Introduction
Myocardial ischemia/reperfusion (I/R) injury is a
major pathophysiological condition associated with cardiopulmonary
bypass surgery and various cardiovascular diseases (CVDs),
including coronary heart disease and myocardial infarction, both of
which have high mortality and morbidity rates (1,2).
Mitochondria are the organelles providing the major source of
reactive oxygen species (ROS) production and are rich in
cardiomyocytes (3,4). Mitochondrial oxidative stress serves
a central role in myocardial I/R injury. Hypoxia induces superoxide
generation via the electron transport chain. High concentrations of
ROS disrupt cell functions, induce apoptosis and cause tissue
damage via inducing oxidative stress. Therefore,
mitochondria-mediated oxidative stress is the key step in
I/R-induced cardiomyocytes injury (5,6). It
is also a main target for designing treatment strategies for
I/R-induced cardiomyocyte injury.
Resveratrol (RES), a natural compound found
primarily in red wine, is a naturally occurring antioxidant. The
potential beneficial effects of RES were first observed in major
CVDs, including myocardial ischemia, atherosclerosis, hypertension,
stroke, aging and heart failure (7). Previous studies have revealed that
the cadioprotective effects of RES include anti-oxidative stress,
antiplatelet activity, preventing endothelial cell damage and
inflammation, and increasing the expression of nitric oxide
synthase. The biological effects of RES, including chemoprevention,
immunomodulatory, antiproliferative and antioxidant effects, have
been reported previously (8–11).
Recently, it was reported that RES exerts its cardioprotective
effect through the AMP-activated protein kinase and
phosphoinositide-3-kinase/Akt/Forkhead box O3a signaling pathways
(12,13). Additionally, it was reported that
pre-treatment with RES may prevent ischemic cerebral damage in rats
by activating sirtuin 1 (Sirt1), one of the ubiquitous histone and
protein deacetylases (14).
RES-mediated autophagy is also involved in cardioprotection
(15). However, the mechanisms
involved in the cardioprotective effect of RES remain to be fully
elucidated. The mitochondria-mediated oxidative stress pathway is
one of the major causes of I/R or hypoxia/reoxygenation (H/R)
(4,16). ROS are generated in the I/R or H/R
myocardium, particularly in mitochondria. I/R injury has been
associated with significant increases in ROS, the release of
lactate dehydrogenase (LDH) and depolarization of the mitochondrial
transmembrane potential (ΔΨm), which are all important indices for
reflecting the status of mitochondrial oxidative stress. Enhanced
mitochondrial oxidative stress induces cell injury and apoptosis
(17,18). Previous findings have also shown
that RES has anti-oxidant properties (19). Hypoxia is an important
pathophysiological progress and induces cell injury through
crosstalk with mitochondria and oxidative stress pathways (20). Sirt1 is also involved in
hypoxia-induced pulmonary artery smooth muscle cell injury
(21). To gain further insight
into the mechanisms underlying the RES-mediated protective effect
against I/R injury, an H/R model of cultured NRCMs was established
in the present study. Using this model, the cardioprotective effect
of RES and the underlying mechanism were examined. The results
showed that RES alleviated H/R injury through inhibiting the
mitochondria-mediated oxidative stress pathway. This provides a
robust scientific basis for the clinical application of RES in the
treatment of cardiac conditions.
Materials and methods
Drugs and reagents
RES (molecular formula:
C14H12O3; CAS no. 501-36) was
purchased from Neptunus Company (Shenzhen, China). A MitoProbe™
JC-1 Assay kit (cat. no. M34152) for measuring the ΔΨm was obtained
from Thermo Fisher Scientific, Inc. (Waltham, MA, USA). Dimethyl
sulfoxide (DMSO) was purchased from Sigma; Merck KGaA (Darmstadt,
Germany). The BCA protein assay (cat. no. 23225) was purchased from
Thermo Fisher Scientific, Inc. All other reagents and chemicals
were obtained commercially and were of analytical grade.
Cell culture and the treatment
The experimental protocols were performed in
accordance with the National Institutes of Health Guide for the
Care and Use of Laboratory Animals (National Institutes of Health,
Bethesda, MD, USA) and with the approval of the Animal Care and Use
Committee of the Southwest Medical University (Sichuan, China).
Neonatal rats (age, 2–3 days; weight, 5–8 g; n=150)
were purchased from The Animal Center of Southwest Medical
University (Luzhou, China). Neonatal rats were maintained at 22±3°C
under a 12-h light/dark cycle. All rats had free access to water
and food. Primary cultured neonatal rat cardiomyocytes (NRCMs) were
cultured as previously described (22). In brief, neonatal rat ventricles
were digested with collagenase II and cardiomyocytes were purified
through adherence to culture plastic for different durations.
Subsequently, 0.1 mM 5-BrdU was added to DMEM to inhibit the
cardiac fibroblasts. The NRCMs were cultured in DMEM (cat. no.
SH30021; HyClone; GE Healthcare Life Sciences, Logan, UT, USA)
containing 10% FBS (cat. no. 10270; Gibco; Thermo Fisher
Scientific, Inc.) and 1% penicillin/streptomycin (cat. no. C0222;
Beyotime Institute of Biotechnology, Haimen, China) at 37°C in a 5%
CO2 incubator. The NRCMs were randomly divided into four
groups: i) Control group (normal DMEM); ii) H/R group: Cells were
subjected to a completely enclosed environment containing a Bio-Bag
(Thermo Fisher Scientific, Inc.) for 8 h in hypoxic medium and then
subjected to reoxygenation under normoxic conditions for another 8
h, following which the hypoxic medium was replaced with fresh
medium (containing 10% FBS) upon reoxygenation; iii) DMSO + H/R
group: 0.2% DMSO was added to the medium during H/R; iv) RES + H/R
group: RES (final concentration of 100 µM) was added to the medium
during H/R.
Measurement of LDH release
LDH release was detected using an LDH assay kit
(cat. no. A020, Nanjing Jiancheng Bioengineering Institute,
Nanjing, China) according to the manufacturer's protocol. In brief,
the NRCMs were subjected to centrifugation at 300 × g for 10 min.
Following this, 60 µl of supernatant was added to 30 µl of LDH
substrate solution and incubated for 30 min at 37°C. The LDH levels
were detected with a microplate reader (Tecan Group, Inc.,
Mannedorf, Switzerland) at 440 nm.
Measurement of ΔΨm
The ΔΨm was measured by flow cytometry using the
MitoProbe™ JC-1 assay kit. The approximate excitation peak of JC-1
is 488 nm. The approximate emission peaks of monomeric and
J-aggregate forms are 529 and 590 nm, respectively. The ratio of
monomeric JC-1 represents the level of ΔΨm. An increased ratio
represents the depolarization of ΔΨm, whereas a decreased ratio
indicates the hyperpolarization of ΔΨm.
Immunofluorescence microscopy
Primary cultured NRCMs were plated on the coverslips
at 60–70% confluency and fixed in cold PBS with paraformaldehyde
for 15 min, permeabilized with 0.1% triton-X 100 for 15 min and
blocked with 5% bovine serum albumin (cat. no. a600332; BBI Life
Sciences Corporation, Shanghai, China) for 1 h at room
temperature. The NRCMs were then incubated with primary mouse
anti-α-actinin 2 antibody (1:100; cat. no. BM4907; Wuhan Boster
Biological Technology, Ltd., Wuhan, China) overnight at 4°C and
then incubated with the DyLight® 488-conjugated donkey
anti-mouse secondary antibody (1:200; cat. no. ab96875; Abcam,
Cambridge, UK) for 1 h at room temperature. In addition, for the
staining of F-actin, the NRCMs were treated with 100 nM rhodamine
phalloidin (cat. no. PHDR1; Cytoskeleton, Inc., Denver, CO, USA)
for 30 min at room temperature. The nuclei were stained with DAPI.
The cells were mounted and images were captured using an Olympus
fluorescence microscope (IX-81, Olympus Corporation, Tokyo, Japan).
The excitation wavelengths for DAPI, DyLight®
488-conjugated and rhodamine phalloidin were 405, 488 and 550 nm,
respectively.
Western blotting
The cells were washed with PBS and lysed with lysis
buffer containing a cocktail of protease inhibitors (cat. no.
87785, Thermo Fisher Scientific, Inc.). Subsequently, 60 µg total
protein for each lane was separated with a 5% stacking gel and 10%
separation gel, and then transferred onto a PVDF membrane (cat. no.
IPVH00010; EMD Millipore, Billerica, MA, USA). The membrane was
incubated in TBST containing 5% non-fat milk for 2 h at room
temperature to block non-specific binding and was then incubated
with primary antibodies (1:1,000) overnight at 4°C. The primary
antibodies against Bax (cat. no. 2772; Cell Signaling Technology,
Inc., Danvers, MA, USA), Bcl-2 (cat. no. ab196495; Absin, Shanghai,
China), Sirt1 (cat. no. 9475; Cell Signaling Technology, Inc.) and
the internal control antibody GAPDH (cat. no. sc-25778; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) were polyclonal antibodies
raised in rabbit. The membrane was then incubated with horseradish
peroxidase (HRP)-conjugated goat anti-rabbit IgG secondary antibody
(cat. no. D110058; BBI Life Sciences, Shanghai, China; 1:1,000) for
1 h at room temperature. The membrane was incubated in
chemiluminescent HRP substrate (cat. no. WBKLS0500; EMD Millipore)
at room temperature for 5 min, following which images were captured
with the Universal Hood II system (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
Measurement of caspase 3 activity
Apoptotic cell death was determined by caspase 3
activation using a Caspase-3 activity assay kit (cat. no. G015,
Nanjing Jiancheng Bioengineering Institute). Briefly, the cells
were harvested using caspase lysis buffer (50 mM HEPES, pH 7.4,
0.1% Chaps, 5 mM dithiothreitol, 0.1 mM EDTA and 0.1% Triton X-100)
for 5 min on ice and centrifuged at 13,000 × g for 10 min at 4°C.
The supernatant (50 µg) was then isolated and incubated with 10 µl
caspase 3 substrate (Ac-DEVDpNA) for 1 h at 37°C. The activity of
caspase 3 was detected with a microplate reader (Tecan Group, Ltd.)
at 400 nm.
Statistical analysis
Data are presented as the mean ± standard error of
the mean and were analyzed by one way analysis of variance using
SPSS 19.0 (IBM Corp., Armonk, NY, USA). The least significant
difference test was used for further multiple group comparisons.
P<0.05 was considered to indicate a statistically significant
difference.
Results
RES protects against H/R
injury-induced structural impairment in NRCMs
Firstly, the present study investigated the effects
of RES on H/R-induced structural impairment in NRCMs using
immunohistological staining techniques. As shown in Fig. 1, NRCMs without H/R injury (control
group) had a clear morphology with a striated pattern (green) and
normal cytoskeletal structure (red). The NRCMs with H/R injury
exhibited significant alterations in morphology and cytoskeletal
structure, characterized by disordered α-actin and F-actin. In
contrast to the NRCMs in the H/R injury group, the impairment of
cell structure induced by H/R injury in the NRCMs was significantly
attenuated by treatment with 100 µM RES. Treatment with 0.2% DMSO,
the final concentration used for diluting RES, did not affect H/R
injury-induced structural impairment.
Effects of RES on H/R injury-induced
oxidative stress of mitochondria
Mitochondrial oxidative stress is a hallmark of I/R
injury in cardiomyocytes. The present study further examined the
effect of RES on mitochondrial oxidative stress injury and cell
apoptosis, including the determination of LDH release, ΔΨm and the
Bcl2/Bax ratio. The Bcl2/Bax ratio in each group was normalized to
the value in the control group. As shown in Fig. 2A, exposure to H/R increased LDH
release by 1.41±0.03-fold compared with that of the control group
(P<0.01, n=4), whereas 100 µM RES significantly attenuated the
increased release of LDH induced by H/R injury in the NRCMs from
1.41±0.03-fold (H/R) to 1.02±0.06-fold (P<0.01, n=4). Treatment
with 0.2% DMSO did not affect LDH release when compared with the
H/R group. Treatment with H/R induced the depolarization of ΔΨm by
shifting the ratio of JC-1 monomers from 40.43±11.21% (control) to
62.39±1.82% (H/R) (P<0.05, n=4), whereas treatment with 100 µM
RES alleviated the H/R-induced depolarization of ΔΨm by shifting
the ratio of JC-1 monomers from 62.39±1.82% (H/R) to 35.31±8.63%
(H/R +100 µM RES) (P<0.05, n=4), as shown in Fig. 2B.
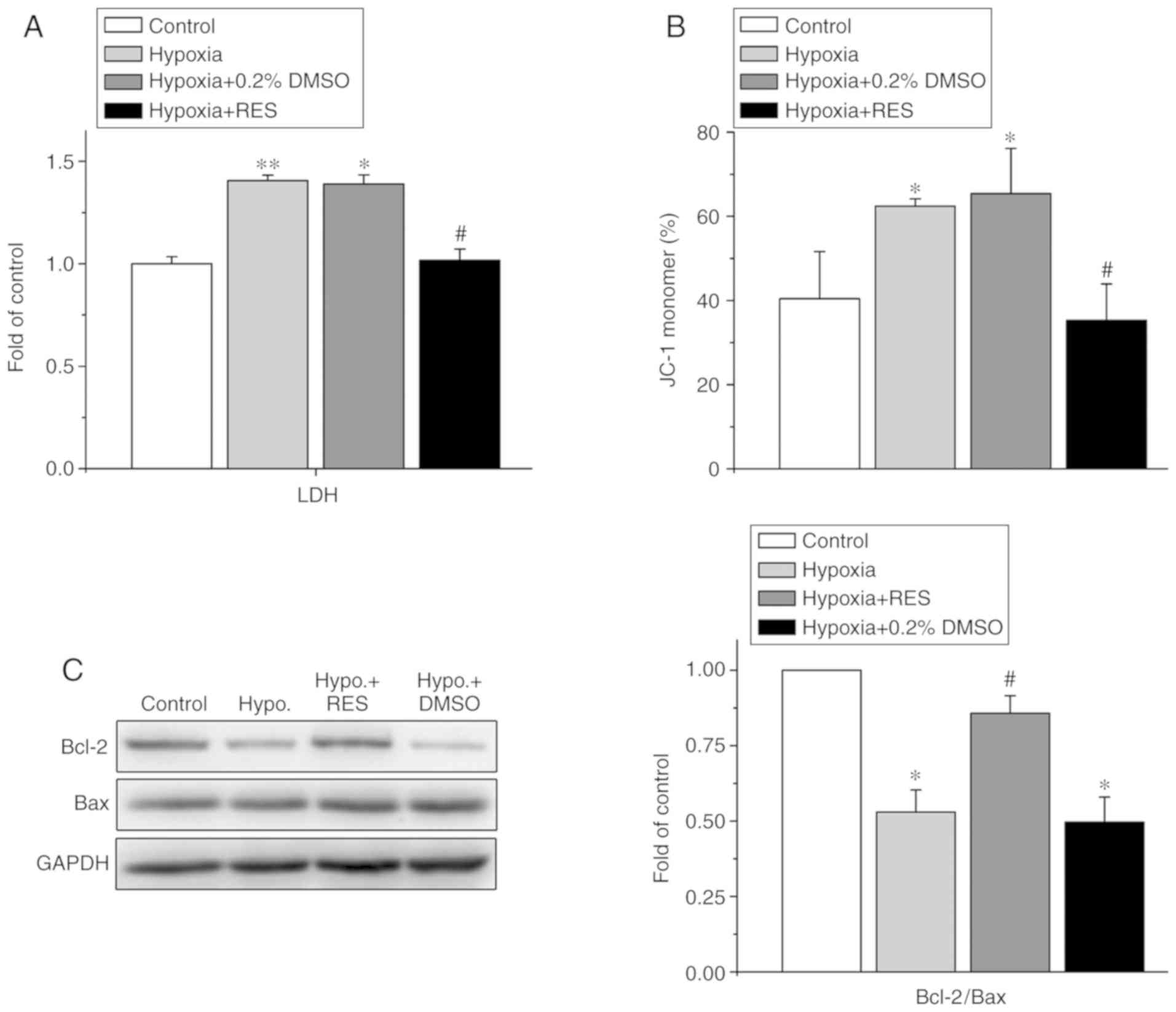 | Figure 2.Effects of RES on H/R injury-induced
mitochondria oxidative stress in neonatal rat cardiomyocytes. (A)
Effect of RES on LDH release (n=4). (B) Effect of RES on the ΔΨm,
as measured with the JC-1 kit. The percentage of monomer JC-1
showed the level of ΔΨm. The increment of monomer JC-1 represents
the depolarization of ΔΨm, whereas the decrement of monomer JC-1
indicates the hyperpolarization of ΔΨm. (C) Western blotting of the
protein levels of Bcl-2 and Bax (left) and the ratio of Bcl-2/Bax
(right) (n=5). *P<0.05, vs. control; #P<0.05, vs.
H/R treatment. RES, resveratrol; H/R, hypoxia/reoxygenation; LDH,
lactate dehydrogenase; ΔΨm, mitochondrial membrane potential;
Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X protein; Hypo,
hypoxia; DMSO, dimethyl sulfoxide. |
Furthermore, the ratio of Bcl2/Bax expression was
examined, as detected by western blotting (Fig. 2C). Exposure to H/R decreased the
Bcl2/Bax ratio to 0.53±0.08-fold of the control (P<0.05, n=5),
whereas treatment with 100 µM RES rescued the H/R-induced decrease
in the Bcl2/Bax ratio to 0.86±0.06-fold (P<0.05, n=5).
Effects of RES on H/R-injury-induced
apoptosis of NRCMs
The present study also detected the effect of RES on
apoptosis induced by H/R injury in NRCMs, as shown in Fig. 3. The results of the flow cytometry
(Fig. 3A and B) showed that RES
attenuated the H/R injury-induced cell apoptosis of NRCMs, with a
decrease in the cell apoptotic rate from 84.25±7.41% (H/R) to
46.39±5.43% (H/R+RES) (P<0.05, n=4). Treatment with 0.2% DMSO
alone did not significantly alter the effect of H/R on the
apoptotic rate of cells (P>0.05, n=4).
In addition, caspase 3 is a common downstream
signaling molecule involved in cell apoptosis induced by different
factors. The present study found that RES alleviated the H/R
injury-induced apoptosis of NRCMs (Fig. 3C). The activity of caspase 3 in
each group was normalized to the value in the control group.
Exposure to H/R increased the activity of caspase 3 to
1.32±0.06-fold compared with the control group (P<0.05, n=5),
whereas treatment with 100 µM RES alleviated the increased activity
of caspase 3 to 1.02±0.04-fold (P<0.05, n=5).
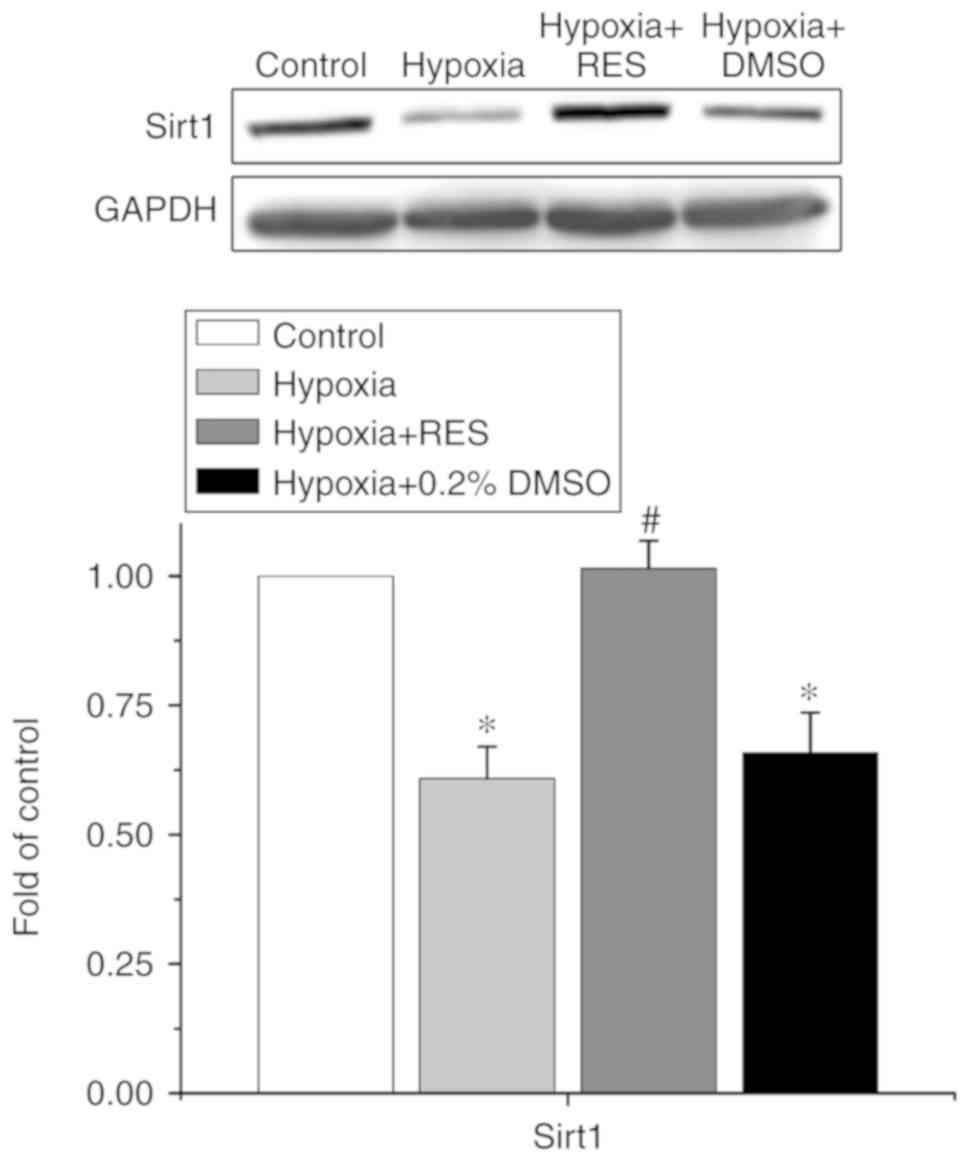
Sirt1 is involved in the effect of RES
on H/R injury in NRCMs
Sirt1 is reportedly an important target of RES,
involved in the cardioprotective effects of several drugs indicated
for cardiovascular diseases. Therefore, the present study examined
whether Sirt1 was involved in the effects of RES on H/R injury in
NRCMs. The data are shown in Fig.
4. The expression of Sirt1 in each group was normalized to the
value in the control group. Treatment of H/R decreased the
expression of Sirt1 to 0.61±0.06-fold compared with that in the
control group (P<0.05, n=5), whereas treatment with RES
alleviated the decrease of Sirt1 induced by H/R injury to
1.01±0.05-fold (P<0.05, n=5).
Discussion
CVDs are the major causes of morbidity and mortality
in most countries. I/R injury is a leading cause of myocardial cell
death following myocardial infarction. Oxidative stress is the main
pathophysiological process involved in I/R injury and other CVDs,
including heart failure and hypertension (23,24).
Active cardiomyocytes consume energy from the aerobic metabolic
pathway, and cardiomyocytes are more sensitive to hypoxia than
other cell types. Cardiomyocytes are more prone to metabolic
dysfunction, as they are decompensated during the acute I/R
condition. Although several studies have verified the
cardioprotective effects of RES through inhibiting inflammation and
platelet aggregation, alleviating endothelial damage and modulating
autophagy, current knowledge is limited regarding the role of
oxidative stress injury on RES cardioprotection (9,25).
In the present study, an in vitro H/R model was established
using NRCMs to imitate I/R injury, and the role of RES on
H/R-induced NRCM injury and underlying mechanism were examined. It
was found that RES alleviated H/R-induced NRCM injury and apoptosis
through attenuating the mitochondria-mediated oxidative stress
pathway.
Dong et al (26) reported that resveratrol protected
against pressure-overload-induced cardiac structure injury, and
exerted beneficial effects on cardiac hypertrophy in a rat model.
In the present study, it was found that treatment with RES
ameliorated H/R-induced cardiomyocyte structural impairment with
F-actin and α-actinin 2, indicating the cytoskeleton and T-tubules
(Fig. 1).
Mitochondria are the main organelle involved in
biological oxidative reactions. Mitochondria are abundant in
cardiomyocytes and the mitochondria-mediated oxidative stress
pathway is involved in the cardiac I/R injury process (5,17).
LDH levels, ΔΨm and the ratio of Bcl-2 to Bax are important indices
for reflecting the mitochondria-mediated oxidative stress status
(27). The present study found
that H/R treatment induced mitochondria oxidative stress, whereas
treatment with RES alleviated H/R-induced mitochondrial injury
through decreasing the release or LDH, inhibiting the
depolarization of ΔΨm and increasing the ratio of Bcl-2 to Bax
(Fig. 2). These data suggest that
RES attenuates H/R-induced cardiomyocyte injury through alleviating
mitochondria-mediated oxidative stress; mitochondria are targets of
RES involved in the cardioprotective effect to attenuate the
H/R-induced injury of NRCMs.
The mitochondria-mediated route is an important
apoptotic pathway in cells. Enhanced oxidative stress induces cell
injury through mitochondria-mediated cell apoptosis. In the present
study, it was found that treatment with RES inhibited the apoptosis
induced by H/R injury through alleviating the cell apoptotic rate
and activity of caspase 3, which reflects the status of cell
apoptosis (Fig. 3). Therefore,
these results support the hypothesis that RES alleviates H/R injury
and exerts cardioprotective effects through the
mitochondria-mediated signaling pathway.
In addition, Sirt1, a member of the conserved
sirtuin family, is an NAD+-dependent histone
deacetylase, which is involved in the various cardiac
pathophysiological process and cardioprotective effects of certain
drugs. Previous studies have suggested that Sirt1 is required for
the RES-mediated cardioprotective effect (28,29).
Sin et al (30) reported
that Sirt1 was involved in the effect of RES for alleviating
doxorubicin-induced cardiotoxicity in aged hearts. Recently, Li
et al (21) reported that
Sirt1 is involved in hypoxia-induced pulmonary artery smooth muscle
cell apoptosis. The data obtained in the present study are
consistent with those of former studies, and also provide novel
evidence that Sirt1 is involved in the cardioprotective effect of
RES with respect to H/R injury.
The present study found that the cardioprotective
effect of RES in H/R-induced cardiomyocyte injury occurs through
alleviating mitochondrial oxidative stress and restoring the
expression of Sirt1. The results further elucidated the underlying
mechanism of RES cardioprotection, and support the widespread
clinical use of RES for cardiovascular disease.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant nos. 31300948 and 81670310) and
the Joint Foundation of Southwest Medical University and Luzhou
city (grant nos. 2015-LZCYD-S03-1/7 and 2016-LZXNYD-T10).
Availability of data and materials
The datasets and materials used and/or analyzed
during the current study are available from the corresponding
author on reasonable request.
Authors' contributions
XQT, TL and LC designed the experiments. TL, LC, YY,
BY and PL performed the experiments and collected the data. XQT, TL
and LC analyzed and interpreted the data. XQT and TL wrote the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The experimental protocols were performed in
accordance with the National Institutes of Health Guide for the
Care and Use of Laboratory Animals (National Institutes of Health,
Bethesda, MD, USA) and with the approval of the Animal Care and Use
Committee of the Southwest Medical University, (Sichuan,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Murphy E and Steenbergen C: Mechanisms
underlying acute protection from cardiac ischemia-reperfusion
injury. Physiol Rev. 88:581–609. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hausenloy DJ and Yellon DM: Myocardial
ischemia-reperfusion injury: A neglected therapeutic target. J Clin
Invest. 123:92–100. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Paradies G, Paradies V, Ruggiero FM and
Petrosillo G: Mitochondrial bioenergetics and cardiolipin
alterations in myocardial ischemia/reperfusion injury. Implications
for pharmacological cardioprotection. Am J Physiol Heart Circ
Physiol. Aug 10–2018.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cadenas S: ROS and redox signaling in
myocardial ischemia-reperfusion injury and cardioprotection. Free
Radic Biol Med. 117:76–89. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Suen DF, Norris KL and Youle RJ:
Mitochondrial dynamics and apoptosis. Genes Dev. 22:1577–1590.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
von Harsdorf R, Li PF and Dietz R:
Signaling pathways in reactive oxygen species-induced cardiomyocyte
apoptosis. Circulation. 99:2934–2941. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bonnefont-Rousselot D: Resveratrol and
cardiovascular diseases. Nutrients. 8(pii): E2502016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nicholson SK, Tucker GA and Brameld JM:
Effects of dietary polyphenols on gene expression in human vascular
endothelial cells. Proc Nutr Soc. 67:42–47. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Petrovski G, Gurusamy N and Das DK:
Resveratrol in cardiovascular health and disease. Ann N Y Acad Sci.
1215:22–33. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dolinsky VW and Dyck JR: Calorie
restriction and resveratrol in cardiovascular health and disease.
Biochim Biophys Acta. 1812:1477–1489. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ge L, Li C, Wang Z, Zhang Y and Chen L:
Suppression of oxidative stress and apoptosis in electrically
stimulated neonatal rat cardiomyocytes by resveratrol and
underlying mechanisms. J Cardiovasc Pharmacol. 70:396–404.
2017.PubMed/NCBI
|
|
12
|
Meng Z, Jing H, Gan L, Li H and Luo B:
Resveratrol attenuated estrogen-deficient-induced cardiac
dysfunction: Role of AMPK, SIRT1, and mitochondrial function. Am J
Transl Res. 8:2641–2649. 2016.PubMed/NCBI
|
|
13
|
Wu Z, Huang A, Yan J, Liu B, Liu Q, Zhang
J, Zhang X, Ou C and Chen M: Resveratrol ameliorates cardiac
dysfunction by inhibiting apoptosis via the PI3K/Akt/FoxO3a pathway
in a rat model of diabetic cardiomyopathy. J Cardiovasc Pharmacol.
70:184–193. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Della-Morte D, Dave KR, DeFazio RA, Bao
YC, Raval AP and Perez-Pinzon MA: Resveratrol pretreatment protects
rat brain from cerebral ischemic damage via a sirtuin 1-uncoupling
protein 2 pathway. Neuroscience. 159:993–1002. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gurusamy N, Lekli I, Mukherjee S, Ray D,
Ahsan MK, Gherghiceanu M, Popescu LM and Das DK: Cardioprotection
by resveratrol: A novel mechanism via autophagy involving the
mTORC2 pathway. Cardiovasc Res. 86:103–112. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ma XQ, Fu RF, Feng GQ, Wang ZJ, Ma SG and
Weng SA: Hypoxia-reoxygenation-induced apoptosis in cultured
neonatal rat cardiomyocyets and the protective effect of
prostaglandin E. Clin Exp Pharmacol Physiol. 32:1124–1130. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Addabbo F, Montagnani M and Goligorsky MS:
Mitochondria and reactive oxygen species. Hypertension. 53:885–892.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu XR, Li T, Cao L, Yu YY, Chen LL, Fan
XH, Yang BB and Tan XQ: Dexmedetomidine attenuates H2O2-induced
neonatal rat cardiomyocytes apoptosis through mitochondria- and
ER-medicated oxidative stress pathways. Mol Med Rep. 17:7258–7264.
2018.PubMed/NCBI
|
|
19
|
Tatlidede E, Sehirli O, Velioğlu-Oğünc A,
Cetinel S, Yeğen BC, Yarat A, Süleymanoğlu S and Sener G:
Resveratrol treatment protects against doxorubicin-induced
cardiotoxicity by alleviating oxidative damage. Free Radic Res.
43:195–205. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bargiela D, Burr SP and Chinnery PF:
Mitochondria and hypoxia: Metabolic crosstalk in cell-fate
decisions. Trends Endocrinol Metab. 29:249–259. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li F, You Y and Zhu H: 15-HETE protects
pulmonary artery smooth muscle cells against apoptosis via SIRT1
regulation during hypoxia. Biomed Pharmacother. 108:325–330. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu XR, Cao L, Li T, Chen LL, Yu YY, Huang
WJ, Liu L and Tan XQ: Propofol attenuates
H2O2-induced oxidative stress and apoptosis
via the mitochondria- and ER-medicated pathways in neonatal rat
cardiomyocytes. Apoptosis. 22:639–646. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Burgoyne JR, Mongue-Din H, Eaton P and
Shah AM: Redox signaling in cardiac physiology and pathology. Circ
Res. 111:1091–1106. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rajendran P, Nandakumar N, Rengarajan T,
Palaniswami R, Gnanadhas EN, Lakshminarasaiah U, Gopas J and
Nishigaki I: Antioxidants and human diseases. Clin Chim Acta.
436:332–347. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang L, Gao M, Chen J, Yang Z, Sun J, Wang
Z, Huang X, Yuan T, Shen X and Xian S: Resveratrol ameliorates
pressure overload-induced cardiac dysfunction and attenuates
autophagy in rats. J Cardiovasc Pharmacol. 66:376–382. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dong Q, Wu Z, Li X, Yan J, Zhao L, Yang C,
Lu J, Deng J and Chen M: Resveratrol ameliorates cardiac
dysfunction induced by pressure overload in rats via structural
protection and modulation of Ca(2+) cycling proteins. J Transl Med.
12:3232014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou L and Chang DC: Dynamics and
structure of the Bax-Bak complex responsible for releasing
mitochondrial proteins during apoptosis. J Cell Sci. 121:2186–2196.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Guo R, Liu W, Liu B, Zhang B, Li W and Xu
Y: SIRT1 suppresses cardiomyocyte apoptosis in diabetic
cardiomyopathy: An insight into endoplasmic reticulum stress
response mechanism. Int J Cardiol. 191:36–45. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen CJ, Yu W, Fu YC, Wang X, Li JL and
Wang W: Resveratrol protects cardiomyocytes from hypoxia-induced
apoptosis through the SIRT1-FoxO1 pathway. Biochem Biophys Res
Commun. 378:389–393. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sin TK, Tam BT, Yung BY, Yip SP, Chan LW,
Wong CS, Ying M, Rudd JA and Siu PM: Resveratrol protects against
doxorubicin-induced cardiotoxicity in aged hearts through the
SIRT1-USP7 axis. J Physiol. 593:1887–1899. 2015. View Article : Google Scholar : PubMed/NCBI
|