Introduction
In the United States, breast cancer is the most
common cancer among women, accounting for 30% of all new cancer
diagnoses in women, and is the second leading cause of
cancer-related deaths of over 40,000 women each year (1). Triple-negative breast cancer (TNBC),
a subtype defined clinically by the lack of estrogen receptor alpha
(ERα), progesterone receptor (PR) and human epidermal growth factor
receptor 2 (HER2), is composed of 15 to 20% of all types of breast
cancers and is associated with highly aggressive biological
characteristics (2). TNBC
frequently presents in younger patients, and often ends with poor
clinical outcome with high rates of recurrence and mortality due in
part to lack of therapeutic options beyond chemotherapy (3,4).
However, although TNBC exhibits an excellent clinical response to
neoadjuvant chemotherapy (NCT) or conventional postoperative
chemotherapy, it often becomes refractory (5,6). It
is reported that >50% of patients will develop recurrent disease
with residual disease after chemotherapy (7). For these reasons, there is an urgent
need to reveal the molecular mechanism and identify novel targeted
therapeutics strategies for TNBC patients.
Recent evidence highlights that TNBC is a complex
disease characterized by molecular and phenotypic heterogeneity
compared with other types (8), and
can be further subdivided into four to six distinct molecular
subtypes for their unique expression signatures and ontologies,
including luminal androgen receptor, basal-like and mesenchymal
subtypes (9,10). Currently, high-throughput
genome-wide gene expression datasets are available freely in public
databases, such as Gene Expression Omnibus (GEO) and The Cancer
Genome Atlas (TCGA) database. By means of comprehensive gene
expression analysis, genome-wide analysis of these public
high-throughput data can provide insight into the molecular
mechanism underlying those complex diseases with different
patterns. However, due to limited sample size and heterogeneous
characteristics, the results of individual gene expression analysis
are often inconsistent or even discrepant among each study, and may
not convincingly predict the functional gene networks involved in
disease. Therefore, a comprehensive integrated analysis of this
enormous volume of data to get credible outcomes is necessary.
The robust rank aggregation (RRA) analysis, a
rigorous and well-accepted approach designed specifically for
comparison of several ranked gene lists, can be used to integrate
multiple genome-wide gene expression datasets and identify key
genes most likely associated in several diseases (11,12).
By means of comparison of several ranked gene lists in an unbiased
manner, RRA is particularly suitable for identification of
statistically significant genes when high-throughput sequencing
experiments are conducted by different platforms covering different
sets of gene probes (11,13). At present, there has been no
attempt to integrate these genome-wide gene expression datasets
regarding TNBC vs. non-TNBC using the RRA method.
In the present study, RRA analysis based on four
high-throughput genome-wide gene profiling datasets was first
performed in the GEO database to identify key genes involved in
TNBC. The differentially expressed genes (DEGs) were firstly
identified by comparing the gene expression profiles between TNBC
and non-TNBC samples. These genes then underwent gene ontology (GO)
and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis to
explore the biological functions. Subsequently, the top-ranked DEGs
in cell lines, clinical tissues and TCGA cohorts were further
randomly validated. Finally, the potential clinical significance of
these genes and a two-gene signature acting as novel molecular
prognostic markers were examined in TCGA cohorts using the
Kaplan-Meier method and ROC analysis. The present study aimed to
provide insights into TNBC pathogenesis and identify some novel
potential biomarkers for TNBC.
Materials and methods
Dataset search and eligibility
criteria
To identify genome-wide gene expression datasets in
TNBC patients, the widely used Gene Expression Omnibus (GEO,
www.ncbi.nlm.nih.gov/geo/) database was
searched.
The search terms were based on a combination of
Triple-Negative Breast Cancer and TNBC. The search results and
relevant datasets were filtrated according to the following
eligibility criteria: i) Genome-wide expression profiling designed
for comparison between TNBC and non-TNBC or involving TNBC and
non-TNBC using high-throughput array or next generation sequencing
were included; ii) databases using only cell lines or peripheral
blood of patients were excluded; iii) the total number of samples
should not be <15; iv) there should not be <10 differentially
expressed genes identified in each gene expression dataset; and v)
the raw data should be provided in selected databases which could
be used for reanalysis. Studies that did not meet the
aforementioned eligibility criteria were excluded.
RRA analysis
The gene expression profiling was firstly annotated
using the annotation document of corresponding platforms, followed
with normalization of the expression data using ‘limma’ package of
Bioconductor (http://www.bioconductor.org/packages/release/bioc/html/limma.html).
Subsequently, the ranked lists of both upregulated and
downregulated genes in each dataset were generated according to
their fold changes. Finally, an R package of ‘Robust Rank
Aggregation’ was utilized for the integrated analysis of these
ranked gene lists (12). P-values
were calculated for each gene, indicating the possibility of
ranking high in the final gene list. In addition, a Bonferroni
correction was also used to reduce false-positive results.
Functional enrichment analysis
To reveal the possible functions of these DEGs
identified by RRA method with Bonferroni's adjusted P<0.05 and
logarithmic fold changes (logFCs)>1, GO functional enrichment
analysis and KEGG pathway enrichment analysis were performed using
‘GO.db’ (http://www.bioconductor.org/packages/release/data/annotation/html/GO.db.html)
and ‘org.Hs.eg.db’ (http://www.bioconductor.org/packages/release/data/annotation/html/org.Hs.eg.db.html)
packages in Bioconductor. The top enriched ontological or pathway
terms with Bonferroni's adjusted P<0.05 were selected.
Cell culture
The TNBC and non-TNBC cell lines, MDA-MB-231 and
MCF-7, were purchased from the Cell Bank of the Chinese Scientific
Academy. These two cell lines were maintained in Roswell Park
Memorial Institute (RMPI)-1640 medium (Gibco; Life Technologies;
Thermo Fisher Scientific, Inc.) with 10% fetal bovine serum (FBS;
Biological Industries) at 37°C and 5% CO2.
Validation of the selected DEGs using
quantitative real-time PCR
To validate the results of RRA analysis,
immunohistochemically diagnosed fresh samples were collected from
20 TNBC patients and 20 non-TNBC patients by experienced surgeons
at the First Affiliated Hospital of Zhejiang University. The
written informed consents were provided from all patients, and the
study was approved by the Institute Ethics Committee of the
hospital. After being surgically resected, the samples were stored
in sterile tubes and frozen immediately in liquid nitrogen.
Total RNA of the cell lines and clinical samples was
extracted using Qiagen RNeasy Kit (QIAGEN, Inc.) and then
reverse-transcribed into complementary DNA (cDNA) using a cDNA
synthesis kit (Takara Biotechnology Co., Ltd.), at 37°C for 15 min
and 85°C for 5 sec, according to the manufacturer's instructions.
Subsequently, six genes randomly selected from the identified 40
top-ranked DEGs through RRA analysis were used to validate the
expression by RT-qPCR using the SYBR Premix Ex Taq (Takara
Biotechnology Co., Ltd.). The 20-µl system included 10 µl SYBR
Premix Ex Taq, 1 µl each primer, 2 µl cDNA and 6 µl double
distilled water. PCR was performed as follows: 10 sec at 95°C for 1
cycle, then 5 sec at 95°C and 20 sec at 60°C for 39 cycles. GAPDH
was used as the reference gene, and the relative expression of each
gene to GAPDH. The specific primers used in the present study were
as follows: ADP-ribosyltransferase 3 (ART3) forward,
5′-GCAACCATGATTCTAGTGGACA-3′ and reverse,
5′-CTTTAGCAGTTGGGGAACGTAT-3′; fatty acid binding protein 7 (FABP7)
forward, 5′-GGCTTTGCCACTAGGCAGG-3′ and reverse,
5′-TGACCACTTTGTCTCCTTCTTGA-3′; HORMA domain containing 1 (HORMAD1)
forward, 5′-GCCCAGTTGCAGAGGACTC-3′ and reverse,
5′-TCTTGTTCCATAAGCGCATTCT-3′; trefoil factor1 (TFF1) forward,
5′-CCCCGTGAAAGACAGAATTGT-3′ and reverse, 5′-GGTGTCGTCGAAACAGCAG-3′;
anterior gradient 2 (AGR2) forward, 5′-GTCAGCATTCTTGCTCCTTGT-3′ and
reverse, 5′-GGGTCGAGAGTCCTTTGTGTC-3′; and forkhead box A1 (FOXA1)
forward, 5-GCAATACTCGCCTTACGGCT-3′ and reverse,
5′-TACACACCTTGGTAGTACGCC-3′. All experiments were performed in
triplicate.
TCGA database
The mRNA expression data and corresponding clinical
information were downloaded from TCGA data portal (https://genome-cancer.ucsc.edu/). After
extracting the ER, PR and HER2 information of each sample, a total
of 99 TNBC cases and 558 non-TNBC cases were identified. Cases with
uncertain status (equivocal, indeterminate or unknown) of any ER,
PR or HER2 were excluded. All the top-ranked DEGs underwent
survival analysis using Kaplan-Meier survival curve analysis.
Differences were considered significant with a P-value
<0.05.
Statistical analyses
All data in the present study were analyzed using
the R statistical package (R version 3.4.4) unless otherwise
stated. The univariate Cox proportional hazard analysis was firstly
conducted in the TCGA cohort to identify genes significantly
(P-value <0.05) correlated with TNBC patient survival as
candidate genes. Then, these genes were further screened to
construct a gene signature using multivariate Cox regression
analysis. Two genes from the candidate genes were selected to
construct a risk score formula by which a prognostic risk score for
each patient was calculated. Subsequently, these patients were
further separated into ‘low-risk’ and ‘high-risk’ groups using the
median risk score as the cutoff point. Kaplan-Meier survival curve
analysis was then applied to compare the differences between the
two groups for survival time. Finally, ROC analysis was conducted
and visualized by SPSS 24.0 (SPSS, Inc.).
Results
Characteristics of included microarray
datasets
After thoroughly searching in the Gene Expression
Omnibus (GEO) database according to the eligibility criteria, a
total of 4 genome-wide gene expression datasets involving TNBC and
non-TNBC were finally included. The characteristics of these 4
datasets are summarized in Table I
including GSE number, involved participants and detection
platforms. Among these datasets, the number of TNBC patients ranged
from 14 to 198, while the number of non-TNBC subjects ranged from 5
to 67. Finally, the pooled dataset included 251 TNBC samples and
158 non-TNBC samples. Various microarray platforms were used in the
studies including GPL570 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL570),
GPL10558 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL10558)
and GPL6244 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL6244).
 | Table I.Summary of 4 genome-wide gene
expression datasets including TNBC and non-TNBC tissues. |
Table I.
Summary of 4 genome-wide gene
expression datasets including TNBC and non-TNBC tissues.
| Dataset ID | GSE number | Samples | Platform |
|---|
| 1 | GSE76275 | 198 TNBC and 67
non-TNBC | GPL570 |
| 2 | GSE36693 | 21 TNBC and 66
non-TNBC | GPL10558 |
| 3 | GSE27447 | 14 TNBC and 5
non-TNBC | GPL6244 |
| 4 | GSE3744 | 18 TNBC and 20
non-TNBC | GPL570 |
RRA analysis
To identify credible aberrantly expressed genes
involved in TNBC vs. non-TNBC, an integrated analysis was performed
using the R package ‘Robust Rank Aggregation’, and upregulated and
downregulated ranked gene lists were successfully generated. A
total of 194 highly ranked differentially expressed genes (DEGs)
with P-values <0.05 were identified in TNBC vs. non-TNBC. Table
SI exhibited the top 100 significant DEGs in TNBC compared with
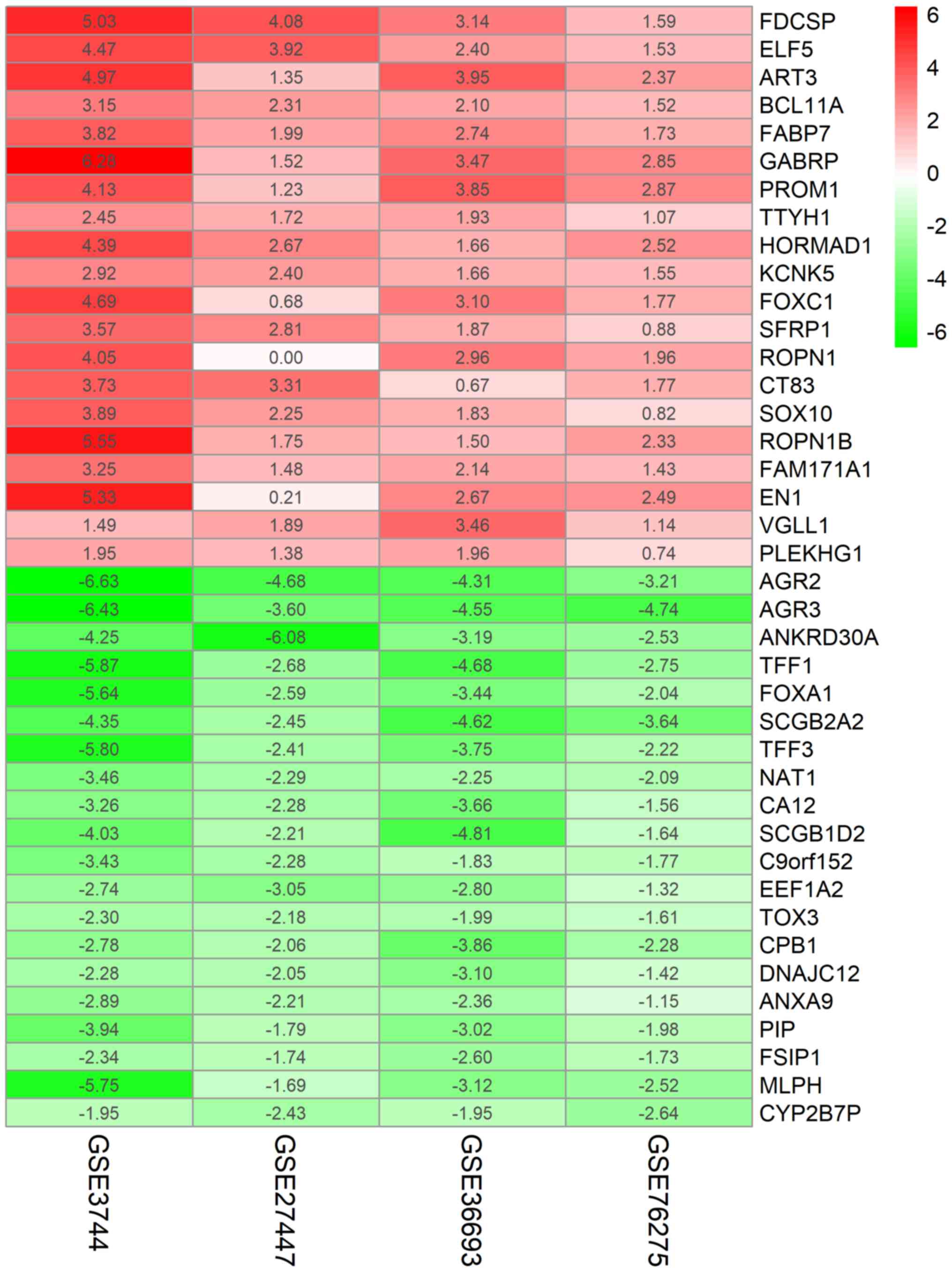
non-TNBC samples. As revealed in Fig.
1, the top 20 upregulated and downregulated genes expressed
consistently across all profiling were identified by the RRA
analysis.
Functional analysis of DEGs
To explore the systematic features and biological
functions of the identified DEGs, GO term enrichment analysis and
KEGG pathway analysis, were performed. For GO annotation, cellular
component (CC), molecular function (MF) and participation in
biological processes (BP) were included. In the BP analysis, it was
revealed that DEGs were significantly enriched in terms of
‘positive regulation of transcription from RNA polymerase II
promoter’ (Fig. 2A). Moreover, as
revealed in Fig. 2A, DEGs were
also involved in negative regulation of apoptotic process, response
to drug, and response to estradiol, indicating that these gene were
closely associated with TNBC. For MF analysis, DEGs were
significantly related with protein homodimerization activity,
transcription factor binding and calcium ion binding (Fig. 2B). CC analysis further revealed
that these genes were mainly localized in extracellular exosome,
integral component of membrane, and extracellular space (Fig. 2C). Furthermore, as revealed in
Fig. 3, KEGG pathway annotation
revealed that genes were significantly enriched in estrogen
signaling pathway, which was closely related with non-TNBC.
Validation of mRNA by RT-qPCR
According to the results analyzed by bioinformatics,
the expression of six genes randomly selected in top upregulated
and downregulated DEGs revealed in the RRA analysis was then
validated. Two cell lines, MDA-MB-231 and MCF-7 belonging to TNBC
and non-TNBC respectively, were selected to determine the
expression of randomly selected key genes including ART3, FABP7,
HORMAD1, TFF1, AGR2 and FOXA1. As revealed in Fig. 4A, the expression of ART3, FABP7 and
HORMAD1 was significantly upregulated whereas the expression of
TFF1, AGR2 and FOXA1 was significantly downregulated in the TNBC
cell line, which was consistent with the results in the RRA
analysis. Next, 20 fresh clinical tissues originating from TNBC or
non-TNBC patients were used to further validate these six randomly
selected genes. Consistent with the results obtained with the cell
lines, it was revealed that ART3, FABP7 and HORMAD1 were
respectively upregulated in TNBC tissues compared to non-TNBC
breast tissues, whereas TFF1, AGR2 and FOXA1 were downregulated
analogously (Fig. 4B).
Furthermore, similar results were also obtained in the TCGA
validation cohort including 99 TNBC samples and 558 non-TNBC
samples (Fig. S1). Collectively, these findings indicated that
reliable analysis results were obtained from the RRA method and
these top DEGs may serve as key regulators of TNBC.
 | Figure 4.Validation of six randomly selected
DEGs through RT-qPCR. (A) Expression of ART3, FABP7, HORMAD1, TFF1,
AGR2 and FOXA1 in TNBC cell lines compared with non-TNBC cell
lines. (B) Expression of ART3, FABP7, HORMAD1, TFF1, AGR2 and FOXA1
in TNBC patients compared with non-TNBC patients. *P<0.05. DEGs,
differentially expressed genes; ART3, ADP-ribosyltransferase 3;
FABP7, fatty acid binding protein 7; HORMAD1, HORMA domain
containing 1; TFF1, trefoil factor1; AGR2, anterior gradient 2;
FOXA1, forkhead box A1; TNBC, triple-negative breast cancer. |
Association between DEGs and clinical
outcome in TNBC
To explore the prognostic values of DEGs in TNBC
patients, the breast cancer data including gene expression and
clinical outcome were extracted and analyzed from TCGA, which has
vigorous criteria for sample collection and processing (14). Next, the association between the
identified top DEGs and overall survival (OS) of 99 TNBC patients
was assessed using the Kaplan-Meier survival curve analysis.
Notably, there were 4 genes, including FABP7, ART3, CT83 and TTYH1,
which were positively correlated to the life expectancy (Fig. S2),
indicating that these genes may serve as prognostic biomarkers.
To further identify key genes in the top 40 DEGs
associated with OS of TNBC patients, univariate Cox proportional
hazard regression analysis (data not shown) was performed using
gene expression as variables in the TCGA dataset. The results
revealed, 5 genes that were significantly associated with the OS of
TNBC patients with P-values <0.05. Subsequently, multivariate
Cox regression with stepwise regression (data not shown) was
performed and screened for these 5 genes. In consequence, a hazard
ratio model consisting of 2 genes, including FABP7 and CT83, was
identified as the optimum prognostic model for predicting the
prognosis of breast cancer patients. Notably, the risk scoring
formula of these 3 genes was obtained as follows: Risk
score=−0.1256×FABP7-0.119×CT83. TNBC patients in TCGA dataset were
divided into a high-risk group or low-risk group using the median
of risk score as the cutoff point, which calculated by the
aforementioned formula. As revealed in Fig. 5, the patients in the high-risk
group suffered a significantly (P<0.05) worse prognosis compared
with the low-risk group. Collectively, these results demonstrated
that the two-gene signature could well differentiate high-risk
patients from low-risk patients, which hinted its potential
clinical application value in the prognostic prediction of TNBC
patients.
Evaluation of the predictive
performance using ROC analysis
To further evaluate the predictive accuracy of the
two-gene signature, the sensitivity and specificity of the two-gene
signature in predicting prognosis were assessed using ROC analysis.
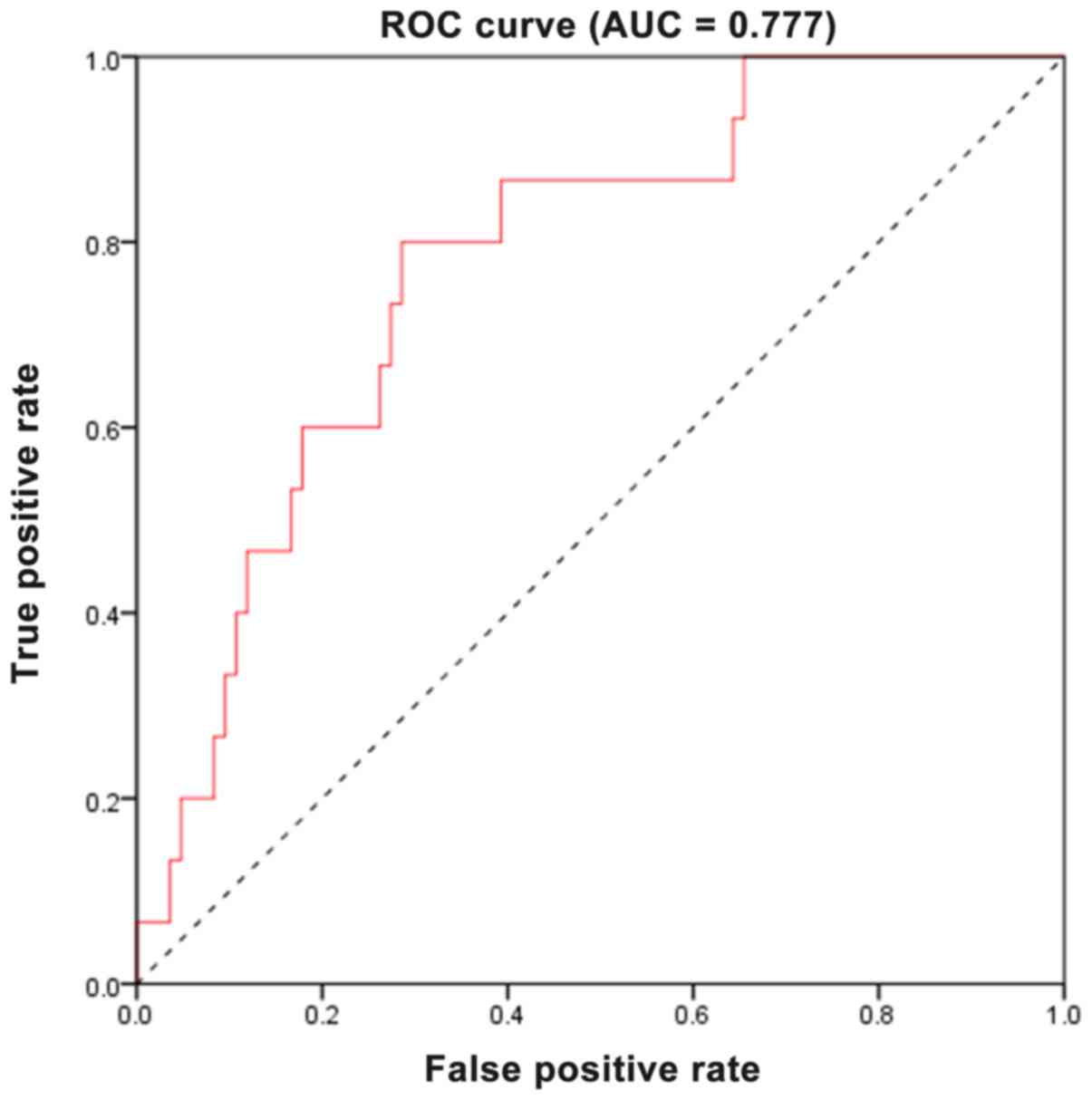
As revealed in Fig. 6, the area
under the curve (AUC) was 0.777, revealing that the two-gene
signature had a relative high sensitivity and specificity.
Consequently, along with the aforementioned results, the two-gene
signature may serve as a potential marker to predict the prognostic
survival of patients with TNBC, possessing significant clinical
application value.
Discussion
TNBC, a subtype of breast cancer comprising 15–20%
of breast cancers, is a highly aggressive cancer with poor
prognosis due to its tendency for recurrence and metastasis
(9,15–17).
Its pathogenesis and underlying mechanisms are still unclear. In
the present study, various genome-wide gene expression datasets
were integrated using the RRA method for the purpose of exploring
the underlying mechanism of TNBC at a systems level. According to
inclusion criteria, four genome-wide datasets were finally
downloaded from the GEO database, which involved a total of 251
TNBC and 158 non-TNBC patients. By means of integrated analysis, a
large cohort of significantly or downregulated genes were
identified, some of which has been documented to be strongly
associated with estrogen sensitivity in breast cancer, such as ELF5
(18,19), TFF1 (20,21),
and TFF3 (22,23). However, there are also some genes
identified to be novel TNBC gene signatures, and their underlying
mechanisms in TNBC are still poorly understood, which require
further exploration in a future study.
Subsequently, GO annotation and KEGG pathway
analysis were performed to elucidate the significance of these
identified aberrantly expressed genes. The GO analysis indicated
that these genes were significantly enriched in the following GO
terms: Positive regulation of transcription from RNA polymerase II
promoter, negative regulation of apoptotic process, response to
drug, and response to estradiol. This indicated that DEGs exerted
their biological function by catalyzing the process of
transcription. In addition, the enriched GO terms also suggested
that DEGs were closely related to the biological behavior of TNBC,
since it has been well documented that TNBC exhibits a distinctly
aggressive nature with higher rates of relapse, resistance to
endocrine therapy (ET) and sensitivity to cytotoxic chemotherapy
(24–26). Moreover, the KEGG pathway analysis
revealed that DEGs were significantly enriched in estrogen
signaling pathway, which is of importance to luminal breast cancer
with positive estrogen receptor (27).
Next, further confirmation of the DEGs analyzed from
the RRA method was performed using genes selected at random. RT-PCR
data revealed that ART3, FABP7 and HORMAD1 were significantly
upregulated whereas TFF1, AGR2 and FOXA1 were significantly
downregulated in both cell lines and clinical samples. Notably, all
these genes were confirmed to be associated with tumorigenesis.
Previous investigation indicated that overexpression of ART3
promoted TNBC via activation of Akt and ERK pathways (28). FABP7 has been reported to act as an
oncogene in TNBC, and the FABP7/RXRβ pathway was revealed to
promote cell proliferation in TNBC (29). HORMAD1 has been demonstrated to
contribute to homologous recombination (HR) deficiency in TNBC and
be associated with response to platinum-based chemotherapy in this
disease (30). A study by
Fritzsche et al (31)
demonstrated that AGR2 was positively correlated with improved
outcomes in breast cancer. In addition, AGR2 was recently revealed
to be linked with FOXA1, and the FOXA1/ERα/AGR2 signaling axis may
be utilized as a therapeutic target for the treatment of breast
cancer (32). Furthermore,
deficiency of TFF1, a small cysteine-rich acidic secreted protein,
was demonstrated to be involved in tumorigenesis of gastric cancer
(33) and breast cancer (34). These results, collectively,
indicated that integrated analysis could provide reliable results
and these DEGs may play pivotal roles in tumorigenesis and
development in breast cancer.
In order to determine whether these identified top
40 DEGs were related to the life expectancy of patients with TNBC,
all the top DEGs underwent survival analysis using TCGA database.
Using Kaplan-Meier survival curve analysis, 4 genes (FABP7, ART3,
CT83 and TTYH1) were identified to be positively correlated to life
expectancy. Moreover, a two-gene signature, including FABP7 and
CT83, was predicted to be significantly associated with the OS of
breast cancer patients using univariate Cox analysis followed with
multivariate Cox analysis. By calculating risk score according to a
formula, the two-gene signature performed well in differentiating
low-risk and high-risk groups, which exhibited significant
prognostic differences. Finally, ROC analysis was performed and a
relatively high AUC of 0.777 was demonstrated, revealing that the
two-gene signature could act as an independent predictor of
survival of patients with TNBC.
To sum up, the present study provides an integrated
analysis of gene expression profiles of TNBC compared with non-TNBC
and identifies a number of key genes involved in the pathogenesis
of TNBC. It is inferred from the present research that these DEGs
may regulate the initiation and progression of TNBC in various
ways. Some of these key genes are novel and their precise roles in
TNBC remain unclear. The present findings in this study may provide
insights into the pathogenesis of TNBC at a systems level, and also
identify some alternative biomarkers and potential therapeutic
targets for TNBC patients. Continuous studies on these key genes
should be performed to elucidate their detailed role in TNBC.
Acknowledgements
Not applicable.
Funding
The present study was financially supported by grant
2017KY019 from the Medical Health Science and Technology Project of
Zhejiang Provincial Health Commission (China).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
GZ, WL and QS conceived and designed the present
study. GZ, WL, QS and KY performed the experiments and analyzed the
data. YZ interpreted the data and wrote the manuscript. All authors
read and approved the final manuscript and agree to be accountable
for all aspects of the research in ensuring that the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the First Affiliated Hospital of Zhejiang University
(Zhejiang, China). Written informed consent was provided by all
patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Foulkes WD, Smith IE and Reis-Filho JS:
Triple-negative breast cancer. N Engl J Med. 363:1938–1948. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dent R, Trudeau M, Pritchard KI, Hanna WM,
Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P and Narod SA:
Triple-negative breast cancer: Clinical features and patterns of
recurrence. Clin Cancer Res. 13:4429–4434. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Carey LA, Perou CM, Livasy CA, Dressler
LG, Cowan D, Conway K, Karaca G, Troester MA, Tse CK, Edmiston S,
et al: Race, breast cancer subtypes, and survival in the Carolina
Breast Cancer Study. JAMA. 295:2492–2502. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Carey L, Winer E, Viale G, Cameron D and
Gianni L: Triple-negative breast cancer: Disease entity or title of
convenience? Nat Rev Clin Oncol. 7:683–692. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rouzier R, Perou CM, Symmans WF, Ibrahim
N, Cristofanilli M, Anderson K, Hess KR, Stec J, Ayers M, Wagner P,
et al: Breast cancer molecular subtypes respond differently to
preoperative chemotherapy. Clin Cancer Res. 11:5678–5685. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
von Minckwitz G, Untch M, Blohmer JU,
Costa SD, Eidtmann H, Fasching PA, Gerber B, Eiermann W, Hilfrich
J, Huober J, et al: Definition and impact of pathologic complete
response on prognosis after neoadjuvant chemotherapy in various
intrinsic breast cancer subtypes. J Clin Oncol. 30:1796–1804. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Byler S, Goldgar S, Heerboth S, Leary M,
Housman G, Moulton K and Sarkar S: Genetic and epigenetic aspects
of breast cancer progression and therapy. Anticancer Res.
34:1071–1077. 2014.PubMed/NCBI
|
|
9
|
Lehmann BD, Bauer JA, Chen X, Sanders ME,
Chakravarthy AB, Shyr Y and Pietenpol JA: Identification of human
triple-negative breast cancer subtypes and preclinical models for
selection of targeted therapies. J Clin Invest. 121:2750–2767.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Burstein MD, Tsimelzon A, Poage GM,
Covington KR, Contreras A, Fuqua SA, Savage MI, Osborne CK,
Hilsenbeck SG, Chang JC, et al: Comprehensive genomic analysis
identifies novel subtypes and targets of triple-negative breast
cancer. Clin Cancer Res. 21:1688–1698. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Võsa U, Kolde R, Vilo J, Metspalu A and
Annilo T: Comprehensive meta-analysis of microRNA expression using
a robust rank aggregation approach. Methods Mol Biol. 1182:361–373.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kolde R, Laur S, Adler P and Vilo J:
Robust rank aggregation for gene list integration and
meta-analysis. Bioinformatics. 28:573–580. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shi KQ, Lin Z, Chen XJ, Song M, Wang YQ,
Cai YJ, Yang NB, Zheng MH, Dong JZ, Zhang L and Chen YP:
Hepatocellular carcinoma associated microRNA expression signature:
Integrated bioinformatics analysis, experimental validation and
clinical significance. Oncotarget. 6:25093–25108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chin L, Andersen JN and Futreal PA: Cancer
genomics: From discovery science to personalized medicine. Nat Med.
17:297–303. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Curtis C, Shah SP, Chin SF, Turashvili G,
Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et
al: The genomic and transcriptomic architecture of 2,000 breast
tumours reveals novel subgroups. Nature. 486:346–352. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Perou CM, Sørlie T, Eisen MB, van de Rijn
M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA,
et al: Molecular portraits of human breast tumours. Nature.
406:747–752. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Anders CK and Carey LA: Biology,
metastatic patterns, and treatment of patients with triple-negative
breast cancer. Clin Breast Cancer. 9 (Suppl 2):S73–S81. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kalyuga M, Gallego-Ortega D, Lee HJ, Roden
DL, Cowley MJ, Caldon CE, Stone A, Allerdice SL, Valdes-Mora F,
Launchbury R, et al: ELF5 suppresses estrogen sensitivity and
underpins the acquisition of antiestrogen resistance in luminal
breast cancer. PLoS Biol. 10:e10014612012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Furth PA, Nakles RE, Millman S, Diaz-Cruz
ES and Cabrera MC: Signal transducer and activator of transcription
5 as a key signaling pathway in normal mammary gland developmental
biology and breast cancer. Breast Cancer Res. 13:2202011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou L, Yan T, Jiang Y, Di G, Shen Z, Shao
Z and Lu J: Prognostic and predictive value of TFF1 for adjuvant
endocrine therapy in Chinese women with early ER positive breast
cancer: Comparing aromatase inhibitors with tamoxifen. Breast.
20:15–20. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Prest SJ, May FE and Westley BR: The
estrogen-regulated protein, TFF1, stimulates migration of human
breast cancer cells. FASEB J. 16:592–594. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
May FE and Westley BR: TFF3 is a valuable
predictive biomarker of endocrine response in metastatic breast
cancer. Endocr Relat Cancer. 22:465–479. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ishibashi Y, Ohtsu H, Ikemura M, Kikuchi
Y, Niwa T, Nishioka K, Uchida Y, Miura H, Aikou S, Gunji T, et al:
Serum TFF1 and TFF3 but not TFF2 are higher in women with breast
cancer than in women without breast cancer. Sci Rep. 7:48462017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Abramson VG, Lehmann BD, Ballinger TJ and
Pietenpol JA: Subtyping of triple-negative breast cancer:
Implications for therapy. Cancer. 121:8–16. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Podo F, Buydens LM, Degani H, Hilhorst R,
Klipp E, Gribbestad IS, Van Huffel S, van Laarhoven HW, Luts J,
Monleon D, et al: Triple-negative breast cancer: Present challenges
and new perspectives. Mol Oncol. 4:209–229. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liedtke C, Mazouni C, Hess KR, André F,
Tordai A, Mejia JA, Symmans WF, Gonzalez-Angulo AM, Hennessy B,
Green M, et al: Response to neoadjuvant therapy and long-term
survival in patients with triple-negative breast cancer. J Clin
Oncol. 26:1275–1281. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Iwamoto T, Bianchini G, Booser D, Qi Y,
Coutant C, Shiang CY, Santarpia L, Matsuoka J, Hortobagyi GN,
Symmans WF, et al: Gene pathways associated with prognosis and
chemotherapy sensitivity in molecular subtypes of breast cancer. J
Natl Cancer Inst. 103:264–272. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tan L, Song X, Sun X, Wang N, Qu Y and Sun
Z: ART3 regulates triple-negative breast cancer cell function via
activation of Akt and ERK pathways. Oncotarget. 7:46589–46602.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu RZ, Graham K, Glubrecht DD, Lai R,
Mackey JR and Godbout R: A fatty acid-binding protein 7/RXRβ
pathway enhances survival and proliferation in triple-negative
breast cancer. J Pathol. 228:310–321. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Watkins J, Weekes D, Shah V, Gazinska P,
Joshi S, Sidhu B, Gillett C, Pinder S, Vanoli F, Jasin M, et al:
Genomic complexity profiling reveals that HORMAD1 overexpression
contributes to homologous recombination deficiency in
triple-negative breast cancers. Cancer Discov. 5:488–505. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fritzsche FR, Dahl E, Pahl S, Burkhardt M,
Luo J, Mayordomo E, Gansukh T, Dankof A, Knuechel R, Denkert C, et
al: Prognostic relevance of AGR2 expression in breast cancer. Clin
Cancer Res. 12:1728–1734. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wright TM, Wardell SE, Jasper JS, Stice
JP, Safi R, Nelson ER and McDonnell DP: Delineation of a
FOXA1/ERalpha/AGR2 regulatory loop that is dysregulated in
endocrine therapy-resistant breast cancer. Mol Cancer Res.
12:1829–1839. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Soutto M, Peng D, Katsha A, Chen Z,
Piazuelo MB, Washington MK, Belkhiri A, Correa P and El-Rifai W:
Activation of β-catenin signalling by TFF1 loss promotes cell
proliferation and gastric tumorigenesis. Gut. 64:1028–1039. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Buache E, Etique N, Alpy F, Stoll I,
Muckensturm M, Reina-San-Martin B, Chenard MP, Tomasetto C and Rio
MC: Deficiency in trefoil factor 1 (TFF1) increases tumorigenicity
of human breast cancer cells and mammary tumor development in
TFF1-knockout mice. Oncogene. 30:3261–3273. 2011. View Article : Google Scholar : PubMed/NCBI
|