transforming growth factor-β stimulates Smad1/5
signaling in pulmonary artery smooth muscle cells and fibroblasts
of the newborn mouse through ALK1. Am J Physiol Lung Cell Mol
Physiol. 313:L615–L627. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhu D, Mackenzie NC, Shanahan CM, Shroff
RC, Farquharson C and MacRae VE: BMP-9 regulates the osteoblastic
differentiation and calcification of vascular smooth muscle cells
through an ALK1 mediated pathway. J Cell Mol Med. 19:165–174. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li L, Shi JY, Zhu GQ and Shi B: MiR-17-92
cluster regulates cell proliferation and collagen synthesis by
targeting TGFB pathway in mouse palatal mesenchymal cells. J Cell
Biochem. 113:1235–1244. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang X, Yan C, Xu X, Dong L, Su H, Hu Y,
Zhang R and Ying K: Long noncoding RNA expression profiles of
hypoxic pulmonary hypertension rat model. Gene. 579:23–28. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Krek A, Grun D, Poy MN, Wolf R, Rosenberg
L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M
and Rajewsky N: Combinatorial microRNA target predictions. Nat
Genet. 37:495–500. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lewis BP, Shih IH, Jones-Rhoades MW,
Bartel DP and Burge CB: Prediction of mammalian microRNA targets.
Cell. 115:787–798. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhou S, Sun L, Cao C, Wu P, Li M, Sun G,
Fei G, Ding X and Wang R: Hypoxia-induced microRNA-26b inhibition
contributes to hypoxic pulmonary hypertension via CTGF. J Cell
Biochem. 119:1942–1952. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee
DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, et al:
Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic
Acids Res. 33:e1792005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang X: A PCR-based platform for microRNA
expression profiling studies. RNA. 15:716–723. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Song Y, Jones JE, Beppu H, Keaney JF Jr,
Loscalzo J and Zhang YY: Increased susceptibility to pulmonary
hypertension in heterozygous BMPR2-mutant mice. Circulation.
112:553–562. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fischer AH, Jacobson KA, Rose J and Zeller
R: Hematoxylin and eosin staining of tissue and cell sections. CSH
Protoc 2008: pdb prot4986. 2008.
|
|
32
|
Xu X, Hu H, Wang X, Ye W, Su H, Hu Y, Dong
L, Zhang R and Ying K: Involvement of CapG in proliferation and
apoptosis of pulmonary arterial smooth muscle cells and in
hypoxia-induced pulmonary hypertension rat model. Exp Lung Res.
42:142–153. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Thum T, Galuppo P, Wolf C, Fiedler J,
Kneitz S, van Laake LW, Doevendans PA, Mummery CL, Borlak J,
Haverich A, et al: MicroRNAs in the human heart: A clue to fetal
gene reprogramming in heart failure. Circulation. 116:258–267.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mann DL: MicroRNAs and the failing heart.
N Eng J Med. 356:2644–2645. 2007. View Article : Google Scholar
|
|
35
|
Sarkar J, Gou D, Turaka P, Viktorova E,
Ramchandran R and Raj JU: MicroRNA-21 plays a role in
hypoxia-mediated pulmonary artery smooth muscle cell proliferation
and migration. Am J Physiol Lung Cell Mol Physiol. 299:L861–L871.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Courboulin A, Paulin R, Giguère NJ,
Saksouk N, Perreault T, Meloche J, Paquet ER, Biardel S, Provencher
S, Côté J, et al: Role for miR-204 in human pulmonary arterial
hypertension. J Exp Med. 208:535–548. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Guo L, Qiu Z, Wei L, Yu X, Gao X, Jiang S,
Tian H, Jiang C and Zhu D: The microRNA-328 regulates hypoxic
pulmonary hypertension by targeting at insulin growth factor 1
receptor and L-type calcium channel-α1C. Hypertension.
59:1006–1013. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Siragam V, Rutnam ZJ, Yang W, Fang L, Luo
L, Yang X, Li M, Deng Z, Qian J, Peng C and Yang BB: MicroRNA
miR-98 inhibits tumor angiogenesis and invasion by targeting
activin receptor-like kinase-4 and matrix metalloproteinase-11.
Oncotarget. 3:1370–1385. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Trembath RC: Mutations in the TGF-beta
type 1 receptor, ALK1, in combined primary pulmonary hypertension
and hereditary haemorrhagic telangiectasia, implies pathway
specificity. J Heart Lung Transplant. 20:1752001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fujiwara M, Yagi H, Matsuoka R, Akimoto K,
Furutani M, Imamura S, Uehara R, Nakayama T, Takao A, Nakazawa M
and Saji T: Implications of mutations of activin receptor-like
kinase 1 gene (ALK1) in addition to bone morphogenetic protein
receptor II gene (BMPR2) in children with pulmonary arterial
hypertension. Circ J. 72:127–133. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jerkic M, Kabir MG, Davies A, Yu LX,
McIntyre BA, Husain NW, Enomoto M, Sotov V, Husain M, Henkelman M,
et al: Pulmonary hypertension in adult Alk1 heterozygous mice due
to oxidative stress. Cardiovasc Res. 92:375–384. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Soubrier F, Chung WK, Machado R, Grünig E,
Aldred M, Geraci M, Loyd JE, Elliott CG, Trembath RC, Newman JH and
Humbert M: Genetics and genomics of pulmonary arterial
hypertension. J Am Coll Cardiol. 62:D13–D21. 2013. View Article : Google Scholar : PubMed/NCBI
|
















