Introduction
Colorectal cancer (CRC) is one of the most common
types of cancer worldwide, second to lung or breast cancer. In
addition, CRC is the third most common type of cancer among men and
the second among women worldwide. Annually, 30,000 new cases of CRC
are identified and there are 90,000 cases of CRC-associated
mortality globally (1). In
addition, CRC is the third leading cause of morbidity and mortality
among all malignant tumors in the United States (2). Despite continuing innovations in
cancer treatment, the survival rate of patients with advanced CRC
is low. The possibility of metastasis and recurrence is
significantly increased once the tumor enters the middle and late
stage, which is the main cause of mortality (3). In CRC, the 5-year survival rate for
cancer in situ is >65%; however, the 5-year survival rate
is between 25 and 60% if lymph node metastasis develops, and the
5-year survival rate remains <7% once tumor cells have
metastasized to distal organs (4).
Conventional chemotherapeutic drugs, including irinotecan,
oxaliplatin and fluorouracil, can improve the efficacy of
metastatic CRC (mCRC) treatment; however, the median survival of
patients remains <2 years (5,6). The
target epidermal growth factor receptor (EGFR) monoclonal antibody
cetuximab, as a single drug therapy or as part of combination
therapy, is the main method used to treat late mCRC (7). However, a number of patients are
still resistant to cetuximab following treatment (8,9).
The tumor suppressor gene Smad4 is an important
transcriptional factor in the transforming growth factor β
signaling pathway. Gene aberration, including chromosome fragment
loss, gene mutation and abnormal gene expression, often occurs in
CRC and other gastrointestinal tumors (10–13).
Smad4 is a member of the Smads protein family, and is located on
chromosome 18q21 (14). Clinical
studies have demonstrated that the risk of Smad4 deletion is
increased in patients with advanced CRC with liver metastasis, and
leads to poor prognosis (15–17).
By contrast, the median survival time of CRC patients with high
Smad4 expression is significantly longer compared with in those
with low Smad4 expression (14).
Previous studies have demonstrated that tumor cells
undergo epithelial-mesenchymal transition (EMT) with increased drug
resistance (18,19). EMT is a biological process in which
epithelial cells gradually transform into cells with an
interstitial phenotype through a specific procedure; this process
may be involved in numerous biological behaviors, including wound
healing and tumor metastasis (20–22).
Its main characteristics are decreased cell adhesion molecule
expression, transformation of the cytoskeleton from a cytokeratin
to vimentin phenotype, and morphological characteristics of
mesenchymal cells (22,23). From classic morphological
observations of CRC, it has been identified that reversible
morphological alterations occur during the process of tumor
invasion and metastasis. Therefore, EMT is considered to serve an
important role in CRC metastasis (24,25).
Although several studies have reported that mutation
or loss of Smad4 in CRC is closely associated with chemoresistance,
these previous studies have mainly focused on conventional
chemotherapeutic drugs, including 5-fluorouracil and oxaliplatin,
and classic pathways including Akt and PI3K signaling (26–29).
The present study aimed to investigate the effects of Smad4 on the
sensitivity of CRC cells to cetuximab, which is an EGFR monoclonal
antibody, and whether the effects were implicated in EMT.
Materials and methods
The Cancer Genome Atlas (TCGA)
database analysis
A total of 629 colorectal adenocarcinoma cases were
downloaded from TCGA database (http://www.cbioportal.org/). The mutations of Smad4,
and the expression of Smad4 in CRC and matched normal tissues were
analyzed.
Cell culture
Normal human colon epithelial cells (CCD 841 CoN
cells) and four CRC cell lines (SW480, LoVo, SW620 and LS174T) were
obtained from Invitrogen; Thermo Fisher Scientific, Inc. The cells
were cultured in RPMI 1640 medium (Gibco; Thermo Fisher Scientific,
Inc.) containing 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.), 0.03% glutamine and 100 µg/ml streptomycin at
37°C in an incubator with 5% CO2. The cells were
digested and subcultured with 0.25% trypsin when confluence reached
80–90%.
Cell transfection and grouping
The Smad4 silencing plasmid (siSmad4) (Forward:
5′-CGAAUACACCAACAAGUAATT-3′, Reverse:
5′-UUACUUGUUGGUGUAUAUUCGTA-3′), Smad4 overexpression plasmid and
empty control plasmid (negative control, NC) were purchased from
Invitrogen; Thermo Fisher Scientific, Inc. SW480 cells were seeded
in a 6-well plate (1.0×105 cells /well) for 24 h before
transfection and divided into three groups: i) the control (0.1%
PBS) subgroup, siNC subgroup, siSmad4 subgroup, NC subgroup and
Smad4 overexpression plasmid subgroup; ii) the control (0.1% PBS)
subgroup, cetuximab (20 µg/ml; Merck KGaA) subgroup, NC + cetuximab
(20 µg/ml) subgroup, siSmad4 + cetuximab (20 µg/ml) subgroup and
siSmad4 subgroup; iii) the control (0.1% PBS) subgroup, NC +
cetuximab (20 µg/ml) subgroup and Smad4 overexpression plasmid +
cetuximab (20 µg/ml) subgroup. Transient transfection was effected
by Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. A total
of 20 µM NC, siSmad4 or Smad4 overexpression plasmid, and 5 µl
Lipofectamine® 2000 were added to Opti-MEM and incubated
at 25°C for 10 min. Lipofectamine® 2000 was then mixed
into each well and the cells were cultured in Opti-MEM. After 6 h
of culturing, the medium was replaced with RPMI-1640 containing 10%
FBS. After 24 h, the cells were used to perform the subsequent
experiments.
Cell Counting kit-8 (CCK-8) viability
assay
In the present study, the CCK-8 assay was conducted
in triplicate. Cells were plated into 96-well plates at a seeding
density of 1×104 cells/well for 24 h. Initially, fresh
medium containing 0.1, 1, 10, 30, 50 or 100 µg/ml Cetuximab was
used to treat the cells for 24 or 48 h at 37°C, after which the
CCK-8 assay was performed. In addition, the CCK-8 assay was
performed to explore the effects of silencing or overexpressing
Smad4 on the viability of cetuximab-treated cells. Briefly, 10 µl
CCK-8 solution was added to the cells for 2 h at 37°C. Optical
density (OD) was measured at 450 nm (Thermo Fisher Scientific,
Inc.).
Cell wound scratch assay
Cells were seeded in 6-well plates (6×104
cells/well) and incubated at 37°C for 24 h. A sterile 100-µl
pipette tip was used to generate a wound in the center of the
plate. PBS was used to gently wash the cells three times, and
serum-free medium was added to the cells. Cell migration was
observed using an inverted light microscope at 24 h. The wound area
was measured using ImageJ software version 1.49 (National
Institutes of Health).
Cell invasion assay
Cell invasion was detected using a Matrigel-coated
Transwell assay. Briefly, 100 µl serum-free medium containing
105 cells was added into the upper chamber (Corning,
Inc.) and 600 µl culture medium containing 10% fetal bovine serum
was added to the lower chamber. The cells were incubated for 24–48
h at 37°C in order to detect invasion into the lower membrane.
Subsequently, the cells were fixed with 95% ethanol for 10 min at
25°C and stained with 0.5% crystal violet solution for 5 min at
room temperature after the Matrigel on the filter membrane was
removed.
Flow cytometry
Cell apoptosis was assessed by flow cytometry. Cells
were harvested, collected and washed with cold PBS twice. The
suspension (5×105 cells/well) in binding buffer was
cultured with Annexin V-APC/7-AAD apoptosis kit [cat. no. AP105,
Hangzhou Multi Sciences (Lianke) Biotech Co., Ltd.] in the dark at
25°C for 20 min. Binding buffer was then added to each well and the
samples were analyzed by flow cytometry within 1 h using BD
CellQuest Pro Software version 1.2; BD Biosciences).
Reverse transcription-quantitative PCR
(RT-qPCR)
Smad4, apoptosis-related genes (Bax and Bcl-2),
E-cadherin and vimentin were detected by RT-qPCR in the different
groups. Total RNA was extracted from cultured cells using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Total RNA was reverse
transcribed with the PrimeScript™ RT reagent kit (Takara Bio, Inc.)
at 37°C for 45 min and then at 85°C for 5 min. cDNA was amplified
using SYBR Fast qPCR Mix (Invitrogen; Thermo Fisher Scientific,
Inc.). For quantitative real-time PCR, the PCR reaction was set at
95°C for 5 min, followed by 33 cycles at 94°C for 30 sec, 55°C for
30 sec, 72°C for 45 sec and 72°C extension for 10 min. The primer
sequences used are listed in Table
I. mRNA expression was quantified using the 2−ΔΔCq
method (30). GAPDH served as an
internal control.
 | Table I.Primers used in reverse
transcription-quantitative PCR. |
Table I.
Primers used in reverse
transcription-quantitative PCR.
| Gene | Sequence |
|---|
| Smad4 | F:
5′-CTGAACTGTTTGTACCTCTGGGCCATATTGC-3′ |
|
| R:
5′-CAAATTTCTGAAGAGTAGGTGATCCGGGTGGAG-3′ |
| Bcl-2 | F:
5′-ATGGCGCACGCTGGGAGAAC-3′ |
|
| R:
5′-CTGGCGGAGGGTCAGGTGGA-3′ |
| Bax | F:
5′-GCCGCCGTGGACACAGACTC-3′ |
|
| R:
5′-CCGCTCCCGGAGGAAGTCCA-3′ |
| E-cadherin | F:
5′-ATCCAAAGCCTCAGGTCATA-3′ |
|
| R:
5′-CAGCAAGAGCAGCAGAAT-3′ |
| Vimentin | F:
5′-GAACTTTGCCGTTGAAGCTG-3′ |
|
| R:
5′-TCTCAATGTCAAGGGCCATC-3′ |
| GAPDH | F:
5′-ACCACAGTCCATGCCATCAC-3′ |
|
| R:
5′-TCCACCACCCTGTTGCTGTA-3′ |
Western blotting
Total protein was extracted from cultured cells
using lysis buffer; RIPA buffer (Thermo Fisher Scientific, Inc.)
was added to each well and cells were centrifuged at 14,000 × g for
15 min at 4°C. Bicinchoninic acid protein quantification (Thermo
Fisher Scientific, Inc.) was used to detect protein concentration.
Proteins (20 µg/lane) were separated by 10% SDS-PAGE and were then
transferred onto polyvinylidene difluoride membranes, which were
blocked in TBS-0.1% Tween-20 (TBST) containing 1% milk at room
temperature for 2 h. The membranes were then incubated with the
following primary antibodies: Rabbit anti-Smad4 (cat. no. ab40759,
1:5,000; Abcam), anti-Bcl-2 (cat. no. ab182858, 1:2,000; Abcam),
anti-Bax (cat. no. ab32503, 1:1,500; Abcam), mouse anti- E-cadherin
(cat. no. ab1416, 1:50; Abcam), anti-vimentin (cat. no. ab8978,
1:1,000; Abcam) and anti-GAPDH (cat. no. ab8245, 1:1,000; Abcam).
The membranes were washed with TBST, and were then incubated with
horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G
(1:2,000; SA00001-2; ProteinTech Group, Inc.) and with horseradish
peroxidase -conjugated goat anti-mouse IgG (1:2,000; sc-516102;
Santa Cruz Biotechnology, Inc.) as secondary antibodies. The blots
were visualized by enhanced chemiluminescence (ECL; Thermo Fisher
Scientific, Inc.). An ECL system (Amersham; GE Healthcare) was used
to detect the bands. The density of the blots was measured using
Quantity One software version 2.4 (Bio-Rad Laboratories, Inc.).
Statistical analysis
Statistical analysis was performed using Prism
GraphPad version 6.0 software (GraphPad Software, Inc.). All data
are presented as the means ± standard deviation. Differences were
analyzed using one-way analysis of variance following Tukey's
multiple comparison post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
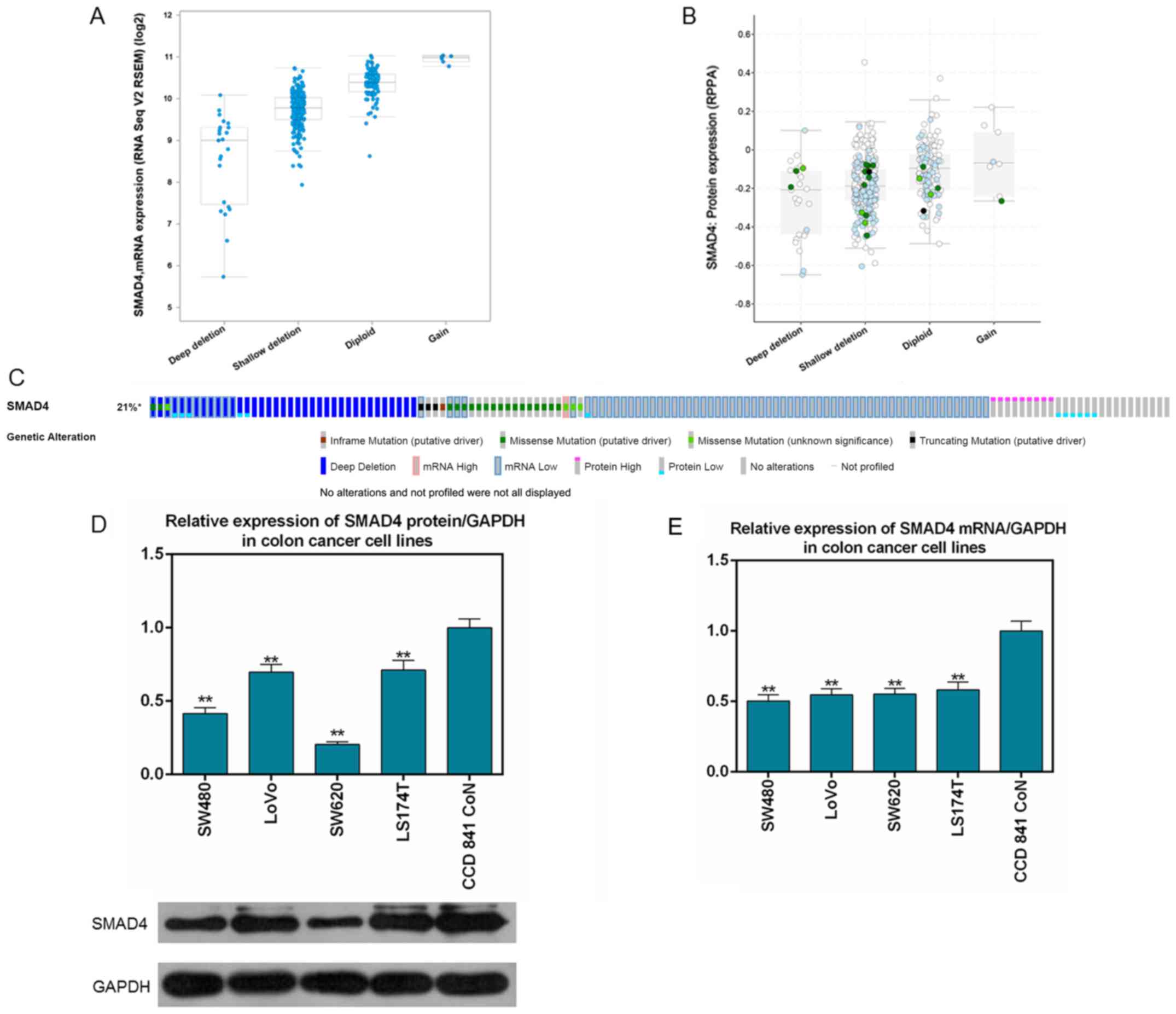
Low expression of Smad4 in CRC tissues
and CRC cell lines
TCGA data (Fig. 1A and
B) demonstrated that Smad4 was mutated in 131 (21%) out of 629
sequenced cases/patients with colorectal adenocarcinoma. The
majority of the mutations were deletions. In addition, mRNA and
protein expression levels of Smad4 in cancer tissues were lower
than those in normal tissues in the cases downloaded from TCGA
(Fig. 1C). The expression levels
of Smad4 in normal colonic epithelial cells and four CRC cell lines
were determined by RT-qPCR and western blot analysis. The results
demonstrated that expression of Smad4 in CRC cell lines (SW480,
LoVo, SW620 and LS174T) was significantly decreased compared with
in CCD 841 CoN cells at the mRNA and protein levels (P<0.01;
Fig. 1D and E). The SW480 cell
line was selected as the subject of subsequent experiments as it
had the lowest expression of Smad4 at the mRNA and protein
levels.
Silencing Smad4 attenuates cell
sensitivity to cetuximab in SW480 CR C cells
SW480 cells were treated with or without different
concentrations (0.1, 1, 10, 30, 50 and 100 µg/ml) of cetuximab.
There was a difference in OD 450 nm with 0 µg/ml Cetuximab since
the cells proliferated after 24 and 48 h. Cell viability was
increased in response to treatment with different concentrations of
cetuximab for 24 and 48 h compared to 0 h (P<0.05; Fig. 2A). The results indicated that
certain cells already had drug resistance, and therefore,
the inhibitory effect of cetuximab was not significant at 24 h.
However, at 48 h, cell viability was downregulated in a
dose-dependent manner until the cells were treated with 20 µg/ml
cetuximab. Therefore, 20 µg/ml cetuximab (48 h) was selected as an
effective concentration in subsequent experiments. The transfection
efficiency of siSmad4 was demonstrated in Fig. 2B and C; the expression levels of
Smad4 were significantly decreased post-transfection with siSmad4.
In addition, treatment with 20 µg/ml cetuximab (P<0.05) or
siSmad4 (P<0.01) significantly inhibited Smad4 protein (Fig. 2D) and mRNA (Fig. 2E) expression. When cetuximab was
used in combination with siSmad4, the inhibitory effect on Smad4
expression was at its most significant (P<0.01). Furthermore,
cetuximab treatment alone and NC + cetuximab markedly downregulated
SW480 cell viability (P<0.01; Fig.
2F). However, the effect of cetuximab treatment was clearly
inhibited when cetuximab was used in combination with siSmad4,
compared with single cetuximab or NC + cetuximab (P<0.01). In
addition, compared with groups treated with cetuximab, siSmad4
alone significantly enhanced cell viability (P<0.01). Silencing
Smad4 not only increased SW480 cell viability, but also partly
reversed the cell viability decrease induced by cetuximab,
indicating that silencing Smad4 may attenuate cell sensitivity to
cetuximab in the SW480 CRC cell line.
 | Figure 2.mRNA and protein expression levels of
Smad4, and cell viability analysis in SW480 colorectal cancer cells
with or without cetuximab treatment. (A) Different concentrations
(0.1, 1, 10, 30, 50 and 100 µg/ml) of cetuximab were used to treat
SW480 cells for 0, 24 or 48 h, and cell viability was detected by
CCK-8 assay. *P<0.05 and **P<0.01 vs. 0 h. (B) Protein
expression levels of Smad4 were assessed post-transfection with
plasmids silencing or overexpressing Smad4 by western blotting in
SW480 cells. (C) mRNA expression levels of Smad4 in SW480 cells
post-transfection with plasmids silencing and overexpressing Smad4
by reverse transcription-quantitative PCR. GAPDH served as an
internal control. Data are expressed as the means ± standard
deviation from three independent experiments. **P<0.01 vs.
control; ##P<0.01 vs. NC. (D) Protein expression
levels of Smad4 were assessed post-transfection with a plasmid
silencing Smad4 using western blot analysis in SW480 cells.
*P<0.05 and **P<0.01 vs. control; ##P<0.01 vs.
cetuximab; ^^P<0.01 vs. NC + cetuximab. (E) mRNA
expression levels of Smad4 were assessed post-transfection by
reverse transcription-quantitative PCR in SW480 cells. GAPDH served
as an internal control. (F) Viability of SW480 CRC cells was
assessed by CCK-8 assay after 48 h. Data are expressed as the means
± standard deviation from three independent experiments. *P<0.05
and **P<0.01 vs. control; ■P<0.05 and
■■P<0.01 vs. siSmad4 + cetuximab;
##P<0.01 vs. cetuximab; ^^P<0.01 vs. NC
+ cetuximab. CCK-8, Cell Counting kit-8; CRC, colorectal cancer;
NC, negative control; OD, optical density; siSmad4, small
interfering RNA-Smad4. |
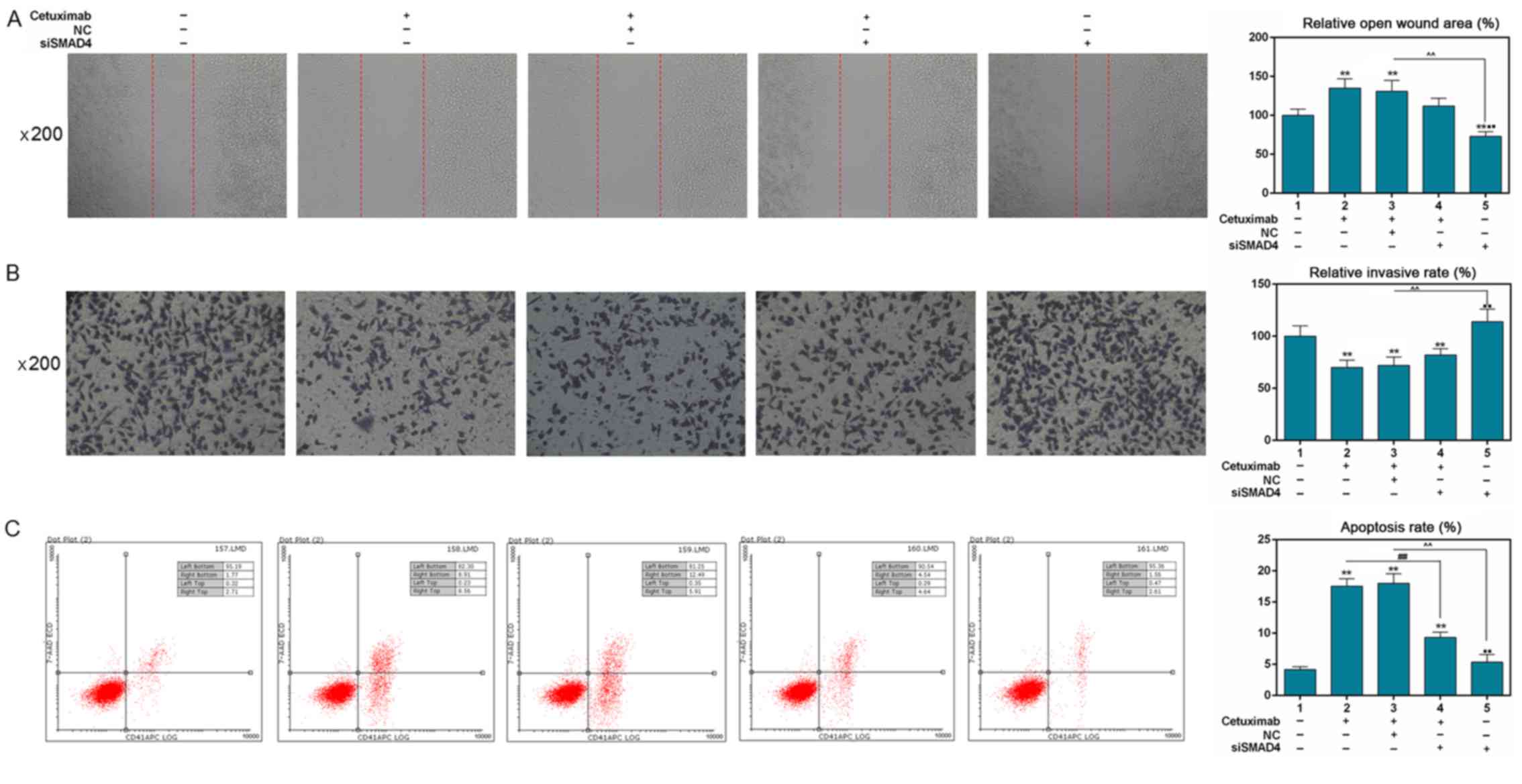
Silencing Smad4 partly reverses the
decrease in cell migration and invasion, and the increase in cell
apoptosis induced by cetuximab
Cell migration was detected using a scratch assay.
The results revealed that cetuximab and NC + cetuximab treatment
significantly increased the open wound area, compared with the
control (P<0.01; Fig. 3A).
Silencing Smand4 markedly reduced the open wound area (P<0.01;
Fig. 3A). The open wound area was
decreased by siSmad4 in combination with cetuximab compared with
siSmad4 treatment alone; however, the decreased effect was not
significant compared with cetuximab or cetuximab + NC. A Transwell
assay was used to determine cell invasion and the result
demonstrated that the invasion rate of SW480 cells was markedly
decreased in groups treated with cetuximab (P<0.01; Fig. 3B). Conversely, invasion of cells
transfected with siSmad4 was significantly increased (P<0.01;
Fig. 3B). However, silencing Smad4
only partly increased the cell invasion attenuated by cetuximab.
The effects of silencing Smad4 on the apoptosis of SW480 cells with
or without cetuximab were detected by flow cytometric analysis. A
noticeable increase in apoptosis (P<0.01; Fig. 3C) was identified in the cetuximab
(17.47%), NC + cetuximab (18.4%) and siSmad4 + cetuximab (9.18%)
groups, compared with in the control group (4.48%). Silencing Smad4
(4.17%) significantly downregulated apoptosis rate (P<0.01)
compared with in the cetuximab (17.47%) or siSmad4 + cetuximab
(9.18%) groups. In addition, silencing Smad4 partly reversed the
increase in cell apoptosis induced by cetuximab (P<0.01).
Effects of silencing Smad4 on
apoptosis- and EMT-related gene expression in SW480 cells with or
without cetuximab treatment
The expression levels of the apoptosis-related genes
Bax and Bcl-2 were detected by RT-qPCR and western blot analysis.
The results also demonstrated that cetuximab downregulated Bcl-2
(P<0.01; Fig. 4A, B and F) and
upregulated Bax protein (P<0.01; Fig. 4A and C) and mRNA expression
(P<0.01; Fig. 4G) compared with
control. Silencing Smad4 partly reversed the decreased expression
of Bcl-2 and the increased expression of Bax (protein, P<0.01)
by cetuximab. Silencing Smad4 treatment alone could attenuate Bax
expression in comparison with siSmad4 + cetuximab (protein,
P<0.05; mRNA, P<0.01). To further determine how silencing
Smad4 could affect EMT in SW480 CRC cells with or without cetuximab
treatment, the expression of EMT-associated genes (E-cadherin and
vimentin) was detected using RT-qPCR and western blot analysis. The
results demonstrated that both protein (Fig. 4A and D) and mRNA levels (Fig. 4H) of E-cadherin were increased in
the cetuximab and NC + cetuximab groups compared with in the
control group (P<0.01). Conversely, the mRNA and protein
expression levels of E-cadherin were significantly downregulated in
the siSmad4 and siSmad4 + cetuximab groups compared with in the
cetuximab or NC + cetuximab groups (P<0.01). The effects of
siSmad4 on vimentin expression demonstrated opposite results to
those on E-cadherin at the mRNA and protein levels (P<0.01;
Fig. 4A, E and I). These results
indicated that silencing Smad4 partly reversed the effects of
cetuximab on the expression of apoptosis- and EMT-related
genes.
Overexpression of Smad4 enhances the
sensitivity of SW480 cells to cetuximab
The transfection efficiency of the Smad4
overexpression plasmid was demonstrated in Fig. 2B and C; Smad4 expression was
significantly increased post-transfection with the overexpression
plasmid. As shown in Fig. 5A-C,
cetuximab significantly inhibited Smad4 protein (P<0.05) and
mRNA expression (P<0.01). Smad4 overexpression partly reversed
the expression of Smad4 decreased by cetuximab (P<0.01). Cell
viability was detected using the CCK-8 assay; the results
demonstrated that NC + cetuximab significantly decreased cell
viability (P<0.01; Fig. 5D).
Cetuximab in combination with Smad4 overexpression further
decreased cell viability compared with the control group
(P<0.01). In addition, cell viability was attenuated in the
Smad4 overexpression + cetuximab group compared with in the NC +
cetuximab group (P<0.05), suggesting that overexpressing Smad4
may enhance the sensitivity of SW480 cells to cetuximab.
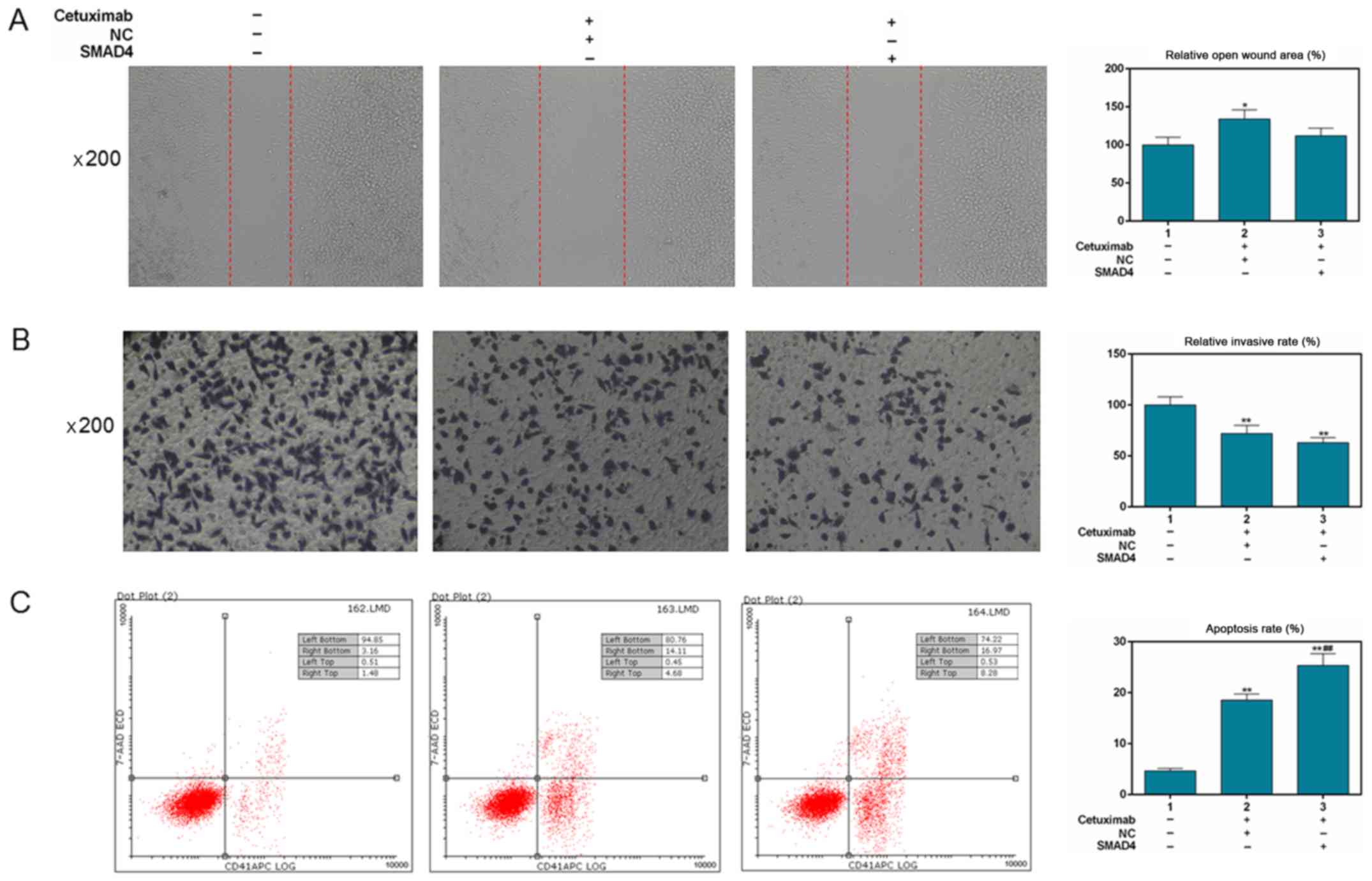
Overexpression of Smad4 enhances the
effects of cetuximab on invasion, migration and apoptosis of SW480
cells
As shown in Fig.
6A, NC + cetuximab slightly increased the wound area compared
with in the control group (P<0.05). Overexpression of Smad4 had
no evident effect on the inhibition of migration induced by
cetuximab (P>0.05). The Transwell assay demonstrated that NC +
cetuximab and Smad4 overexpression + cetuximab significantly
downregulated invasion rate (P<0.01; Fig. 6B). For cell apoptosis, the results
demonstrated that cetuximab treatment alone (18.79%), and also
Smad4 overexpression + cetuximab (25.25%) significantly upregulated
the apoptosis rate compared with in the control group (4.64%,
P<0.01; Fig. 6C). Additionally,
the apoptotic effect of Smad4 overexpression + cetuximab was
significantly different compared with treatment with cetuximab
alone (P<0.01).
Effects of Smad4 overexpression on
apoptosis- and EMT-related gene expression in SW480 cells treated
with cetuximab
The results revealed that the anti-apoptosis gene
Bcl-2 was markedly decreased in the Smad4 overexpression +
cetuximab group compared with in the control and NC + cetuximab
groups, at the mRNA and protein levels (P<0.01; Fig. 7A, B and F). At the protein level,
the expression of Bax was significantly increased in the Smad4
overexpression + cetuximab and NC + cetuximab groups compared with
in the control group (P<0.01; Fig.
7A and C). Notably, the increase in Bax expression was more
noticeable in the Smad4 overexpression + cetuximab group compared
with in the NC + cetuximab group. The effects of treatments on the
mRNA expression levels of Bax were consistent with those on protein
expression (P<0.01; Fig. 7G).
Furthermore, the protein and mRNA expression levels of E-cadherin
were significantly upregulated in the Smad4 overexpression +
cetuximab group compared with in the control and NC + cetuximab
groups (P<0.01; Fig. 7A, D and
H). Conversely, the protein expression levels of vimentin were
significantly decreased in the Smad4 overexpression + cetuximab
group compared with in the control and NC + cetuximab groups
(P<0.01; Fig. 7A and E). At the
mRNA level, the Smad4 overexpression + cetuximab group exhibited no
clear difference in vimentin expression compared with the NC +
cetuximab group (P>0.05; Fig.
7I). However, the mRNA expression levels of vimentin were
markedly downregulated compared with in the control group
(P<0.01; Fig. 7I).
Discussion
Smad4 is absent or mutated in numerous tumor types,
including pancreatic, colon, gastric and liver cancer, and
particularly in digestive system tumors. Therefore, it is thought
to function as a tumor suppressor gene (31–34).
One of the major causes of CRC progression is chromosomal
instability, including APC, p53 and DCC/Smad4 deficiency (35–37).
Therefore, the roles of Smad4 cannot be neglected in the
development of CRC (38,39). Surgery is the main method for
treating early CRC; however, recurrence and metastasis often occur
following surgery. With the increasing use of adjuvant therapy
using chemicals such as cetuximab, drug resistance of tumor cells
is a pivotal cause of poor prognosis among patients with CRC
(6).
The present study revealed that cetuximab treatment
alone inhibited SW480 cell viability, migration and invasion, and
promoted cell apoptosis. Although Mei et al (40) reported that Smad4 and neurofibromin
1 mutations may be potential biomarkers for poor responses to
cetuximab-based therapy, related molecular mechanisms have not been
studied. The present study suggested that silencing Smad4 in
combination with cetuximab treatment enhanced SW480 cell viability,
migration and invasion, and inhibited cell apoptosis. Conversely,
the sensitivity of SW480 cells to cetuximab was enhanced when SW480
cells were transfected with a plasmid overexpressing Smad4. The
results indicated that the expression of Smad4 may serve a role in
the response of SW480 cells to cetuximab. Previous studies have
reported that downregulation of Smad4 is associated with a poor
prognosis in response to 5-fluorouracil chemotherapy among patients
with advanced CRC (15,41). Evidence also demonstrated that
Smad4 is associated with chemotherapy and radiotherapy resistance
in cancer. For example, Smad4 knockdown promotes cetuximab
resistance in head and neck squamous cell carcinoma by inducing
mitogen-activated protein kinases/JNK activation (42,43).
Wang et al (44) suggested
that Smad4 mutations are responsible for the resistance of
pancreatic cancer to radiotherapy. Sun et al (45) also reported that microRNA-574
promotes doxorubicin resistance of MCF-7 cells by downregulating
Smad4 in breast cancer cells.
A number of studies have reported that low
expression of Smad4 is associated with EMT and poor prognosis of
colon cancer (46–48). Therefore it was speculated that
this may be the underlying mechanism by which Smad4 affects the
sensitivity of SW480 CRC cells to cetuximab. In the present study,
silencing Smad4 partly reversed the expression of E-cadherin
increased by cetuximab and the expression of vimentin decreased by
cetuximab. EMT commonly occurs in the heart or palate, and is
associated with placentation (49–53).
In recent years, EMT has been proposed to be involved in the
invasion and metastasis of tumor cells (49). Accordingly, fibroblastoid or
‘spindle-cell’ tumors of epithelial origin have been characterized
as highly malignant and invasive (54–56).
E-cadherin is an important component of epithelial intercellular
junctions and vimentin is an interstitial cell-specific protein;
the two proteins are typical characteristic markers of EMT
(57–60). Fuchs et al (61) suggested that EMT serves an
important role in cetuximab resistance to hepatocellular carcinoma.
Liu et al has also demonstrated that Smad4 is implicated
with cancer cell resistance caused by EMT (62).
Studies of the definite mechanism underlying the
effect rendered by Smad4 may also be beneficial. Various factors,
including Wnt, Hedgehog and Notch signaling, may regulate the
association between EMT and cancer cell resistance (63–65).
However, there remain a number of shortcomings in the present
study, including the lack of in vivo experiments. We aim to
conduct a further detailed investigation into the association of
Smad4 protein levels with E-cadherin or vimentin protein levels in
CRC tissues or matched normal tissues, and also to perform
validation experiments in one more cell line and in vivo
mice models.
In conclusion, Smad4 may serve a vital role in the
sensitivity of CRC cells to chemotherapy via EMT. Furthermore, a
high expression of Smad4 may clinically benefit cetuximab-based
treatment.
Acknowledgements
Not applicable.
Funding
The present study was supported by The National Key
Clinical Specialist Construction Program of China; and Medical
Innovation Project of Fujian Health Department (grant no.
2017-CX-29).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZL made substantial contributions to conception and
design; LZ, JZho and JZhe performed data acquisition, data analysis
and interpretation. All authors drafted the article or critically
revised it for important intellectual content and All authors
approved the final version to be published. All authors agreed to
be accountable for all aspects of the work in ensuring that
questions related to the accuracy or integrity of the work were
appropriately investigated and resolved:
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
O'Connell MJ, Campbell ME, Goldberg RM,
Grothey A, Seitz JF, Benedetti JK, André T, Haller DG and Sargent
DJ: Survival following recurrence in stage II and III colon cancer:
Findings from the ACCENT data set. J Clin Oncol. 26:2336–2341.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Weitz J, Koch M, Debus J, Höhler T, Galle
PR and Büchler MW: Colorectal cancer. Lancet. 365:153–165. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Van Cutsem E, Köhne CH, Láng I, Folprecht
G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D,
Tejpar S, et al: Cetuximab plus irinotecan, fluorouracil, and
leucovorin as first-line treatment for metastatic colorectal
cancer: Updated analysis of overall survival according to tumor
KRAS and BRAF mutation status. J Clin Oncol. 29:2011–2019. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Meyerhardt JA and Mayer RJ: Systemic
therapy for colorectal cancer. N Engl J Med. 352:476–487. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cunningham D, Humblet Y, Siena S, Khayat
D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype
C, et al: Cetuximab monotherapy and cetuximab plus irinotecan in
irinotecan-refractory metastatic colorectal cancer. N Engl J Med.
351:337–345. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Misale S, Di Nicolantonio F,
Sartore-Bianchi A, Siena S and Bardelli A: Resistance to anti-EGFR
therapy in colorectal cancer: From heterogeneity to convergent
evolution. Cancer Discov. 4:1269–1280. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bronte G, Silvestris N, Castiglia M,
Galvano A, Passiglia F, Sortino G, Cicero G, Rolfo C, Peeters M,
Bazan V, et al: New findings on primary and acquired resistance to
anti-EGFR therapy in metastatic colorectal cancer: Do all roads
lead to RAS? Oncotarget. 6:24780–24796. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Demagny H and De Robertis EM: Point
mutations in the tumor suppressor Smad4/DPC4 enhance its
phosphorylation by GSK3 and reversibly inactivate TGF-β signaling.
Mol Cell Oncol. 3:e10251812015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Inamoto S, Itatani Y, Yamamoto T,
Minamiguchi S, Hirai H, Iwamoto M, Hasegawa S, Taketo MM, Sakai Y
and Kawada K: Loss of SMAD4 promotes colorectal cancer progression
by accumulation of myeloid-derived suppressor cells through the
CCL15-CCR1 chemokine axis. Clin Cancer Res. 22:492–501. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Voorneveld PW, Kodach LL, Jacobs RJ, Liv
N, Zonnevylle AC, Hoogenboom JP, Biemond I, Verspaget HW, Hommes
DW, de Rooij K, et al: Loss of SMAD4 alters BMP signaling to
promote colorectal cancer cell metastasis via activation of Rho and
ROCK. Gastroenterology. 147:196–208.e113. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu L, Nie J, Chen L, Dong G, Du X, Wu X,
Tang Y and Han W: The oncogenic role of microRNA-130a/301a/454 in
human colorectal cancer via targeting Smad4 expression. PLoS One.
8:e555322013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Alazzouzi H, Alhopuro P, Salovaara R,
Sammalkorpi H, Järvinen H, Mecklin JP, Hemminki A, Schwartz S Jr,
Aaltonen LA and Arango D: SMAD4 as a prognostic marker in
colorectal cancer. Clin Cancer Res. 11:2606–2611. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Alhopuro P, Alazzouzi H, Sammalkorpi H,
Dávalos V, Salovaara R, Hemminki A, Järvinen H, Mecklin JP,
Schwartz S Jr, Aaltonen LA and Arango D: SMAD4 levels and response
to 5-fluorouracil in colorectal cancer. Clin Cancer Res.
11:6311–6316. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Losi L, Bouzourene H and Benhattar J: Loss
of Smad4 expression predicts liver metastasis in human colorectal
cancer. Oncol Rep. 17:1095–1099. 2007.PubMed/NCBI
|
|
17
|
Miyaki M, Iijima T, Konishi M, Sakai K,
Ishii A, Yasuno M, Hishima T, Koike M, Shitara N, Iwama T, et al:
Higher frequency of Smad4 gene mutation in human colorectal cancer
with distant metastasis. Oncogene. 18:3098–3103. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shintani Y, Okimura A, Sato K, Nakagiri T,
Kadota Y, Inoue M, Sawabata N, Minami M, Ikeda N, Kawahara K, et
al: Epithelial to mesenchymal transition is a determinant of
sensitivity to chemoradiotherapy in non-small cell lung cancer. Ann
Thorac Surg. 92:1794–1804. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tomono T, Yano K and Ogihara T:
Snail-induced epithelial-to-mesenchymal transition enhances
P-gp-mediated multidrug resistance in HCC827 cells. J Pharm Sci.
106:2642–2649. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shi Q, Diao Y, Jin F and Ding Z:
Antimetastatic effects of Aidi on human esophageal squamous cell
carcinoma by inhibiting epithelialmesenchymal transition and
angiogenesis. Mol Med Rep. 18:131–138. 2018.PubMed/NCBI
|
|
21
|
Zheng J, Zhang M, Zhang L, Ding X, Li W
and Lu S: HSPC159 promotes proliferation and metastasis via
inducing EMT and activating PI3K/Akt pathway in breast cancer.
Cancer Sci. 109:2153–2163. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kalluri R: EMT: When epithelial cells
decide to become mesenchymal-like cells. J Clin Invest.
119:1417–1419. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Greenburg G and Hay ED: Epithelia
suspended in collagen gels can lose polarity and express
characteristics of migrating mesenchymal cells. J Cell Biol.
95:333–339. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Brabletz T, Hlubek F, Spaderna S,
Schmalhofer O, Hiendlmeyer E, Jung A and Kirchner T: Invasion and
metastasis in colorectal cancer: Epithelial-mesenchymal transition,
mesenchymal-epithelial transition, stem cells and beta-catenin.
Cells Tissues Organs. 179:56–65. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Polyak K and Weinberg RA: Transitions
between epithelial and mesenchymal states: Acquisition of malignant
and stem cell traits. Nat Rev Cancer. 9:265–273. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Moon SU, Kang MH, Sung JH, Kim JW, Lee JO,
Kim YJ, Lee KW, Bang SM, Lee JS and Kim JH: Effect of Smad3/4 on
chemotherapeutic drug sensitivity in colorectal cancer cells. Oncol
Rep. 33:185–192. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang B, Zhang B, Chen X, Bae S, Singh K,
Washington MK and Datta PK: Loss of Smad4 in colorectal cancer
induces resistance to 5-fluorouracil through activating Akt
pathway. Br J Cancer. 110:946–957. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Papageorgis P, Cheng K, Ozturk S, Gong Y,
Lambert AW, Abdolmaleky HM, Zhou JR and Thiagalingam S: Smad4
inactivation promotes malignancy and drug resistance of colon
cancer. Cancer Res. 71:998–1008. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sun C, Wang FJ, Zhang HG, Xu XZ, Jia RC,
Yao L and Qiao PF: miR-34a mediates oxaliplatin resistance of
colorectal cancer cells by inhibiting macroautophagy via
transforming growth factor-β/Smad4 pathway. World J Gastroenterol.
23:1816–1827. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhao S, Venkatasubbarao K, Lazor JW,
Sperry J, Jin C, Cao L and Freeman JW: Inhibition of STAT3 Tyr705
phosphorylation by Smad4 suppresses transforming growth factor
beta-mediated invasion and metastasis in pancreatic cancer cells.
Cancer Res. 68:4221–4228. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang H, Rajan S, Liu G and Chakrabarty S:
Transforming growth factor beta suppresses beta-catenin/Wnt
signaling and stimulates an adhesion response in human colon
carcinoma cells in a Smad4/DPC4 independent manner. Cancer Lett.
264:281–287. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Barros R, Pereira B, Duluc I, Azevedo M,
Mendes N, Camilo V, Jacobs RJ, Paulo P, Santos-Silva F, van
Seuningen I, et al: Key elements of the BMP/SMAD pathway
co-localize with CDX2 in intestinal metaplasia and regulate CDX2
expression in human gastric cell lines. J Pathol. 215:411–420.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huang S, Zhang F, Miao L, Zhang H, Fan Z,
Wang X and Ji G: Lentiviral-mediated Smad4 RNAi induced
anti-proliferation by p16 up-regulation and apoptosis by caspase 3
down-regulation in hepatoma SMMC-7721 cells. Oncol Rep.
20:1053–1059. 2008.PubMed/NCBI
|
|
35
|
Druliner BR, Ruan X, Sicotte H, O'Brien D,
Liu H, Kocher JA and Boardman L: Early genetic aberrations in
patients with sporadic colorectal cancer. Mol Carcinog. 57:114–124.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Al-Shamsi HO, Jones J, Fahmawi Y, Dahbour
I, Tabash A, Abdel-Wahab R, Abousamra AO, Shaw KR, Xiao L, Hassan
MM, et al: Molecular spectrum of KRAS, NRAS, BRAF, PIK3CA, TP53,
and APC somatic gene mutations in Arab patients with colorectal
cancer: Determination of frequency and distribution pattern. J
Gastrointest Oncol. 7:882–902. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Müller MF, Ibrahim AE and Arends MJ:
Molecular pathological classification of colorectal cancer.
Virchows Arch. 469:125–134. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mehrvarz Sarshekeh A, Advani S, Overman
MJ, Manyam G, Kee BK, Fogelman DR, Dasari A, Raghav K, Vilar E,
Manuel S, et al: Association of SMAD4 mutation with patient
demographics, tumor characteristics, and clinical outcomes in
colorectal cancer. PLoS One. 12:e01733452017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang C, Zhou Y, Ruan R, Zheng M, Han W and
Liao L: High expression of COUP-TF II cooperated with negative
Smad4 expression predicts poor prognosis in patients with
colorectal cancer. Int J Clin Exp Pathol. 8:7112–7121.
2015.PubMed/NCBI
|
|
40
|
Mei Z, Shao YW, Lin P, Cai X, Wang B, Ding
Y, Ma X, Wu X, Xia Y, Zhu D, et al: SMAD4 and NF1 mutations as
potential biomarkers for poor prognosis to cetuximab-based therapy
in Chinese metastatic colorectal cancer patients. BMC Cancer.
18:4792018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang B, Leng C, Wu C, Zhang Z, Dou L, Luo
X, Zhang B and Chen X: Smad4 sensitizes colorectal cancer to
5-fluorouracil through cell cycle arrest by inhibiting the
PI3K/Akt/CDC2/survivin cascade. Oncol Rep. 35:1807–1815. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ozawa H, Ranaweera RS, Izumchenko E,
Makarev E, Zhavoronkov A, Fertig EJ, Howard JD, Markovic A, Bedi A,
Ravi R, et al: SMAD4 loss is associated with cetuximab resistance
and induction of MAPK/JNK activation in head and neck cancer cells.
Clin Cancer Res. 23:5162–5175. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cheng H, Fertig EJ, Ozawa H, Hatakeyama H,
Howard JD, Perez J, Considine M, Thakar M, Ranaweera R, Krigsfeld G
and Chung CH: Decreased SMAD4 expression is associated with
induction of epithelial-to-mesenchymal transition and cetuximab
resistance in head and neck squamous cell carcinoma. Cancer Biol
Ther. 16:1252–1258. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang F, Xia X, Yang C, Shen J, Mai J, Kim
HC, Kirui D, Kang Y, Fleming JB, Koay EJ, et al: SMAD4 gene
mutation renders pancreatic cancer resistance to radiotherapy
through promotion of autophagy. Clin Cancer Res. 24:3176–3185.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sun FD, Wang PC, Luan RL, Zou SH and Du X:
MicroRNA-574 enhances doxorubicin resistance through
down-regulating SMAD4 in breast cancer cells. Eur Rev Med Pharmacol
Sci. 22:1342–1350. 2018.PubMed/NCBI
|
|
46
|
Shiou SR, Singh AB, Moorthy K, Datta PK,
Washington MK, Beauchamp RD and Dhawan P: Smad4 regulates claudin-1
expression in a transforming growth factor-beta-independent manner
in colon cancer cells. Cancer Res. 67:1571–1579. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Xiao DS, Wen JF, Li JH, Hu ZL, Zheng H and
Fu CY: Effect of deleted pancreatic cancer locus 4 gene
transfection on biological behaviors of human colorectal carcinoma
cells. World J Gastroenterol. 11:348–352. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liu SQ, Xu CY, Wu WH, Fu ZH, He SW, Qin MB
and Huang JA: Sphingosine kinase 1 promotes the metastasis of
colorectal cancer by inducing the epithelialmesenchymal transition
mediated by the FAK/AKT/MMPs axis. Int J Oncol. 54:41–52.
2019.PubMed/NCBI
|
|
49
|
Thiery JP and Chopin D: Epithelial cell
plasticity in development and tumor progression. Cancer Metastasis
Rev. 18:31–42. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Potts JD and Runyan RB:
Epithelial-mesenchymal cell transformation in the embryonic heart
can be mediated, in part, by transforming growth factor beta. Dev
Biol. 134:392–401. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Brown CB, Boyer AS, Runyan RB and Barnett
JV: Requirement of type III TGF-beta receptor for endocardial cell
transformation in the heart. Science. 283:2080–2082. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Proetzel G, Pawlowski SA, Wiles MV, Yin M,
Boivin GP, Howles PN, Ding J, Ferguson MW and Doetschman T:
Transforming growth factor-beta 3 is required for secondary palate
fusion. Nat Genet. 11:409–414. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kaartinen V, Voncken JW, Shuler C,
Warburton D, Bu D, Heisterkamp N and Groffen J: Abnormal lung
development and cleft palate in mice lacking TGF-beta 3 indicates
defects of epithelial-mesenchymal interaction. Nat Genet.
11:415–421. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Oft M, Peli J, Rudaz C, Schwarz H, Beug H
and Reichmann E: TGF-beta1 and Ha-Ras collaborate in modulating the
phenotypic plasticity and invasiveness of epithelial tumor cells.
Genes Dev. 10:2462–2477. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Battifora H: Spindle cell carcinoma:
Ultrastructural evidence of squamous origin and collagen production
by the tumor cells. Cancer. 37:2275–2282. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Buchmann A, Ruggeri B, Klein-Szanto AJ and
Balmain A: Progression of squamous carcinoma cells to spindle
carcinomas of mouse skin is associated with an imbalance of H-ras
alleles on chromosome 7. Cancer Res. 51:4097–4101. 1991.PubMed/NCBI
|
|
57
|
Geiger T, Sabanay H, Kravchenko-Balasha N,
Geiger B and Levitzki A: Anomalous features of EMT during
keratinocyte transformation. PLoS One. 3:e15742008. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Huber MA, Kraut N and Beug H: Molecular
requirements for epithelial-mesenchymal transition during tumor
progression. Curr Opin Cell Biol. 17:548–558. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Larue L and Bellacosa A:
Epithelial-mesenchymal transition in development and cancer: Role
of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene.
24:7443–7454. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Thompson EW, Newgreen DF and Tarin D:
Carcinoma invasion and metastasis: A role for
epithelial-mesenchymal transition? Cancer Res. 65:5991–5995. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Fuchs BC, Fujii T, Dorfman JD, Goodwin JM,
Zhu AX, Lanuti M and Tanabe KK: Epithelial-to-mesenchymal
transition and integrin-linked kinase mediate sensitivity to
epidermal growth factor receptor inhibition in human hepatoma
cells. Cancer Res. 68:2391–2399. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Liu Y, Li Y, Wang R, Qin S, Liu J, Su F,
Yang Y, Zhao F, Wang Z and Wu Q: MiR-130a-3p regulates cell
migration and invasion via inhibition of Smad4 in gemcitabine
resistant hepatoma cells. J Exp Clin Cancer Res. 35:192016.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wen Z, Feng S, Wei L, Wang Z, Hong D and
Wang Q: Evodiamine, a novel inhibitor of the Wnt pathway, inhibits
the self-renewal of gastric cancer stem cells. Int J Mol Med.
36:1657–1663. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Della Corte CM, Bellevicine C, Vicidomini
G, Vitagliano D, Malapelle U, Accardo M, Fabozzi A, Fiorelli A,
Fasano M, Papaccio F, et al: SMO gene amplification and activation
of the hedgehog pathway as novel mechanisms of resistance to
anti-epidermal growth factor receptor drugs in human lung cancer.
Clin Cancer Res. 21:4686–4697. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Güngör C, Zander H, Effenberger KE,
Vashist YK, Kalinina T, Izbicki JR, Yekebas E and Bockhorn M: Notch
signaling activated by replication stress-induced expression of
midkine drives epithelial-mesenchymal transition and
chemoresistance in pancreatic cancer. Cancer Res. 71:5009–5019.
2011. View Article : Google Scholar : PubMed/NCBI
|