Introduction
Phosphatase and tensin homolog (PTEN) is a
multifunctional gene, which encodes a dual-specific phosphatase
that dephosphorylates phosphatidylinositol (3,4,5)-triphosphate (PIP3) to
phosphatidylinositol 4,5-bisohosphate and inhibits the
phosphorylation of Akt (1).
Mutations in the PTEN gene are common in human cancer (2–5). The
protein expression levels of PTEN notably affect a variety of
cellular processes, including cell death, proliferation, migration
and metabolism (6,7). In addition, previous studies have
indicated that PTEN serves fundamental functions in maintaining
chromosomal stability (8–12). Mutations in the PTEN gene usually
lead to aneuploidy (8,13,14);
however, how PTEN controls the stability of chromosomes remains
unknown.
Mitotic arrest deficient 2 (MAD2) protein is
essential for the function of the spindle assembly checkpoint
(SAC). At the beginning of mitosis, MAD2 maintains the activity of
the SAC complex until all kinetochores are correctly connected to
spindle microtubules (15). MAD2,
and other proteins (cell division cycle 20, budding uninhibited by
benzimidazole-related 1, BUB3 mitotic checkpoint protein and MAD3),
constitute the mitotic checkpoint complex, which may inhibit the
activity of Anaphase promoting complex/cyclosome (APC/C) to ensure
cells remain in metaphase (16,17).
Once the chromosomes have properly attached to the spindle, the
APC/C complex facilitates the degradation of securin and cyclin B1,
promoting cells to enter anaphase (15,18,19).
Accumulating evidence has suggested that human tumors with
chromosome instability exhibit abnormal MAD2 expression.
Investigations using MAD2+/− mice revealed an increased
frequency of aneuploidy (20–22);
however the mechanism underlying the regulation of MAD2 remains
unknown.
The present study demonstrated that depletion of
PTEN induced cell cycle arrest and inhibited SAC, which led to
aberrant chromosome segregation and abnormal chromosome number,
suggesting that PTEN is important for chromosome stability. In
addition, it was determined that PTEN deficiency decreased the
stability of MAD2, which resulted in increased protein degradation
via the ubiquitin-proteasome pathway, whereas the recovery of MAD2
expression partially rescued aberrant chromosome segregation in
PTEN-knockdown cells. The results of the present study indicated
that MAD2 function may be mediated by a mechanism in which PTEN
regulates chromosome segregation.
Materials and methods
Cell culture, short hairpin RNA
(shRNA), plasmids and chemicals
HeLa cells were obtained from The Cell Bank of Type
Culture Collection of Chinese Academy of Sciences. Mouse embryonic
fibroblasts (MEFs) were a gift from Dr Yuxin Yin of the Department
of Pathology, Peking University. The cells were grown in Dulbecco's
modified Eagle's medium (Gibco; Thermo Fisher Scientific, Inc.)
containing 10% fetal bovine serum (Sijiqing; Zhejiang Tianhang
Biotechnology Co., Ltd.), and 1X penicillin-streptomycin and
cultured in 5% CO2 incubator at 37°C.
PTEN-targeted shRNA (5′-GACAAAGCCAACCGATACTTT-3′)
and control shRNA (5′-TTCTCCGAACGTGTCACGT-3′), and MAD2
overexpression and control vectors were obtained from Shanghai
GenePharma Co., Ltd. The ubiquitin expression vector was purchased
from OriGene Technologies, Inc. In total, 25 µl shRNA containing
lentivirus (1×108 transducing U/ml) was used for
transfection. Plasmids were transfected into cells using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturers' protocol. The
cells were cultured for a further 48 h before they were collected
for subsequent experiments. MG132 and nocodazole were purchased
from Sigma-Aldrich; Merck KGaA, and cycloheximide (CHX) was
obtained from VWR International, LLC.
Western blot analysis and
ubiquitination assay
Total HeLa cell extracts were prepared using a
radioimmunoprecipitation assay buffer (Beyotime Institute of
Biotechnology); proteins were quantified with a BCA kit (Beyotime
Institute of Biotechnology). A total of 30 µg protein were
separated using 10% SDS-PAGE and then transferred to a
polyvinylidene difluoride membrane (EMD Millipore). The membrane
was blocked with 5% skimmed milk in TBS + 0.5% Tween-20 (TBS-T) for
2 h at room temperature, and then incubated with primary antibodies
at 4°C overnight. Following 3 washes with TBS-T, the membrane was
incubated with a horseradish peroxidase (HRP)-labeled secondary
antibody for 2 h at room temperature. Subsequently, the membrane
was washed 3 times and the bands were detected using an Enhanced
Chemiluminescent substrate (Pierce; Thermo Fisher Scientific, Inc.)
and analyzed using ImageJ (version 1.48; National Institute of
Health). The following primary antibodies were used: Rabbit
anti-human PTEN (1:2,000; Cell Signaling Technology, Inc.; cat. no.
9188L), rabbit anti-human MAD2 (1:1,000) and cyclin B1 (1:4,000;
Santa Cruz Biotechnology, Inc.; cat. nos. sc-28261 and sc-752,
respectively), and mouse anti-GAPDH (1:20,000; ProteinTech Group,
Inc.; cat. no. 60004-1-Ig) were used. HRP-labelled goat anti-rabbit
IgG (1:10,000) and rabbit anti-mouse IgG (1:10,000) were obtained
from Santa Cruz Biotechnology, Inc. (cat. nos. sc-2004 and sc-2005,
respectively). GAPDH was used as an internal control.
For the ubiquitination assay, HeLa cells were
incubated with 20 µM MG132 for 4 h and collected 48 h after
transfection of the ubiquitin expression plasmid; cells were then
lysed in Triton lysis buffer (50 mM Tris-HCl pH 7.4, 0.25 M NaCl,
0.1% Triton X-100, 50 mM NaF). Cell lysates were pre-cleared using
Protein A/G Agarose (cat. no., sc-2003; Santa Cruz Biotechnology,
Inc.) and then incubated with anti-MAD2 antibody overnight at 4°C;
Protein A/G Agarose was added 2 h prior to centrifugation (1,000 ×
g for 3 min at 4°C) to obtain cell lysates. Triton lysis buffer was
used to wash the immunocomplexes 5 times. Finally, the samples were
eluted by resuspending in loading buffer and boiling for 5 min.
Protein expression from total extracts and immunoprecipitates were
then determined by western blot analysis, as aforementioned.
Immunofluorescence and confocal
microscopy
HeLa cells were cultured to 80% confluence prior to
fixation with 4% formaldehyde at room temperature for 10 min,
following which the cells were blocked with 5% bovine serum albumin
(VWR International, LLC) for 1 h at room temperature.
Anti-α-tubulin antibody (1:200; cat. no. 11224-1-AP; ProteinTech
Group, Inc.) was then applied to the cells at 4°C overnight.
Following washing with PBS three times, the cells were incubated
with fluorescently-labeled secondary antibodies (Alexa
Fluor® 594; 1:400; cat. no. A-11012; Invitrogen; Thermo
Fisher Scientific, Inc.) for 2 h at room temperature. Subsequently,
the cells were washed with PBS three times and stained with DAPI (1
µg/ml; Nanjing KeyGen Biotech Co., Ltd.) at room temperature for 3
min. Images were obtained at ×600 magnification and cells were
analyzed with a confocal microscope (Leica Microsystems, Inc.).
Cell survival assay
HeLa cells were cultured to 50% confluence and then
incubated with nocodazole (1 µM) for 36 h. Afterwards, cells were
collected by centrifugation (200 × g for 5 min at room temperature)
and resuspended in PBS. Cell viability was determined using a
trypan blue exclusion assay as follows: 0.1 ml trypan blue solution
(0.4%; Gibco; Thermo Fisher Scientific, Inc.) was added to 0.9 ml
cell suspension, mixed and loaded into a hemocytometer, the cells
were immediately examined under a microscope (Olympus IX53; Olympus
Corporation) and the number of total cells and stained cells were
counted. The cell viability was calculated as [1.00-(number of
stained cells/number of total cells)] ×100%.
Cell cycle analysis
Cell cycle analysis was performed using a kit from
Nanjing KeyGen Biotech Co., Ltd. HeLa cells in the exponential
growth phase were collected by centrifugation (200 × g for 5 min at
room temperature) and fixed in ice-cold 75% ethanol for 2 h at 4°C.
Then, the cells were treated with propidium iodide (50 µg/ml) and
RNase A (100 µg/ml) at room temperature for 30 min and analyzed
using a flow cytometer (BD FACSCalibur; BD Biosciences). The cell
cycle was analyzed with MODFIT LT 3.0 software (Verity Software
House, Inc.).
Metaphase spread preparation
MEFs in the exponential growth phase were treated
with colcemid (0.1 µg/ml; VWR International, LLC) for 2 h. Then,
the cells were trypsinized and harvested by centrifugation at 200 ×
g at room temperature for 5 min. The collected cells were suspended
in 8 ml 75 mM KCl solution at 37°C for 20 min. Following
pre-fixation with 1 ml fresh pre-cold fixation solution (methanol:
Acetic acid at a ratio of 3:1) for 2 min at room temperature, the
cells were collected by centrifugation at 200 × g for 5 min at room
temperature. The cells were resuspended and fixed in 15 ml cold
fixation solution for 20 min at room temperature three times.
Following centrifugation (200 × g for 8 min at room temperature),
the cells were resuspended in 400 µl fixation solution, and 50 µl
cell suspension was applied to slides and air-dried. The slides
were stained with DAPI (1 µg/ml) at room temperature for 3 min.
Images were obtained at ×1000 magnification and cells were analyzed
with a fluorescence microscope (Carl Zeiss AG).
Statistical analysis
All statistical analyses were performed using
GraphPad Prism v.5.0 (GraphPad Software, Inc.). Data were presented
as the mean ± standard deviation. A paired Student's t-test was
used for comparisons of two groups and one-way analysis of variance
for comparisons of multiple groups. The post-hoc Newman-Keuls test
was used for pairwise comparisons in multiple groups. A
χ2 test was used to compare chromosome numbers: The
metaphase spread was prepared and the chromosome numbers of each
cell were counted in two categories (PTEN+ and
PTEN− MEFs). The chromosome numbers were then divided
into two different categories [euploid (40) vs. aneuploid (≠40) or tetraploid
(80) vs. non-tetraploid (≠80)]. The cell numbers of each category
were counted and then used for statistical analysis. The data were
considered to be categorical variables, and were obtained by
counting the frequency of the different categories. P<0.05 was
considered to indicate a statistically significant difference.
Results
PTEN-deficient cells undergo G2/M
arrest and are resistant to spindle disruption
PTEN serves various functions in different stages of
the cell cycle, including modulating G1 cell cycle progression,
controlling DNA replication and regulating chromosome congression
(11,23–25).
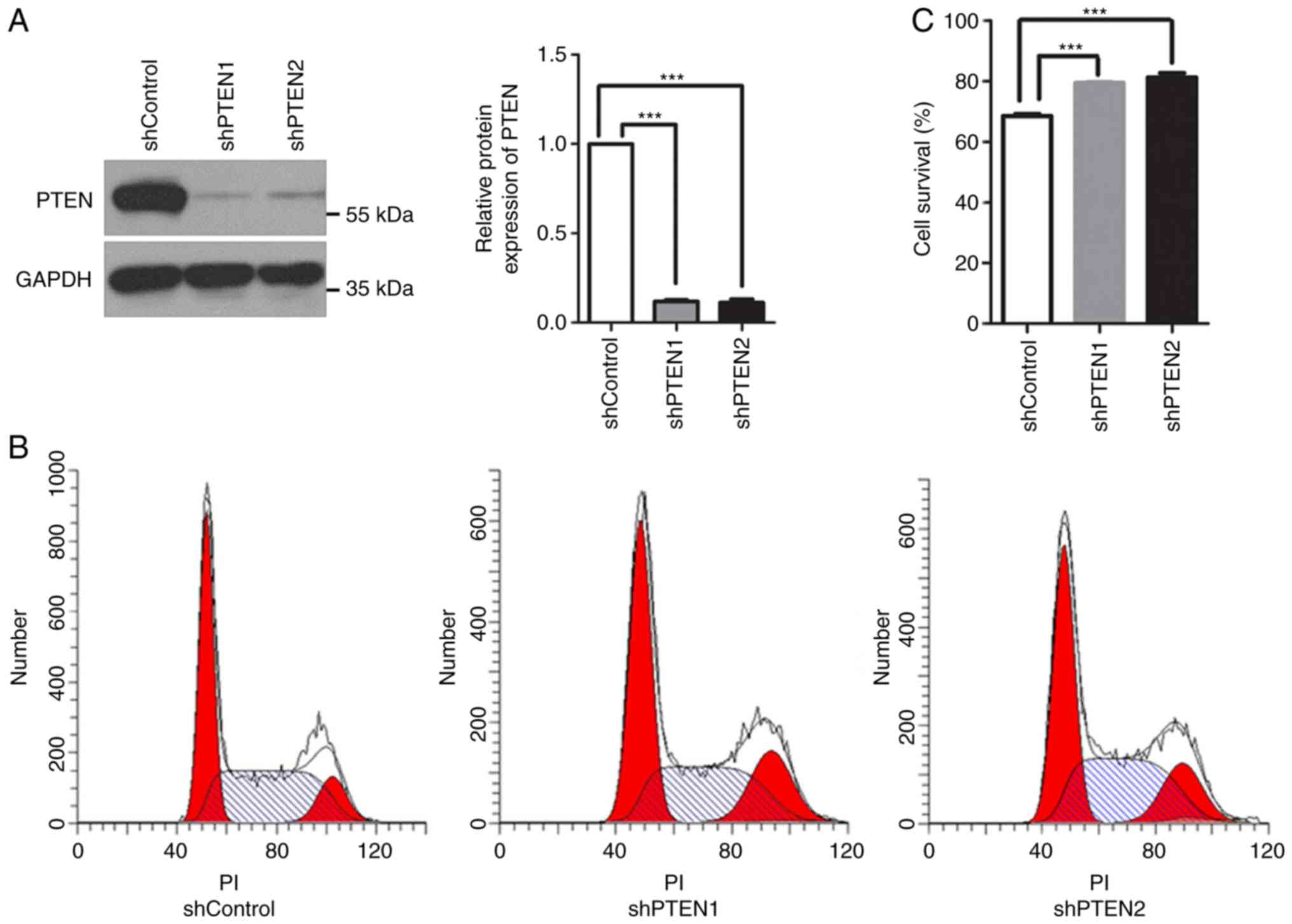
To additionally investigate the function of PTEN in mitosis, the
expression of PTEN in HeLa cells was knocked down using 2
PTEN-targeted shRNAs, shPTEN1 and shPTEN2. Western blot analysis
revealed that the protein expression levels of PTEN decreased
significantly in these cells (Fig.
1A). Additionally, PTEN knockdown arrested the cells in G2/M
phase; the population of cells in G2/M phase increased from
12.88±0.40% in the control group to 19.20±0.88 and 17.64±1.61%, in
the shPTEN1 and shPTEN2 PTEN knockdown groups, respectively
(P<0.05; Fig. 1B). This
suggested potential defects in the cell cycle checkpoints within
PTEN-deficient cells.
The SAC is responsible for metaphase-to-anaphase
transition. Once the SAC is activated, cells are arrested in
metaphase until all sister chromatids have correctly attached to
microtubules. To evaluate the function of the SAC in PTEN-knockdown
cells, cell viability following nocodazole treatment was analyzed.
Nocodazole is an anti-microtubule agent that halts cells at
prometaphase by activating the SAC; prolonged nocodazole treatment
may result in cell death by apoptosis (26,27).
As presented in Fig. 1C,
nocodazole suppressed the viability of control cells, while
PTEN-knockdown cells were markedly resistant to nocodazole
treatment. The results revealed that PTEN knockdown alleviated the
effects of nocodazole, suggesting that the SAC was inhibited. These
results indicated that PTEN deficiency may induce G2/M arrest,
which may be associated with dysregulations in the assembly spindle
checkpoint. As both of the PTEN-targeted shRNAs had similar
effects, only shPTEN1 was selected for subsequent analyses.
Loss of PTEN leads to aberrant
chromosome segregation
To additionally investigate the effects of PTEN loss
on mitosis, chromosome segregation during mitosis was analyzed via
DAPI staining. Examination of the mitotic cells revealed that the
PTEN-knockdown cells exerted notable chromosome mis-segregation,
which was associated with lagging chromosomes and the formation of
anaphase bridges. In addition, deletion of PTEN significantly
increased the percentage of lagging chromosomes in anaphase from
10.84±1.74 to 20.37±2.87%. Furthermore, the percentage of anaphase
bridges increased from 10.39±1.37 to 21.66±4.09%; tripolar
segregation was also observed in PTEN-knockdown cells (Fig. 2A). The results indicated that PTEN
silencing impeded normal chromosome segregation.
Proper spindle assembly is important for accurate
chromosome segregation. Therefore, the spindle apparatus in
PTEN-knockdown cells was analyzed via immunofluorescence staining.
Numerous abnormal spindles (asymmetric or incomplete) were observed
in PTEN-knockdown cells (Fig. 2B).
Aberrant mitosis usually results in erroneous chromosome
inheritance. Therefore, the effects of PTEN loss on chromosome
numbers were determined in PTEN+/+ and
PTEN−/− MEFs. Metaphase chromosome spreading was
induced, following which the number of chromosomes was determined.
Compared with wild-type MEFs, the frequency of tetraploidy in the
PTEN−/− MEFs was notably increased. Aneuploidy was also
detected in the PTEN−/− MEFs, but not in wild-type MEFs
(Fig. 2C). Additionally,
PTEN−/− MEFs exhibited abnormal morphology and premature
sister chromatid separation (Fig.
2D). These results indicated that PTEN deficiency may induce
aberrant spindle formation and chromosome segregation, leading to
inaccurate chromosome inheritance.
MAD2 is downregulated in
PTEN-deficient cells
MAD2 is essential for proper spindle formation and
correct chromosome segregation. Several studies have indicated that
the dysregulated expression of MAD2 may induce cell cycle arrest,
aberrant chromosomal segregation, premature sister chromatid
separation, aneuploidy and polyploidy (28–30).
Therefore, immunostaining of the control and PTEN-knockdown HeLa
cells was performed using MAD2 antibodies to determine whether PTEN
silencing affected protein expression. Western blot analysis
revealed that the protein expression levels of MAD2 were notably
decreased in asynchronous PTEN-deficient cells (Fig. 3A). Then, cells were treated with
nocodazole for 20 h to synchronize the cells. Under this condition,
the protein level of PTEN was not changed significantly (Fig. S1A), and significant decreases in
the protein expression levels of MAD2 were detected in the cells
transfected with PTEN shRNA (Fig.
3B). These results indicated that PTEN deficiency may decrease
the expression of MAD2 protein in asynchronous and synchronous
cells, suggesting that the underlying mechanism may be independent
of the cell cycle. Additionally, the protein expression levels of
cyclin B1, a downstream target of MAD2 and important factor
involved in the initiation of anaphase, were analyzed. The results
of the western blot analysis demonstrated that cyclin B1 expression
was decreased in PTEN-knockdown asynchronous cells, but also in
nocodazole-treated cells (Fig.
3B), which may disrupt the spindle checkpoint and lead to
impaired mitosis. These data additionally confirmed that the loss
of PTEN may induce mitotic defects by disrupting spindle checkpoint
activity.
 | Figure 3.MAD2 is downregulated in
PTEN-deficient cells. (A) MAD2 expression was evaluated using cell
lysates from control and PTEN-knockdown cells by western blot
analysis. (B) The protein expression levels of MAD2 and cyclin B1
in control and PTEN-knockdown HeLa cells cultured with or without
nocodazole were detected by western analysis. (C) Control and
PTEN-knockdown HeLa cells cultured with or without CHX were
harvested at 0, 3 and 6 h after treatment. Then, the protein
expression levels of MAD2 were evaluated using western blot
analysis. (D) Control and PTEN-knockdown HeLa cells cultured with
or without MG132 were analyzed for MAD2 expression by western blot
analysis. (E) Ubiquitin was overexpressed in control and
PTEN-knockdown HeLa cells. Following MG132 treatment, the cells
were harvested for analysis via a ubiquitination assay. All data
were obtained from 3 independent experiments. *P<0.05,
**P<0.01 and ***P<0.001. MAD2, mitotic arrest deficient 2;
PTEN, phosphatase and tensin homolog; sh, short hairpin RNA; Ub,
ubiquitin; CHX, cycloheximide; IP, immunoprecipitates; IB,
immunoblot. |
PTEN knockdown leads to instability of
MAD2 protein
To investigate how PTEN regulates MAD2 protein
expression, cells were treated with CHX, a protein synthesis
inhibitor, following which the expression of MAD2 was examined.
Western blot analysis revealed that, following CHX treatment, the
expression levels of MAD2 were decreased in cells transfected with
PTEN shRNA, but not in the control cells (Fig. 3C), indicating that MAD2 was prone
to degradation in PTEN-knockdown cells. In addition, cells were
treated with MG132, a proteasome inhibitor. The expression levels
of MAD2 was notably recovered following treatment with MG132 for 4
h in PTEN-knockdown cells, while the protein level of PTEN was not
altered following MG132 treatment (Figs. 3D and S1B). The ubiquitination levels of MAD2
were also investigated using a ubiquitination assay. The results
indicated that PTEN knockdown increased the ubiquitination levels
of MAD2 compared with control cells (Fig. 3E). Collectively, these results
suggested that PTEN deficiency may induce MAD2 degradation via the
ubiquitin-proteasome pathway.
Recovery of MAD2 expression partially
ameliorates impaired mitosis
To investigate whether PTEN loss-induced MAD2
degradation was responsible for aberrant mitosis, MAD2 was
overexpressed in PTEN-knockdown cells. A His-tagged MAD2 expression
plasmid was transfected into HeLa cells and PTEN-knockdown cells;
western blot analysis confirmed that MAD2 was markedly
overexpressed (Figs. 4A and
S2). The protein expression
levels of cyclin B1 were also increased in MAD2-overexpression
cells, suggesting recovery of the spindle assembly checkpoint
(Fig. 4A). Additionally, ectopic
expression of MAD2 decreased cell viability following nocodazole
treatment compared with the shPTEN group (Fig. 4B). Furthermore, restoration of MAD2
expression significantly decreased the number of binucleated and
multinucleated cells observed in the PTEN-knockdown groups, but to
a lesser extent compared with in the control group (Fig. 4C), which indicated that MAD2 may
decrease the polyploidization caused by PTEN loss. These data
suggested that abnormal chromosome segregation caused by PTEN
knockdown may be mediated by MAD2.
 | Figure 4.Recovery of MAD2 expression partially
ameliorates impaired mitosis. (A) PTEN-knockdown HeLa cells were
transfected with His-tagged MAD2 expression plasmids prior to the
analysis of MAD2 and cyclin B1 expression by western blot analysis.
(B) The rate of cell survival for control, PTEN-knockdown and
His-MAD2-overexpression PTEN-knockdown HeLa cells incubated with
nocodazole for 36 h was calculated. (C) (Left panel) Representative
images indicating the general morphology of mononucleated,
binucleated and multinucleated cells at high magnification. (Middle
panel) Representative images demonstrating cells of the shControl,
shPTEN and shPTEN-MAD2 groups stained by DAPI. Binucleated cells
(arrowhead) and multinucleated cells (arrow) are marked in each
image. (Right panel) Statistical analysis of the percentage of
binucleated or multinucleated cells in the different groups. A
total of 500 cells were counted. Among these cells, binucleated or
multinucleated cells were counted. The percentage was calculated as
binucleated cell number (or multinucleated cell number)/total cell
number. All data were obtained from 3 independent experiments,
*P<0.05, **P<0.01 and ***P<0.001. BF, bright field; MAD2,
mitotic arrest deficient 2; PTEN, phosphatase and tensin homolog;
sh, short hairpin RNA; his-MAD2, His-tagged MAD2 expression
plasmid. |
Discussion
PTEN is important in chromosome protection and tumor
suppression (5,8,14,31).
In the present study, it was demonstrated that the basal expression
of PTEN was essential for MAD2 stability and chromosome
segregation. The ectopic expression of MAD2 ameliorated PTEN
loss-induced mitotic defects, which additionally confirmed the
important role of MAD2 in mitosis. These data indicated that PTEN
serves a key role in chromosome segregation by maintaining accurate
mitosis. The results of the present study also revealed a novel
function of PTEN in regulating the expression of MAD2 in
mitosis.
Regulation upstream of MAD2 has been studied
previously: It has been demonstrated that MAD2 may be regulated by
E2F1, Myc and RE1 silencing transcription factor at the
transcriptional level (32,33);
however, how the cellular expression of MAD2 is regulated at the
post-transcriptional level requires further investigation.
Withaferin A was suggested to induce the degradation of MAD2, yet
the molecular mechanisms remain unknown (34). SMAD specific E3 ubiquitin protein
ligase 2 (Smurf2) has been suggested to inhibit the ubiquitination
of MAD2 (35), but the underlying
mechanism requires additional study. In the present study, the
regulator PTEN was examined, and was determined to maintain the
stability of MAD2. The mechanism underlying the regulation of MAD2
degradation mediated by PTEN was not explored, but Akt has been
recently demonstrated to induce the phosphorylation and degradation
of Smurf2 (36). Therefore, we
hypothesized that the deletion of PTEN may activate Akt, resulting
in decreased Smurf2 expression, which may lead to the degradation
of MAD2. Further investigation is required to validate this
hypothesis regarding the PTEN-Smurf2-MAD2 axis and other
mechanisms.
The results of the present study suggested that
restoration of MAD2 may partially rescue the mitotic defects
induced by PTEN loss, which indicated that MAD2 may not be the only
protein responsible for the PTEN-associated functions in mitosis.
PTEN may target different protein substrates that control mitosis.
Notably, it has been demonstrated that PTEN may control the spindle
architecture and chromosome congression by regulating Eg5 (25). Polo like kinase 1 is another target
of PTEN involved in the regulation of chromosome stability during
mitosis (37). In addition, PTEN
serves a role in cytokinesis by dephosphorylating PIP3 (38). Additional targets of PTEN may be
identified in the future, which may improve understanding regarding
the function of PTEN in mitosis.
PTEN is a multifunctional tumor suppressor gene.
Numerous studies have indicated that PTEN may regulate the cell
cycle through different mechanisms (39–41).
In the present study, PTEN knockdown was determined to cause G2/M
arrest in HeLa cells by downregulating MAD2 and cyclin B1.
Conversely, Song et al (9)
revealed that nuclear PTEN interacted with APC/C and enhanced the
activity of APC-fizzy-related protein homolog (CDH1), which
facilitated the ubiquitination of cyclin B1, decreasing its
expression. It appears that the results of the present study are
contradictory to these data, but this may be due to the various
functions of PTEN. For example, PTEN may regulate cyclin B1 through
a variety of mechanisms in different cell cycle phases. In
metaphase, PTEN maintains the levels of cyclin B1 by stabilizing
MAD2 to ensure accurate chromosome segregation. In addition, from
anaphase to G1 phase, PTEN downregulates cyclin B1 by enhancing
APC-CDH1 activity to prevent the abnormal accumulation of cyclin
B1. These contradictory results suggest the different functions of
PTEN in the various phases of the cell cycle. Furthermore, Song
et al (9) investigated the
function of nuclear PTEN and indicated that the functions of this
protein were independent of its phosphatase activity, whereas the
present study explored the function of total PTEN. The present
study proposed that PTEN functions may be attributed to the
dephosphorylation of p-Akt. This suggests the different functions
of nuclear PTEN, based on C-terminal protein binding activity, and
cytoplasmic PTEN, based on N-terminal phosphatase activity.
Abnormal mitosis is a hallmark of cancer, which
makes it a common target for anticancer therapies. For example,
microtubule-targeting agents (MTAs) are now applied to treat cancer
clinically; MTAs arrest cancer cells in mitosis, resulting in their
death (42,43); however, a proportion of cancer
cells may develop resistance to MTAs, which restricts its
application (44). The present
study revealed that PTEN deficiency may disrupt the function of SAC
and protect cells against the damage caused by MTAs. As a result,
MTAs may be less effective to cancer cells with PTEN deficiency.
Cyclin B1/cyclin-dependent kinase 1 (CDK1) is an additional target
for cancer therapy (45,46); agents against these targets may
inhibit their activity and induce cell cycle arrest. However, the
results of the present study indicated that PTEN knockdown may lead
to the downregulation of cyclin B1, which may also attenuate the
anti-cancer activity of cyclin B1/CDK1 inhibitors. Therefore, the
expression levels of PTEN may be associated with the effectiveness
of these types of anti-cancer drugs. Therefore, to ensure the
effectiveness of treatment, the expression of PTEN in patients may
be evaluated prior to the administration of these agents. The
results of the present study may aid the selection of anti-mitotic
therapeutic agents under conditions of PTEN deficiency.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by National Natural
Science Foundation of China (grant nos. 81502387 and 81372479),
Natural Science Foundation of the Jiangsu Higher Education
Institutions (grant no. 18KJB310014), Key Research and Development
Program (grant no. BE2017635) and Young Medical talents of Jiangsu
Province (grant no. QNRC2016386).
Availability of data and materials
The datasets used and/or analyzed during the curernt
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZSu and JL performed the majority of the
experiments, analyzed the data and prepared the manuscript. MW and
ML performed experiments. LB and ZSh analyzed the data. LH and YW
participated in the conception and design of the study. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chalhoub N and Baker SJ: PTEN and the
PI3-kinase pathway in cancer. Annu Rev Pathol. 4:127–150. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Steck PA, Pershouse MA, Jasser SA, Yung
WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T,
et al: Identification of a candidate tumour suppressor gene, MMAC1,
at chromosome 10q23.3 that is mutated in multiple advanced cancers.
Nat Genet. 15:356–362. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Di Cristofano A and Pandolfi PP: The
multiple roles of PTEN in tumor suppression. Cell. 100:387–390.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Salmena L, Carracedo A and Pandolfi PP:
Tenets of PTEN tumor suppression. Cell. 133:403–414. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yin Y and Shen WH: PTEN: A new guardian of
the genome. Oncogene. 27:5443–5453. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Carracedo A and Pandolfi PP: The PTEN-PI3K
pathway: Of feedbacks and cross-talks. Oncogene. 27:5527–5541.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Berger AH and Pandolfi PP:
Haplo-insufficiency: A driving force in cancer. J Pathol.
223:137–146. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shen WH, Balajee AS, Wang J, Wu H, Eng C,
Pandolfi PP and Yin Y: Essential role for nuclear PTEN in
maintaining chromosomal integrity. Cell. 128:157–170. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Song MS, Carracedo A, Salmena L, Song SJ,
Egia A, Malumbres M and Pandolfi PP: Nuclear PTEN regulates the
APC-CDH1 tumor-suppressive complex in a phosphatase-independent
manner. Cell. 144:187–199. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen ZH, Zhu M, Yang J, Liang H, He J, He
S, Wang P, Kang X, McNutt MA, Yin Y and Shen WH: PTEN interacts
with histone H1 and controls chromatin condensation. Cell Rep.
8:2003–2014. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
He J, Kang X, Yin Y, Chao KS and Shen WH:
PTEN regulates DNA replication progression and stalled fork
recovery. Nat Commun. 6:76202015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kang X, Song C, Du X, Zhang C, Liu Y,
Liang L, He J, Lamb K, Shen WH and Yin Y: PTEN stabilizes TOP2A and
regulates the DNA decatenation. Sci Rep. 5:178732015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Puc J, Keniry M, Li HS, Pandita TK,
Choudhury AD, Memeo L, Mansukhani M, Murty VV, Gaciong Z, Meek SE,
et al: Lack of PTEN sequesters CHK1 and initiates genetic
instability. Cancer Cell. 7:193–204. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sun Z, Huang C, He J, Lamb KL, Kang X, Gu
T, Shen WH and Yin Y: PTEN C-terminal deletion causes genomic
instability and tumor development. Cell Rep. 6:844–854. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schuyler SC, Wu YF and Kuan VJ: The
Mad1-Mad2 balancing act-a damaged spindle checkpoint in chromosome
instability and cancer. J Cell Sci. 125:4197–4206. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Musacchio A: The molecular biology of
spindle assembly checkpoint signaling dynamics. Curr Biol.
25:R1002–R1018. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shandilya J and Roberts SG: A role of WT1
in cell division and genomic stability. Cell Cycle. 14:1358–1364.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Han JS, Holland AJ, Fachinetti D, Kulukian
A, Cetin B and Cleveland DW: Catalytic assembly of the mitotic
checkpoint inhibitor BubR1-Cdc20 by a Mad2-induced functional
switch in Cdc20. Mol Cell. 51:92–104. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lau DT and Murray AW: Mad2 and Mad3
cooperate to arrest budding yeast in mitosis. Curr Biol.
22:180–190. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Burds AA, Lutum AS and Sorger PK:
Generating chromosome instability through the simultaneous deletion
of Mad2 and p53. Proc Natl Acad Sci USA. 102:11296–11301. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rao CV, Yamada HY, Yao Y and Dai W:
Enhanced genomic instabilities caused by deregulated microtubule
dynamics and chromosome segregation: A perspective from genetic
studies in mice. Carcinogenesis. 30:1469–1474. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schvartzman JM, Duijf PH, Sotillo R, Coker
C and Benezra R: Mad2 is a critical mediator of the chromosome
instability observed upon Rb and p53 pathway inhibition. Cancer
Cell. 19:701–714. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Feng J, Liang J, Li J, Li Y, Liang H, Zhao
X, McNutt MA and Yin Y: PTEN controls the DNA replication process
through MCM2 in response to replicative stress. Cell Rep.
13:1295–1303. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ramaswamy S, Nakamura N, Vazquez F, Batt
DB, Perera S, Roberts TM and Sellers WR: Regulation of G1
progression by the PTEN tumor suppressor protein is linked to
inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc
Natl Acad Sci USA. 96:2110–2115. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
He J, Zhang Z, Ouyang M, Yang F, Hao H,
Lamb KL, Yang J, Yin Y and Shen WH: PTEN regulates EG5 to control
spindle architecture and chromosome congression during mitosis. Nat
Commun. 7:123552016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang LG, Liu XM, Kreis W and Budman DR:
The effect of antimicrotubule agents on signal transduction
pathways of apoptosis: A review. Cancer Chemother Pharmacol.
44:355–361. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang TH, Wang HS and Soong YK:
Paclitaxel-induced cell death: Where the cell cycle and apoptosis
come together. Cancer. 88:2619–2628. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shandilya J, Toska E, Richard DJ, Medler
KF and Roberts SG: WT1 interacts with MAD2 and regulates mitotic
checkpoint function. Nat Commun. 5:49032014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Homer HA, McDougall A, Levasseur M, Yallop
K, Murdoch AP and Herbert M: Mad2 prevents aneuploidy and premature
proteolysis of cyclin B and securin during meiosis I in mouse
oocytes. Genes Dev. 19:202–207. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Michel LS, Liberal V, Chatterjee A,
Kirchwegger R, Pasche B, Gerald W, Dobles M, Sorger PK, Murty VV
and Benezra R: MAD2 haplo-insufficiency causes premature anaphase
and chromosome instability in mammalian cells. Nature. 409:355–359.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Trotman LC, Wang X, Alimonti A, Chen Z,
Teruya-Feldstein J, Yang H, Pavletich NP, Carver BS, Cordon-Cardo
C, Erdjument-Bromage H, et al: Ubiquitination regulates PTEN
nuclear import and tumor suppression. Cell. 128:141–156. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hernando E, Nahlé Z, Juan G,
Diaz-Rodriguez E, Alaminos M, Hemann M, Michel L, Mittal V, Gerald
W, Benezra R, et al: Rb inactivation promotes genomic instability
by uncoupling cell cycle progression from mitotic control. Nature.
430:797–802. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Guardavaccaro D, Frescas D, Dorrello NV,
Peschiaroli A, Multani AS, Cardozo T, Lasorella A, Iavarone A,
Chang S, Hernando E and Pagano M: Control of chromosome stability
by the beta-TrCP-REST-Mad2 axis. Nature. 452:365–369. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Das T, Roy KS, Chakrabarti T, Mukhopadhyay
S and Roychoudhury S: Withaferin A modulates the Spindle assembly
checkpoint by degradation of Mad2-Cdc20 complex in colorectal
cancer cell lines. Biochem Pharmacol. 91:31–39. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Osmundson EC, Ray D, Moore FE, Gao Q,
Thomsen GH and Kiyokawa H: The HECT E3 ligase Smurf2 is required
for Mad2-dependent spindle assembly checkpoint. J Cell Biol.
183:267–277. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Choi YH, Kim YJ, Jeong HM, Jin YH, Yeo CY
and Lee KY: Akt enhances Runx2 protein stability by regulating
Smurf2 function during osteoblast differentiation. FEBS J.
281:3656–3666. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang Z, Hou SQ, He J, Gu T, Yin Y and
Shen WH: PTEN regulates PLK1 and controls chromosomal stability
during cell division. Cell Cycle. 15:2476–2485. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sagona AP, Nezis IP, Pedersen NM, Liestøl
K, Poulton J, Rusten TE, Skotheim RI, Raiborg C and Stenmark H:
PtdIns(3)P controls cytokinesis through KIF13A-mediated recruitment
of FYVE-CENT to the midbody. Nat Cell Biol. 12:362–371. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Radu A, Neubauer V, Akagi T, Hanafusa H
and Georgescu MM: PTEN induces cell cycle arrest by decreasing the
level and nuclear localization of cyclin D1. Mol Cell Biol.
23:6139–6149. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang L, Yang L, Lu Y, Chen Y, Liu T, Peng
Y, Zhou Y, Cao Y, Bi Z, Liu T, et al: Osthole induces cell cycle
arrest and inhibits migration and invasion via PTEN/Akt pathways in
osteosarcoma. Cell Physiol Biochem. 38:2173–2182. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xiong X, Ren HZ, Li MH, Mei JH, Wen JF and
Zheng CL: Down-regulated miRNA-214 induces a cell cycle G1 arrest
in gastric cancer cells by up-regulating the PTEN protein. Pathol
Oncol Res. 17:931–937. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bolanos-Garcia VM: Assessment of the
mitotic spindle assembly checkpoint (SAC) as the target of
anticancer therapies. Curr Cancer Drug Targets. 9:131–141. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Dominguez-Brauer C, Thu KL, Mason JM,
Blaser H, Bray MR and Mak TW: Targeting mitosis in cancer: Emerging
strategies. Mol Cell. 60:524–536. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Huang HC, Shi J, Orth JD and Mitchison TJ:
Evidence that mitotic exit is a better cancer therapeutic target
than spindle assembly. Cancer Cell. 16:347–358. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: A changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cicenas J, Kalyan K, Sorokinas A, Jatulyte
A, Valiunas D, Kaupinis A and Valius M: Highlights of the latest
advances in research on CDK inhibitors. Cancers (Basel).
6:2224–2242. 2014. View Article : Google Scholar : PubMed/NCBI
|