Introduction
Alzheimer's disease (AD) is the most common
neurodegenerative disorder of the central nervous system.
Worldwide, >46.8 million patients have been diagnosed with AD,
with 50% of those patients >85 years of age. The number of
individuals with AD is expected to double by 2030 (1–3).
Previous studies have reported a correlation between diabetes
mellitus (DM) and the risk of developing AD (4,5).
Certain scholars have even suggested that AD should be considered
as a third type of DM (6–9).
Advanced glycation end products (AGEs) affect the
functions of normal tissues through different pathways and play
important roles in the occurrence and development of various
diseases, including diabetic complications, osteoporosis, AD,
tumorigenesis, aging and cardiovascular disease (10–13).
AGEs can induce apoptosis in neurons, leading to the early onset of
AD (14). Additionally,
substantial quantities of carboxymethyllysine, pyrraline and
pentosidine have been detected in the senile plaques and
neurofibrillary tangles (NFTs) of patients with AD; these AGEs may
promote the development of AD by affecting the metabolism of
amyloid β (Aβ) (15).
Macrophage migration inhibitory factor (MIF) is a
cytokine involved in the occurrence of various inflammatory and
immune diseases (16,17). The inhibitor of MIF,
(S,R)-3-(4-hydroxyphenyl)-4,5-dihydro-5-isoxazole acetic acid
methyl ester (ISO-1), can significantly inhibit the activity of MIF
and affect its pathophysiological functions (18). MIF activity is one of the risk
factors for type I diabetes and type II diabetes (T2D) (17,19).
A previous clinical study by Bacher et al (20) reported significantly increased MIF
levels in the cerebrospinal fluid of patients with AD and mild
cognitive impairment (MCI) compared with controls of the same age.
Oyama et al (21) reported
that MIF bound to Aβ in the brains of patients with AD, and thus,
the toxicity of Aβ was directly attributed to the upregulation of
MIF expression. MIF deficiency was also reported to attenuate the
hyperphosphorylation of tau proteins (22).
Previous studies have identified a close link
between DM and AD (4,5); however, the exact molecular link
between these two diseases remains unclear. AGEs promote the
deposition of Aβ and the hyperphosphorylation of tau protein
(14,15). Whether MIF also serves a role in
the AGEs-induced development of AD requires further investigation.
The present study investigated the neuroinflammatory mechanisms
underlying the pathogenesis of AD and assessed the effect of the
pathogenic factor MIF on AD. It was hypothesized that AGEs would
affect the levels of neuroinflammation observed in AD by inducing
MIF expression, and that the levels of neuroinflammation could be
reduced using ISO-1.
Materials and methods
Cell culture and treatment
PC12 cells were purchased from the Type Culture
Collection of the Chinese Academy of Sciences. PC12 cells were
cultured in DMEM (HyClone; GE Healthcare Life Sciences)
supplemented with 10% FBS (Zhejiang Tianhang Biotechnology Co.,
Ltd.) at 37°C in a humidified atmosphere of 5% CO2/95%
air. PC12 cells in the logarithmic phase of growth were plated in
6-well plates at the density of 5×105/ml and divided
into three treatment groups: A blank control group, a BSA (300
µg/ml; Beijing ComWin Biotech Co., Ltd.) control group and an AGEs
(Biorbyt, Ltd.) group. The AGEs group were treated with different
concentrations (100, 200 or 300 µg/ml) of AGEs for 24 h at 37°C,
and then subsequent assays were performed. As AGEs were prepared in
a non-enzymatic glycosylation reaction using BSA as a substrate,
BSA (300 µg/ml) was also used as a control.
Reverse transcription-quantitative
(RT-q)PCR
Total RNA was extracted from cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) following the aforementioned treatments, according to the
manufacturer's protocol. For mRNA analysis, cDNAs were synthesized
using a TIANScript First Strand cDNA Synthesis kit (Tiangen Biotech
Co., Ltd.), according to the manufacturer's protocol. The
temperature protocol for the reverse transcription reaction was
37°C for 60 min. qPCR was performed using a SuperReal PreMix Color
(SYBR-Green; Tiangen Biotech Co., Ltd.) in an Mx3000P system
(Agilent Technologies, Inc.). The cycling conditions included a
pre-denaturation step at 95°C for 15 min, followed by 40 cycles of
denaturation at 95°C for 10 sec, annealing at 55°C for 20 sec and
extension at 72°C for 32 sec. The primer sequences used in this
study are presented in Table I.
Quantification cycle (Cq) values for the target genes and the
internal control gene, GAPDH, were measured. The expression levels
of each mRNA were determined from three independent experiments,
and the fold change was calculated using the 2−ΔΔCq
method (23).
 | Table I.Oligonucleotide primer sets used for
reverse transcription-quantitative PCR. |
Table I.
Oligonucleotide primer sets used for
reverse transcription-quantitative PCR.
| Name | Sequence
(5′-3′) | Length (bp) |
|---|
| MIF | F:
TACGACATGAACGCAGCCAACG | 22 |
|
| R:
GAACAGCGGTGCAGGTAAGTGAG | 23 |
| IL-1β | F:
ATCTCACAGCAGCATCTCGACAAG | 24 |
|
| R:
CACACTAGCAGGTCGTCATCATCC | 24 |
| IL-6 | F:
ATGATGAGAAACGAGCCAATTG | 22 |
|
| R:
GCTTTGGCTTCTTTCTTACGAG | 22 |
| TNF-α | F:
TTGGGTTATGCCAAAGATGTTG | 22 |
|
| R:
GCTGTGTACGGCTTATTTTCAA | 22 |
| GAPDH | F:
ACGGCAAGTTCAACGGCACAG | 21 |
|
| R:
CGACATACTCAGCACCAGCATCAC | 24 |
Western blot analysis
Protein levels in PC12 cells were determined after
treatment using western blot analysis. Cells were lysed with RIPA
lysis buffer (Sigma-Aldrich; Merck KGaA) containing a protease
inhibitor cocktail (Sigma-Aldrich; Merck KGaA). Samples were
incubated on ice for 30 min and then centrifuged at 16,000 × g for
15 min at 4°C. The supernatant was removed and the protein
concentration was determined using the bicinchoninic acid method.
Total protein (50 µg) was separated on 12% SDS-PAGE gels and
transferred to PVDF membranes. Membranes were blocked with 5% dried
skimmed milk/TBS 0.5% Tween-20 (TBST) for 1 h at room temperature.
Membranes were incubated overnight at 4°C with a monoclonal rabbit
anti-MIF antibody (1:100; cat. no. ab172730; Abcam) or anti-GAPDH
antibody (1:1,000; cat. no. CW0101S; Beijing ComWin Biotech Co.,
Ltd.), and then washed three times with TBST for 10 min each.
Subsequently, horseradish peroxidase-conjugated goat anti-rabbit
immunoglobulin G (1:10,000; cat. no. D110058; BBI Life Sciences
Corporation) secondary antibody was incubated with the membranes
for 1 h at room temperature, which were then washed three times
with TBST for 10 min each. The bands were visualized with an ECL
reagent (Tanon Science and Technology Co., Ltd.) and detected with
the Tanon Imaging System (Tanon Science and Technology Co., Ltd.).
The relative intensity of the protein bands was quantified using
Image-Pro Plus 6.0 software (Media Cybernetics, Inc.). Experiments
were repeated three times.
Screening for the optimum
concentration of AGEs and Aβ1–40 in the AD cell
model
PC12 cells were plated in 96-well plates at the
density of 5×105/ml, and the cells were exposed to
various concentrations of AGEs (100–400 µg/ml) at 37°C for 24 h,
with 5 replicates/group; equivalent concentrations were applied to
the BSA control group. All groups were treated with 10 µl of Cell
Counting Kit-8 (CCK-8) reagent (Dojindo Molecular Technologies,
Inc.) and incubated for 1–4 h at 37°C before the absorbance was
measured at 450 nm using a microplate reader to detect surviving
cells.
Aβ1–40 (Shanghai Aladdin Biochemical
Technology Co., Ltd.) was dissolved in sterile PBS and incubated at
37°C for 7 days to induce aggregation prior to treatment. PC12
cells were plated in 96-well plates at the density of
5×105/ml. The blank control group was treated with 100
µl of fresh culture medium and the AD model groups were incubated
with 100 µl of medium containing different concentrations (10–40
µg/ml) of Aβ1–40 at 37°C for 2, 6, 12 or 24 h, with 3
replicates/group (24). All groups
were treated with 5 mg/ml MTT solution 10 µl at 37°C for 4 h, the
medium was then discarded and 100 µl dimethyl sulfoxide was added
to each well to dissolve the formazan crystal product. The
absorbance was measured at 570 nm using a microplate reader.
Observation of cell number and
morphology under an inverted microscope, and detection of cell
activity using the MTT method
PC12 cells were plated in 12-well plates at the
density of 5×105/ml. The cells were exposed to 300 µg/ml
AGEs at 37°C for 24 h and then treated with 20 µg/ml
Aβ1–40 at 37°C for 12 h. To assess the effect of AGEs on
the expression of inflammatory mediators through MIF and to
determine the effect of ISO-1, cells were pre-treated with 7 µM
ISO-1 (MedChemExpress LLC) at 37°C for 1 h. The blank control group
and the Aβ1–40 model group were used as control groups,
with 3 replicates/group. Following treatment, the number and
morphology of the cells were observed under an inverted microscope
(magnification ×100). Each sample was analyzed in five randomly
selected fields. No significant differences were observed between
the BSA control group and the blank control group; therefore, BSA
was no longer used as a control.
PC12 cells were plated in 96-well plates at the
density of 5×105/ml. The cell groupings and treatments
were the same as for the aforementioned microscopy assay, and the
activity of cells was determined using the MTT method, according
the aforementioned protocol.
Determining the expression of
neuroinflammatory cytokines and MIF mRNA using the RT-qPCR
assay
PC12 cells were plated in 12-well plates at the
density of 5×105/ml. Cells were divided into four groups
and exposed to the aforementioned treatments. After treatment,
total RNA was extracted and RT-qPCR was conducted using the primers
presented in Table I, according
the aforementioned protocol.
Kyoto Encyclopedia of Genes and
Genomes (KEGG) database search for AGEs signal transduction
pathways
KEGG (www.kegg.jp) is
a database resource that integrates genomic, chemical and systemic
functional information (25). By
searching for AGEs-associated signaling pathways, map04933 was
identified; AGEs-mediated proinflammatory signaling was
investigated using this map.
Statistical analysis
Data are presented as the mean ± SD. Differences
among groups were evaluated using one-way ANOVA followed by Tukey's
post hoc test using SPSS version 19.0 software (IBM Corp.) and
Prism version 5.0 software (GraphPad Software, Inc.). P<0.05 was
considered to indicate a statistically significant difference.
Results
Effect of AGEs on the expression of
MIF mRNA and protein
A significant difference in MIF mRNA expression was
not observed between the BSA control group and the blank control
group; however, compared with the blank control group, the
treatment of cells with 200 or 300 µg/ml AGEs produced
statistically significant differences, with MIF mRNA expression
significantly increased (Fig. 1A).
Cells treated with 100 or 300 µg/ml AGEs exhibited significantly
increased levels of MIF protein expression compared with the blank
control group (Fig. 1B and C). It
was hypothesized that a direct intermolecular relationship existed
between AGEs and MIF, and that AGEs could induce the expression of
MIF mRNA and protein.
Optimal concentration of AGEs and the
viability of PC-12 cells following treatment with
Aβ1–40
To screen for the optimum concentration of AGEs,
PC12 cells were treated with different concentrations of AGEs, and
the survival rate was determined using a CCK-8 assay. The results
revealed that the viability of cells decreased with increasing
concentrations of AGEs. The half-maximal effective concentration
was calculated to be 335.0 µg/ml using GraphPad Prism 5 software
(Fig. 2A). The BSA control group
was administered an equivalent concentration of BSA to the optimal
concentration of AGEs for PC12 cells (300 µg/ml); no significant
difference was observed between the BSA control group and the blank
control group (data not shown). The inhibition rate of cells was
measured using an MTT assay. As presented in Fig. 2B, the time at which each
concentration of Aβ1–40 inhibited cell survival to the
greatest extent was 12–24 h; however, at 24 h, the cells were
poorly adherent and a small number of cells were detached.
Therefore, cells were treated with Aβ1–40 for 12 h.
Using GraphPad Prism 5 software, it was calculated that the
half-maximal inhibitory concentration of PC12 cells induced by
Aβ1–40 was 22.17 µg/ml (Fig. 2C). The optimal concentration of
Aβ1–40 treatment and exposure time to establish an AD
model was determined to be 20 µg/ml for 12 h.
 | Figure 2.Finding the optimal concentration of
AGEs and determining the inhibition of PC12 cell survival induced
by Aβ1–40. (A) PC12 cells were treated with different
concentrations of AGEs, and the percentage of surviving cells was
determined using a Cell Counting Kit-8 assay; the percentage of
surviving cells decreased as the AGE concentration increased. The
half-maximal effective concentration was calculated to be 335.0
µg/ml using GraphPad Prism 5 software; therefore, the optimal
concentration of AGEs for PC12 cell treatment was determined to be
300 µg/ml. (B) PC12 cells were treated with different
concentrations of Aβ1–40 for various periods of time.
The percent inhibition of cell growth was measured using an MTT
assay to determine the most appropriate interval and concentration
to create a cell model of AD. Compared with Aβ1–40 (0
µg/ml), the growth of a greater percentage of cells was inhibited
as the Aβ1–40 concentration increased. (C) Using
GraphPad Prism 5 software, it was calculated that the
IC50 of Aβ1–40 in PC12 cells was 22.17 µg/ml.
The optimal concentration and exposure time of Aβ1–40 to
establish the AD model was determined to be 20 µg/ml for 12 h. Data
are presented as the mean ± SD; n=3. *P<0.05, **P<0.01,
***P<0.001 vs. 0 µg/ml AGEs/Aβ1–40. AGEs, advanced
glycation end products; Aβ1–40, amyloid β 1–40;
IC50, half-maximal inhibitory concentration; AD,
Alzheimer's disease. |
Cell morphology and activity
Under an inverted microscope (magnification ×100),
PC12 cells in the blank control group displayed numerous long
dendrites. The blank control cells adhered tightly to the bottom of
the culture plate, with dense, clear and visible nucleoli, and
exhibited a rapid rate of proliferation (Fig. 3A). Compared with the blank control
group, the proliferation of cells in the Aβ1–40 model
group was inhibited, as determined by the decreased number of
cells, relaxed intercellular junctions and partial retraction of
dendrites (Fig. 3B). Following
AGEs treatment, cell proliferation was further inhibited, a large
number of cells were detached and more cell fragments were
observed; the remaining adherent cells lost their characteristic
neuronal features (Fig. 3C).
Following pre-treatment with ISO-1, the decrease in cell
proliferation was notably attenuated. The number of adherent cells
increased, the number of detached cells decreased, cells were
strongly adherent, and formed more intercellular junctions with
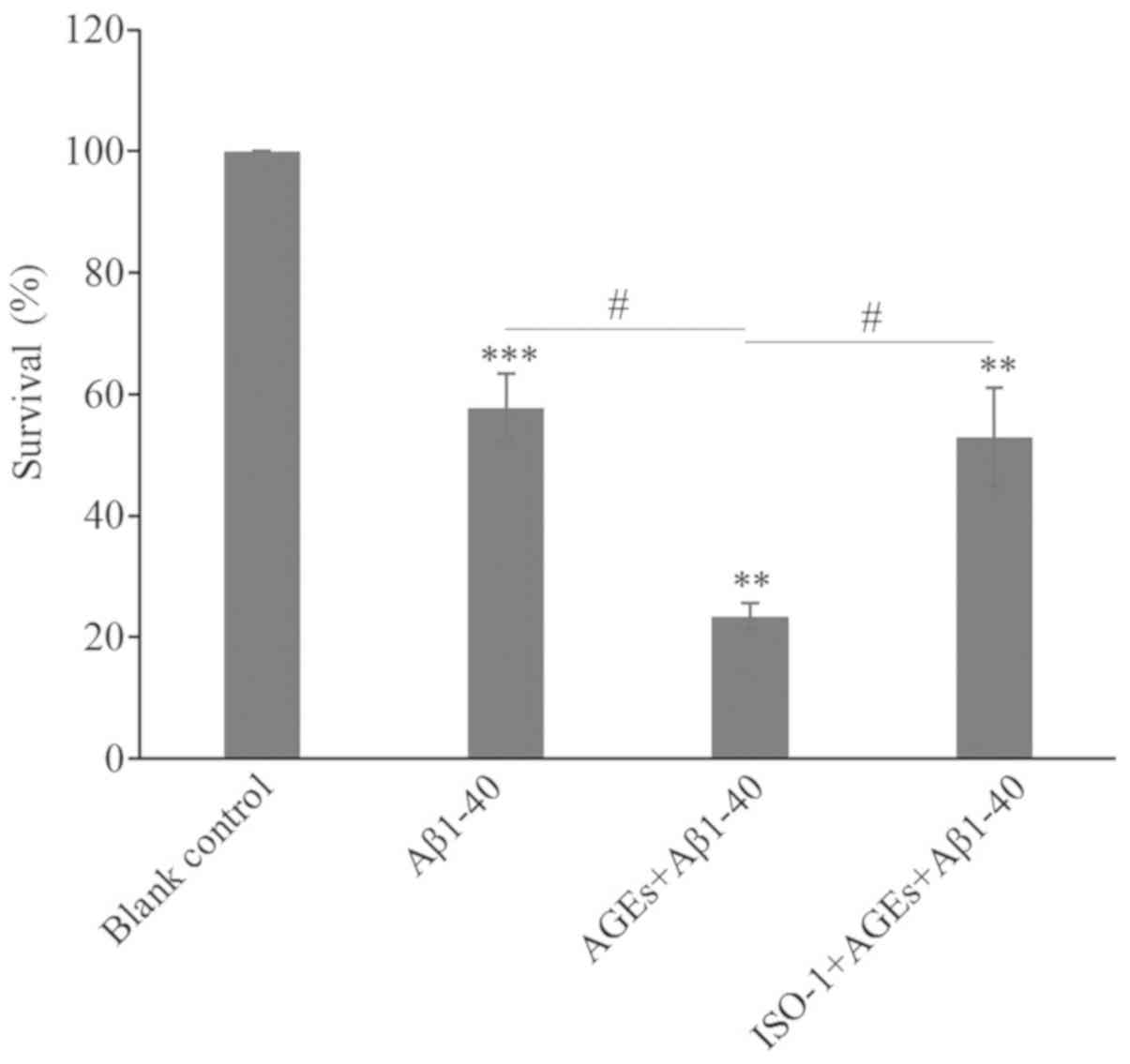
longer and more numerous dendrites (Fig. 3D). Compared with the blank control
group, the viability of cells in the Aβ1–40 model group
significantly decreased; the addition of AGEs further decreased
cell viability. Following pre-treatment with ISO-1, the survival
rate was significantly improved compared with AGEs and
Aβ1–40 alone (Fig. 4).
Based on these results, Aβ1–40 successfully established
an AD cell model. AGEs were found to aggravate the cytotoxicity of
Aβ1–40 in the AD cell model and this effect was
significantly alleviated by exposure to the MIF inhibitor
ISO-1.
 | Figure 3.AGEs promote apoptosis by inducing
MIF expression. Changes to the number and morphology of cells in
the (A) blank group, (B) AD cell model group, (C) AGEs-treated AD
model group and (D) ISO-1-pretreated group were observed under an
inverted microscope (magnification, ×100). AGEs inhibited the
proliferation of cells in the AD model; following pre-treatment
with the MIF inhibitor ISO-1, the decrease in cell proliferation
was notably attenuated. Scale bar, 100 µm. AGEs, advanced glycation
end products; Aβ1–40, amyloid β 1–40; AD, Alzheimer's
disease; MIF, macrophage migration inhibitory factor; ISO-1,
(S,R)-3-(4-hydroxyphenyl)-4,5-dihydro-5-isoxazole acetic acid
methyl ester. |
Expression of neuroinflammatory
cytokines and MIF
Compared with the blank control group, the
expression levels of interleukin (IL)-1β, IL-6, tumor necrosis
factor-α (TNF-α) and MIF mRNA in cells were significantly increased
following treatment with Aβ1–40. The effects of
Aβ1–40 on MIF expression were markedly smaller compared
with the effects on IL expression; it is possible that other
factors in the AD cell model may also regulate the expression of
MIF. The expression levels of IL-1β and TNF-α were notably further
increased by treatment with AGEs, and the expression of MIF and
IL-6 was significantly increased. Compared with the group treated
with AGEs, pre-treatment of cells with ISO-1 resulted in a
significant decrease in the mRNA expression levels of IL-1β, IL-6,
TNF-α and MIF (Fig. 5). Based on
these results, AGEs aggravated neuroinflammation in the AD model by
inducing MIF expression, whereas ISO-1 attenuated this damage.
 | Figure 5.AGEs promote neuroinflammation in the
AD cell model by inducing MIF expression. The mRNA expression of
(A) IL-1β, (B) IL-6, (C) TNF-α and (D) MIF were determined in the
blank, AD cell model, AGEs-treated AD model and the
ISO-1-pretreated groups via reverse transcription-quantitative PCR.
AGEs promoted neuroinflammation in the AD cell model by inducing
MIF expression. Data are presented as the mean ± SD; n=4.
*P<0.05, **P<0.01, ***P<0.001 vs. Blank control;
#P<0.05, ##P<0.01. AGEs, advanced
glycation end products; Aβ1–40, amyloid β 1–40; AD,
Alzheimer's disease; ISO-1,
(S,R)-3-(4-hydroxyphenyl)-4,5-dihydro-5-isoxazole acetic acid
methyl ester; IL, interleukin; TNF-α, tumor necrosis factor-α. |
Discussion
Diabetes has been reported to promote the occurrence
of AD by disrupting the insulin signal transduction pathway.
Insulin resistance and T2D may be associated with an increased
incidence of AD (6). AGEs,
important pathogenic factors involved in DM and its complications,
induce neurotoxicity by promoting the deposition of Aβ, the
hyperphosphorylation of tau protein and the expression of
proinflammatory cytokines in glial cells (14,15).
MIF inhibits macrophage migration and chemotaxis, and promotes the
aggregation of white blood cells at inflammatory sites, serving an
important role in a number of diseases, including rheumatoid
arthritis, septic shock, inflammatory lung diseases, cancer and
delayed-type hypersensitivity (16). MIF promotes the development of
diabetes by disrupting the mechanism that regulates the production
of inflammatory cytokines, causing insulin resistance and
inflammatory injury to islet cells (26). In the present study, the
association between AGEs and MIF, and its effects on
neuroinflammation in AD, was investigated. AGEs induced the
expression of MIF mRNA and protein in PC12 cells, and further
aggravated the occurrence of neuroinflammation in the AD cell
model. The results showed that the effects of AGEs on the
expression of MIF mRNA and protein were not in parallel; the
maximal expression of MIF mRNA was observed following treatment
with 200 µg/ml AGEs, whereas the expression of MIF protein further
increased following treatment with 300 µg/ml AGEs. This phenomenon
of non-parallel expression requires further investigation.
AD is a neurodegenerative disease. Hippocampal
neuronal injury is an important component of AD pathology and is
closely related to the occurrence of neuritis, the deposition of Aβ
and the hyperphosphorylation of tau proteins (27). A previous study into the causal
relationship between neuroinflammation and AD reported that
neuroinflammation may be involved in the pathogenesis of AD, and
that an increase in the levels neuroinflammatory mediators was
observed in AD (28). Thus,
neuroinflammation serves an important role in the development of
AD. Inflammatory mediators cause cognitive impairment through
cytokine-mediated glial cell activation and neuronal toxicity
(29). In addition, AD has been
associated with the upregulation of proinflammatory cytokines,
which promote neuronal degeneration (30). These cytokines include IL-1β, IL-6
and TNF-α, which are regarded as important mediators of
neuroinflammation and leukocyte infiltration (31). Notable roles of these cytokines
include the promotion of Aβ deposition and activation of glial
cells (32). In the present study,
cells were pre-treated with the MIF inhibitor ISO-1 to determine if
the increased expression of IL-1β, IL-6 and TNF-α was a result of
AGE-induced MIF expression. Cells in each group were observed under
an inverted microscope, and the effects of a MIF inhibitor on the
expression of mRNAs encoding inflammatory mediators were analyzed.
AGEs were revealed to mediate neuroinflammation in the AD cell
model by inducing MIF expression. MIF inhibitors block the
hyperactivation of microglia, and exert anti-inflammatory and
neuroprotective effects (33). It
was observed that ISO-1 protected the AD cell model from
AGEs-mediated damage. The results of the present study may have
important theoretical and practical significance in understanding
the pathogenesis and prevention of neuroinflammation in AD, and
improving treatment.
Previous studies have shown that MIF promotes the
development of AD by activating glial cells, initiating cascade
reactions, and promoting the occurrence of neuroinflammation, and
the formation of senile plaques and NFTs (19–21,33).
In the present study, it was demonstrated that AGEs promoted the
expression of MIF and aggravated the neuroinflammatory response at
the cell level. Future studies should investigate the significance
of AGEs in animal models and at the clinical level. Whether AGEs
promote MIF expression in glial cells, not only in nerve cells,
should also be addressed in future studies using animal models. It
should also be investigated as to whether AGEs induce the
activation of glial cells by promoting MIF expression, and if AGEs
then aggregate around nerve cells, leading to neuroinflammation and
the toxic effects of neuronal phagocytosis.
In the present study of the interaction between AGEs
and MIF, the molecular signaling pathways affected by AGEs were not
discussed in detail. The KEGG database (map04933) revealed that
AGEs promoted inflammation via a number of signalling pathways,
including the NF-κB and PI3K pathways (map not shown). The
signaling pathways induced by AGEs that promote the expression of
MIF will be investigated in subsequent studies. Novel ideas and
methods for the treatment of AD may be discovered with a more
complete understanding of the molecular mechanisms underlying
AD.
Acknowledgements
The authors would like to thank Professor Yu Ming
(Department of Neurology, Affiliated Hospital of Jiangsu
University; Jiangsu University) for providing comprehensive
guidance and suggestions on the present study, and the Central
Laboratory of The Affiliated Hospital of Jiangsu University for
providing a good environment for experiments and high-quality
experimental instruments. The authors would also like to thank Dr
Shu Yang (Central Laboratory of The Affiliated Hospital of Jiangsu
University), for providing technical assistance and support during
the present study.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81871343), and The
Jiangsu Provincial Key Research and Development Plan (grant nos.
BE2017699 and BE2017698).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MY and DZ conceived and designed the study, and
drafted the manuscript. DZ conducted the experimental protocols and
analyzed the experimental results. YX, JM and SQ contributed to the
analysis of data and revised the manuscript critically. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Alzheimer's Association: 2017 Alzheimer's
disease facts and figures. Alzheimer's Dementia. 13:325–373. 2017.
View Article : Google Scholar
|
|
2
|
Mushtaq G, Khan JA, Kumosani TA and Kamal
MA: Alzheimer's disease and type 2 diabetes via chronic
inflammatory mechanisms. Saudi J Biol Sci. 22:4–13. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Prince M, Bryce R, Albanese E, Wimo A,
Ribeiro W and Ferri CP: The global prevalence of dementia: A
systematic review and meta analysis. Alzheimers Dement. 9:63–75.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rawlings AM, Sharrett AR, Schneider AL,
Coresh J, Albert M, Couper D, Griswold M, Gottesman RF, Wagenknecht
LE, Windham BG and Selvin E: Diabetes in midlife and cognitive
change over 20 years: A cohort study. Ann Intern Med. 161:785–793.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ohara T, Doi Y, Ninomiya T, Hirakawa Y,
Hata J, Iwaki T, Kanba S and Kiyohara Y: Glucose tolerance status
and risk of dementia in the community: The hisayama study.
Neurology. 77:1126–1134. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kandimalla R, Thirumala V and Reddy PH: Is
Alzheimer's disease a type 3 diabetes? A critical appraisal.
Biochim Biophys Acta Mol Basis Dis. 1863:1078–1089. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
de la Monte SM and Wands JR: Alzheimer's
disease is type 3 diabetes-evidence reviewed. J Diabetes Sci
Technol. 2:1101–1113. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Steen E, Terry BM, Rivera EJ, Cannon JL,
Neely TR, Tavares R, Xu XJ, Wands JR and de la Monte SM: Impaired
insulin and insulin-like growth factor expression and signaling
mechanisms in Alzheimer's disease-is this type 3 diabetes? J
Alzheimers Dis. 7:63–80. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rivera EJ, Goldin A, Fulmer N, Tavares R,
Wands JR and de la Monte SM: Insulin and insulin-like growth factor
expression and function deteriorate with progressionof Alzheimer's
disease: Link to brain reductions in acet ylcholine. J Alzheimers
Dis. 8:247–268. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Goh SY and Cooper ME: Clinical review: The
role of advanced glycation end products in progression and
complications of diabetes. J Clin Endocrinol Metab. 93:1143–1152.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tanaka K, Yamaguchi T, Kanazawa I and
Sugimoto T: Effects of high glucose and advanced glycation end
products on the expressions of sclerostin and RANKL as well as
apoptosis in osteocyte-like MLO-Y4-A2 cells. Biochem Biophys Res
Commun. 461:193–199. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chang CC, Chen CY, Chang GD, Chen TH, Chen
WL, Wen HC, Huang C and Chang CH: Hyperglycemia and advanced
glycation end products (AGEs) suppress the differentiation of
3T3-L1 preadipocytes. Oncotarget. 8:55039–55050. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lin JA, Wu CH, Lu CC, Hsia SM and Yen GC:
Glycative stress from advanced glycation end products (AGEs) and
dicarbonyls: An emerging biological factor in cancer onset and
progression. Mol Nutr Food Res. 60:1850–1864. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sato T, Shimogaito N, Wu X, Kikuchi S,
Yamagishi S and Takeuchi M: Toxic advanced glycation end products
(TAGE) theory in Alzheimer's disease. Am J A1zheimers Dis Other
Demen 2l. 197–208. 2006. View Article : Google Scholar
|
|
15
|
Vitek MP, Bhattacharya K, Glendening JM,
Stopa E, Vlassara H, Bucala R, Manogue K and Cerami A: Advanced
glycation end products contribute to amyloidosis in alzheimer
disease. Proc Natl Acad Sci USA. 91:4766–4770. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lue H, Kleemann R, Calandra T, Roger T and
Bernhagen J: Macrophage migration inhibitory factor (MIF):
Mechanisms of action and role in disease. Microbes Infection.
4:449–460. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Grieb G, Merk M, Bernhagen J and Bucala R:
Macrophage migration inhibitory factor(MIF): A promising biomarker.
Drug News Perspect. 23:257–264. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Abed Y, Dabideen D, Aljabari B, Valster A,
Messmer D, Ochani M, Tanovic M, Ochani K, Bacher M, Nicoletti F, et
al: ISO-1 binding to the tautomerase activesite of MIF inhibits its
pro-inflammatory activity and increases survival in severe sepsis.
J Biol Chem. 280:36541–36544. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vujicic M, Senerovic L, Nikolic I, Saksida
T, Stosic-Grujicic S and Stojanovic I: The critical role of
macrophage migration inhibitory factor in insulin activity.
Cytokine. 69:39–46. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bacher M, Deuster O, Aljabari B,
Egensperger R, Neff F, Jessen F, Popp J, Noelker C, Reese JP,
Al-Abed Y and Dode R: The role of macrophage migration inhibitory
factor in Alzheimer's disease. Mol Med. 16:116–121. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Oyama R, Yamamoto H and Titani K:
Glutamine synthetase, hemoglobin alpha-chain, and macrophage
migration inhibitory factor binding to amyloid beta-protein: Their
identification in rat brain by a novel affinity chromatography and
in Alzheimer's disease brain by immunoprecipitation. Biochim
Biophys Acta. 1479:91–102. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li SQ, Yu Y, Han JZ, Wang D, Liu J, Qian
F, Fan GH, Bucala R and Ye RD: Deficiency of macrophage migration
inhibitory factor attenuates tau hyperphosphorylation in mouse
models of Alzheimer's disease. J Neuroinflammation. 12:1–11. 2015.
View Article : Google Scholar
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Luo XD, Xu B, Zhou J and Tian GP:
Protective effect of secretory factors of bone marrow mesenchymal
stem cells on beta-amyloid 1–40 induced apoptosis in PC12 cells. J
Clin Rehabilitat Tissue Eng Res. 13:7937–7941. 2009.
|
|
25
|
Kanehisa M, Furumichi M, Tanabe M, Sato Y
and Morishima K: KEGG: New perspectives on genomes, pathways,
diseases and drugs. Nucleic Acids Res. 45:D353–D361. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Benigni F, Atsunmi T, Calandra T, Metz C,
Echtenacher B, Peng T and Bucala R: The proinflammatory mediator
macrophage migration inhibitory factor induces glucose cataboilsm
in muscle. J Clin Invest. 106:1291–1300. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Blennow K, de Leon MJ and Zetterberg H:
Alzheimer's disease. Lancet. 368:387–403. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nagae T, Araki K, Shimoda Y, Sue LI, Beach
TG and Konishi Y: Cytokines and cytokine receptors involved in the
pathogenesis of Alzheimer's disease. J Clin Cell Immunol.
7:4412016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Heneka MT, Carson MJ, EI Khoury J,
Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T,
Vitorica J, Ransohoff RM, et al: Neuroinflammation in Alzheimer's
disease. Lancet Neurol. 14:388–405. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Azizi G and Mirshafiey A: The potential
role of proinflammatory and antiinflammatory cytokines in Alzheimer
disease pathogenesis. Immunopharmacol Immunotoxicol. 34:881–895.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Azizi G, Navabi SS, Al-Shukaili A,
Seyedzadeh MH, Yazdani R and Mirshafiey A: The role of inflammatory
mediators in the pathogenesis of Alzheimer's disease. Sultan Qaboos
Univ Med J. 15:e305–e316. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Azizi G, Khannazer N and Mirshafiey A: The
potential role of chemokines in Alzheimer's disease pathogenesis.
Am J Alzheimers Dis Other Demen. 29:415–425. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang Y, Gu R, Jia J, Hou T, Zheng LT and
Zhen X: Inhibition of macrophage migration inhibitory factor (MIF)
tautomerase activity suppresses microglia-mediated inflammatory
responses. Clin Exp Pharmacol Physiol. 43:1134–1144. 2016.
View Article : Google Scholar : PubMed/NCBI
|