Introduction
Congenital talipes equinovarus (CTEV), also known as
clubfoot, is one of the most common inborn musculoskeletal
abnormalities, with a worldwide incidence of 1 in 1,000 livebirths
(1). CTEV is identified by four
clinical foot characteristics: Forefoot adduction, hindfoot varus,
midfoot cavus and hindfoot equinus (2).A total of ~80% of CTEV cases are
idiopathic, namely ICTEV (3), with
the remaining 20% being characterized as secondary or syndromic
CTEV (4). ICTEV affects males more
than females (5), with a
male-to-female ratio of 2:1 across different ethnic groups
(1,6).
Although a number of studies on familial
characteristics, especially twins, have strongly suggested that
genetic factors play an important role in the pathogenesis of ICTEV
(4,7–9), the
particular locality of the etiologyis still unclear compared with
syndromic CTEV (5).
Previous studies on the association between ICTEV
and genetic factors have identified several causative gene families
and genes, including Hox (10,11),
caspase (2,11), paired like homeodomain 1 (7–9,12),
T-box transcription factors and GLI family zinc finger 3 (10,13)
genes. However, a major candidate gene remains to be identified
(5). Previous studies have focused
on the interaction between genetic and environmental factors,
displaying the multifactorial identity of the disease (4,5,14,15).
At present, multifactorial identity remains the most validated
theory (5). Additionally, numerous
other phenotypes of syndromic clubfoot in newborns are not
recognizable and can be easily misdiagnosed as ICTEV (16,17).
Accordingly, these patients require the development of improved
molecular methods for accurate differential diagnosis.
In the present study, whole-exome sequencing (WES)
was used to investigate nine neonates/infants with CTEV, among
which seven displayed isolated CTEV and two displayed CTEV combined
with other abnormalities.
Materials and methods
Subjects
The present study was approved by the Ethics
Committee of Shijiazhuang Obstetrics and Gynecology Hospital
(approval no. 20180015) and written informed consent was obtained
from the parents of all patients. Patients with suspected CTEV,
identified by routine clinical and ultrasound diagnostic criteria
(2) without clear genetic
diagnosis, were recruited for the present study. Patients with
isolated (both bilateral and unilateral) and combined CTEV were
included in the present study. The exclusion criteria were as
follows: i) Clear prenatal genetic diagnosis; ii) mechanical injury
during labor. A total of nine neonates or infants with CTEV were
recruited from the Center of Prenatal Diagnosis in Shijiazhuang
Obstetrics and Gynecology Hospital and the Department of Pediatric
Orthopedic in The Third Hospital of Hebei Medical University
between January 2016 and December 2018. Patient characteristics are
shown in Table I. Peripheral blood
(5 ml) was collected from each subject for genetic testing.
 | Table I.Clinical data of the recruited
subjects. |
Table I.
Clinical data of the recruited
subjects.
| Subject | Sex | Sampling time (after
birth) | Main
manifestations | Family history |
|---|
| Case 1 | Male | 2 months | Bilateral CTEV; VSD
and patent ductus arteriosusat 4 month follow-up | G1P1, no family
history of CTEV |
| Case 2 | Female | 1 month | Bilateral CTEV | G2P1,
onemiscarriage at early pregnancy (without genetic testing), no
family history of CTEV |
| Case 3 | Female | 1 month | Bilateral CTEV | G1P1, no family
history of CTEV |
| Case 4 | Male | 5 days | Unilateral (left)
CTEV | G1P1, no family
history of CTEV |
| Case 5 | Female | 15 days | Unilateral (left)
CTEV | G2P2, one
3-year-old healthy son, no family history of CTEV |
| Case 6 | Male | 17 days | Unilateral (right)
CTEV; arthrogryposis at 3 month follow-up | G1P1, no family
history of CTEV |
| Case 7 | Male | 1 month | Bilateral CTEV | G2P2, one
4-year-old healthy daughter, no family history of CTEV |
| Case 8 | Female | 25 days | Unilateral (right)
CTEV | G1P1, no family
history of CTEV |
| Case 9 | Male | 1 month | Unilateral (right)
CTEV | G1P1, no family
history of CTEV |
Chromosome karyotyping
Conventional chromosome karyotyping by G-banding was
performed on the peripheral blood samples to detect overall
chromosomal anomalies, as previously described (18). The karyotype results were analyzed
in accordance with the International System for Human Cytogenomic
Nomenclature (2016 edition) (19).
DNA extraction
Total genomic DNA (1 µg) was extracted from 200 µl
peripheral blood using the DNA Blood Midi/Mini kit (Qiagen GmbH),
according to the manufacturer's protocol.
WES analysis
DNA library construction, quality testing and WES
experiments were performed as previously described (18); however, the ‘proband only’ analysis
strategy was adopted for the identification of causative variants.
WES analysis was performed using the Novaseq6000 platform
(Illumina). Sequencing reads were mapped to the human reference
genome (hg19/GRCh37) and underwent standard quality control
screening. The Verita Trekker® Variants Detection system
(Berry Genomics, Inc.) was used to identify single-nucleotide
polymorphisms, insertion and deletions, copy number variants
(CNVs), mitochondrial gene variants and runs of homozygosity.
Subsequently, the Enliven® Variants Annotation
Interpretation (Berry Genomics, Inc.) system was used to fulfill
the annotation and interpretation progress referring to multiple
databases. The overall workflow is presented in Fig. 1. After the variation filtering
process, several disease-associated variants were identified and
selected for familial validation using Sanger or CNV sequencing as
previously described (20).
 | Figure 1.Data analysis and variation
identification work flow. The green block represented the variation
calling process. SNP, single nucleotide polymorphism; INDEL,
insertion/deletion; CNV, copy number variation; Mt, mitochondrial
variation; ROH, runs of homozygosity; WES, whole-exome sequencing;
Red words represent patented software of Berry Genomics; QC,
quality control; ACMG, American College of Medical Genetics and
Genomics; HGMD, Human Gene Mutation Database; NLpearl™, Natural
Language Pearl processing and analysis system by Berry Genoimcs,
Inc. |
To identify the novel missense variation,
homological analysis among species was performed using the NCBI
blast online software (blast.ncbi.nlm.nih.gov/Blast.cgi; 2019/Oct/29). In
silico analysis was performed using Sorting Intolerant from
Tolerant (SIFT; sift.bii.a-star.edu.sg; 2019/Oct/29) and Polymorphism
Phenotyping (PolyPhen; version 2; genetics.bwh.harvard.edu/pph2) software to calculate
the respective pathogenicity indices. Biophysical analysis was
conducted using Modeller software (version 9.21; salilab.org/modeller).
Results
Clinical data
The main clinical data are presented in Table I. All the subjects were patients
with ICTEV with no family history of the disease (Table I). The parents had no adverse
habits, for example, smoking. Representative clinical and
ultrasonic illustrations are presented in Fig. 2.
Genetic analysis
The karyotyping results were normal for each of the
subjects. However, two potentially causative variations were
identified by WES and were further investigated by familial
validation. The two potential causative variations identified were:
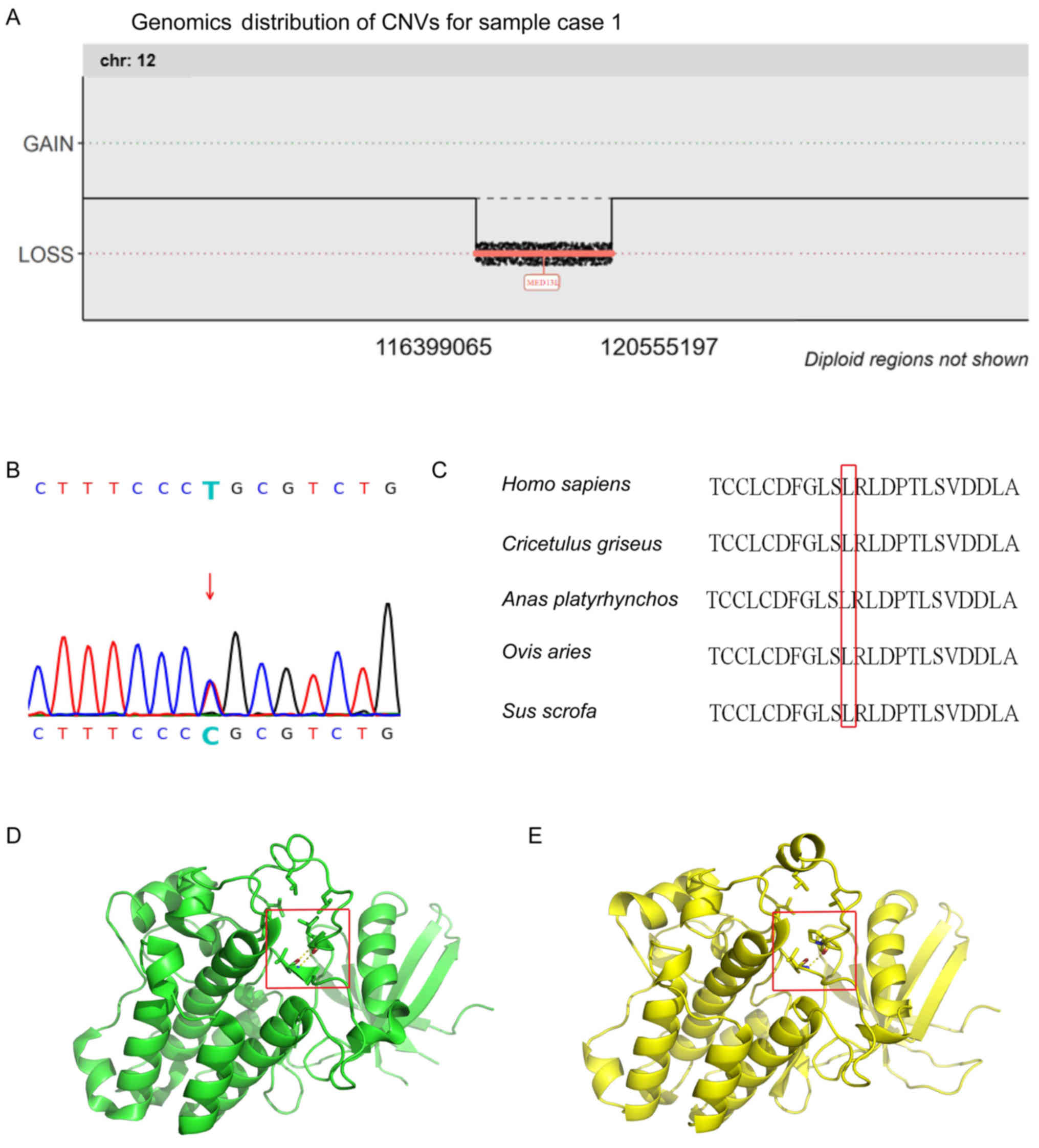
seq[GRCh37]del(12)(q24.21-q24.23)g.116399065-120555197 encompassing
the mediator complex subunit 13L (MED13L) gene in case 1 and
transforming growth factor-β receptor 2
(TGFBR2):NM_001024847.2:exon5:c.1280T>C:p.L427P in case 3
(Fig. 3A and B). Several suspected
variations were detected in other cases, but did not match the
inheritance pattern of the corresponding disease-causing genes
(data not shown).
It was indicated that the novel de novo
missense variation, TGFBR2: p.L427P, was highly conserved among
multiple species (Fig. 3C).
Furthermore, in silico analysis results obtained using the
SIFT (with the index of ‘0’) and PolyPhenV2 (with the index of
‘0.996’) softwares identified the variation as
‘deleterious/probably damaging’. Additionally, biophysical analysis
suggested that the helix in the kinase domain was damaged by the
missense variant, the hydrogen bond force was reduced and the
hydrophobic pocket was affected to some extent (Fig. 3D and E).
According to the 2019 American College of Medical
Genetics and Genomics (ACMG) & Clinical Genome Resource
guideline for CNV interpretation (21), the CNV detected,
seq[GRCh37]del(12)(q24.21-q24.23)g.116399065-120555197, was
interpreted as pathogenic with a score >1 (variant
classification, 2A). Similarly, in accordance with the 2015ACMG
guideline for sequence variant interpretation (22), the
TGFBR2:NM_001024847.2:exon5:c.1280T>C variant was deemed to be
likely pathogenic (variant classification evidence,
PS2+PM2+PP2+PP3).
Additionally, two pathogenic variations not
associated with CTEV were detected, which were a paternal
MYBPC3:NM_000256.3:exon6: c.769C>T: p.H257Y variation in case 4
and a maternal BRCA1:NM_007300.3:exon14:c.4497G>T: p.E1499D
variation in case 5 (data not shown).
Discussion
CTEV seriously affects the aesthetics and functions
of patients. The incidence and pathogenesis of the condition varies
among different ethnicities, suggesting that genetic factors play
an important role (23). ICTEV
only follows the Mendelian inheritance pattern in minorities,
including Polynesians (24), and
therefore further investigation into the multifactorial
pathogenesis of ICTEV in other ethnicities is required. Previous
studies identified certain potential pathogenic genes primarily by
single-nucleotide polymorphism typing combined with statistical
analysis (2,11,25).
Subsequently, further studies performed chromosomal microarray
analysis and other methods to detect clinically significant CNVs
containing important genes (7,26).
However, few attempts have been made to apply next generation
sequencing methods, including WES, to the identification of CTEV
genetic variations (16,27). A study conducted by Yang et
al (28) identified variants
in the filamin B gene, which is consistent with the findings of the
present study (17). In the
present study, the detection rate of variants was low for the
majority of ICTEV cases, indicating that the pathogenesis of ICTEV
in the Chinese population is relatively complicated and further
suggesting multifactorial inheritance. Therefore, to explore the
pathogenesis of ICTEV by detecting rare variants, an effective
method would be to increase the sample size and subsequently
perform gene ontology enrichment analysis (29).
The MED13L gene on chromosome 12q24 encodes a
subunit of the large mediator complex that functions with
DNA-binding transcription factors and RNA polymerase II for gene
activation or repression (30).
The gene is part of the evolutionarily conserved Thyroid Hormone
Receptor Associated Protein gene family, which encode proteins that
regulate embryonic development (31). Several studies have indicated that
heterozygous loss of function variations resulting in haploin
sufficiency could cause Mental Retardation and Distinctive Facial
Features with or without Cardiac Defects [MRFACD; Mendelian
Inheritance in Man (MIM), #616789] (32–35),
and clubfoot was a notable phenotype in these cases. In the present
study, it was hypothesized that the microdeletion of 12q in case 1
would result in MRFACD, particularly on the basis of the
aforementioned evidence. Therefore, the mental and cardiovascular
development of this infant should be closely monitored.
The TGFBR2(MIM, *190182) gene on chromosome 3p24
belongs to the serine-threonine kinase family (36). The activities of TGF-β1 (TGFB1;
MIM,*190180) in regulating cell proliferation, differentiation and
extracellular matrix production are mediated via these receptors.
In the present study, the variant in case 3 was located in the
protein kinase domain of TGFBR2. Previous studies have reported
that missense variants occurring in this domain can cause
Loeys-Dietz syndrome type II (37,38).
Therefore, based on the results of genetic analysis, the child
incase3 should receive long-term follow-up in order to determine
whether they display other phenotypes, as well as to provide
further counseling and guidance for the subsequent pregnancies
within the family. Moreover, although biophysical analysis
predicted how the variant may alter the structure of TGFBR2,
further studies are required to determine how the interaction
between TGFBR2 and its receptor maybe affected.
According to the present study, the use of WES as a
first-line method for the detection of ICTEV in children may not be
efficient. However, WES provided ample sensitivity for the
detection of rare syndromic CTEVs and consequently may be
beneficial for the accurate differential diagnosis. The main
limitations of the present study were the small sample size and the
heterogeneity of CTEV. To validate the results of the present
study, studies including larger and more well defined patient
cohorts without congenital co-morbidities are required.
Additionally, reporting the detection of pathogenic variants
unrelated to proposed phenotypes presents an ethical challenge,
which must be well communicated with patients and clearly reflected
in informed consent.
In conclusion, the present study suggested that WES
may serve as a comprehensive method for the detection of rare
variants in coding sequences. In particular, WES displayed the
advantage of detecting syndromic CTEV, which has a phenotype that
is not easy to determine.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Project of
Introduced Foreign Students Foundation of Hebei Province (grant no.
C201606055).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JZ and YL designed the current study. JZ and SL
analyzed the data and drafted the manuscript. SM and YLiu recruited
the case studies and performed the experiments. XW performed the
experiments. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Shijiazhuang Obstetrics and Gynecology Hospital
(approval no. 20180015) and written informed consent was obtained
from the parents of all patients.
Patient consent for publication
This study has followed the principles of anonymity;
no direct or indirect identifiers of our participants were used for
publication. Written informed consent was obtained for
publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wynne-Davies R: Genetic and environmental
factors in the etiology of talipes equinovarus. Clin Orthop Relat
Res. 84:9–13. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ester AR, Tyerman G, Wise CA, Blanton SH
and Hecht JT: Apoptotic gene analysis in idiopathic talipes
equinovarus (clubfoot). Clin Orthop Relat Res. 462:32–37. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wynne-Davies R: Family studies and the
cause of congenital club foot. Talipes Equinovarus, Talipes
Calcaneo-Valgus and Metatarsus Varus. J Bone Joint Surg Br.
46:445–463. 1964. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pavone V, Chisari E, Vescio A, Lucenti L,
Sessa G and Testas G: The etiology of idiopathic congenital talipes
equinovarus: A systematic review. J Orthop Surg Res. 13:2062018.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Basit S and Khoshhal KI: Genetics of
clubfoot; recent progress and future perspectives. Eur J Med Genet.
61:107–113. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ching GH, Chung CS and Nemechek RW:
Genetic and epidemiological studies of clubfoot in Hawaii:
Ascertainment and incidence. Am J Hum Genet. 21:566–580.
1969.PubMed/NCBI
|
|
7
|
Alvarado DM, Buchan JG, Frick SL,
Herzenberg JE, Dobbs MB and Gurnett CA: Copy number analysis of 413
isolated talipes equinovarus patients suggests role for
transcriptional regulators of early limb development. Eur J Hum
Genet. 21:373–380. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Alvarado DM, Aferol H, McCall K, Huang JB,
Techy M, Buchan J, Cady J, Gonzales PR, Dobbs MB and Gurnett CA:
Familial isolated clubfoot is associated with recurrent chromosome
17q23.1q23.2 microduplications containing TBX4. Am J Hum Genet.
87:154–160. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Alvarado DM, McCall K, Aferol H, Silva MJ,
Garbow JR, Spees WM, Patel T, Siegel M, Dobbs MB and Gurnett CA:
Pitx1 haploinsufficiency causes clubfoot in humans and a
clubfoot-like phenotype in mice. Hum Mol Genet. 20:3943–3952. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cao D, Jin C, Ren M, Lin C, Zhang X and
Zhao N: The expression of Gli3, regulated by HOXD13, may play a
role in idiopathic congenital talipes equinovarus. BMC
Musculoskelet Disord. 10:1422009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ester AR, Weymouth KS, Burt A, Wise CA,
Scott A, Gurnett CA, Dobbs MB, Blanton SH and Hecht JT: Altered
transmission of HOX and apoptotic SNPs identify a potential common
pathway for clubfoot. Am J Med Genet A. 149A:2745–2752. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gurnett CA, Alaee F, Kruse LM, Desruisseau
DM, Hecht JT, Wise CA, Bowcock AM and Dobbs MB: Asymmetric
lower-limb malformations in individuals with homeobox PITX1 gene
mutation. Am J Hum Genet. 83:616–622. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Y, Sun Y, Huang Y, Pan Y, Shi B, Ma
J, Ma L, Lan F, Zhou Y, Shi J, et al: The association study of
nonsyndromic cleft lip with or without cleft palate identified risk
variants of the GLI3 gene in a Chinese population. J Genet.
96:687–693. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dickinson KC, Meyer RE and Kotch J:
Maternal smoking and the risk for clubfoot in infants. Birth
Defects Res A Clin Mol Teratol. 82:86–91. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Parker SE, Mai CT, Strickland MJ, Olney
RS, Rickard R, Marengo L, Wang Y, Hashmi SS and Meyer RE; National
Birth Defects Prevention Network, : Multistate study of the
epidemiology of clubfoot. Birth Defects Res A Clin Mol Teratol.
85:897–904. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang Z, Kong Z, Zhu M, Lu W, Ni L, Bai Y
and Lou Y: Whole genome sequencing identifies ANXA3 and MTHFR
mutations in a large family with an unknown equinus deformity
associated genetic disorder. Mol Biol Rep. 43:1147–1155. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang K, Shen M, Yan Y, Tan Y, Zhang J, Wu
J, Yang G, Li S, Wang J, Ren Z, et al: Genetic analysis in fetal
skeletal dysplasias by trio whole-exome sequencing. BioMed Res Int.
2019:24925902019.PubMed/NCBI
|
|
18
|
Arsham MS, Barch MJ and Lawce HJ: The AGT
Cytogenetics Laboratory Manual. John Wiley & Sons Inc.
(Hoboken, NJ). 2017. View Article : Google Scholar
|
|
19
|
McGowan-Jordan J, Simons A and Schmid M:
An International System for Human Cytogenomic Nomenclature. Karger.
(Basel, Switzerland). 2016.
|
|
20
|
Chen Y, Bartanus J, Liang D, Zhu H, Breman
AM, Smith JL, Wang H, Ren Z, Patel A, Stankiewicz P, et al:
Characterization of chromosomal abnormalities in pregnancy losses
reveals critical genes and loci for human early development. Hum
Mutat. 38:669–677. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Riggs ER, Andersen EF, Cherry AM, Kantarci
S, Kearney H, Patel A, Raca G, Ritter DI, South ST, Thorland EC, et
al: Technical standards for the interpretation and reporting of
constitutional copy-number variants: a joint consensus
recommendation of the American College of Medical Genetics and
Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet
Med. 22:245–257. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al: ACMG
Laboratory Quality Assurance Committee: Standards and guidelines
for the interpretation of sequence variants: A joint consensus
recommendation of the American College of Medical Genetics and
Genomics and the Association for Molecular Pathology. Genet Med.
17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang JH, Palmer RM and Chung CS: The role
of major gene in clubfoot. Am J Hum Genet. 42:772–776.
1988.PubMed/NCBI
|
|
24
|
Chapman C, Stott NS, Port RV and Nicol RO:
Genetics of club foot in Maori and Pacific people. J Med Genet.
37:680–683. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhao XL, Wang YJ, Wu YL and Han WH: Role
of COL9A1 genetic polymorphisms in development of congenital
talipes equinovarus in a Chinese population. Genet Mol Res. Nov
3–2016.(Epub ahead of print). doi: 10.4238/gmr15048773. View Article : Google Scholar :
|
|
26
|
Lu W, Bacino CA, Richards BS, Alvarez C,
VanderMeer JE, Vella M, Ahituv N, Sikka N, Dietz FR, Blanton SH, et
al: Studies of TBX4 and chromosome 17q23.1q23.2: An uncommon cause
of nonsyndromic clubfoot. Am J Med Genet A. 158A:1620–1627. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Alvarado DM, Buchan JG, Gurnett CA and
Dobbs MB: Exome sequencing identifies an MYH3 mutation in a family
with distal arthrogryposis type 1. J Bone Joint Surg Am.
93:1045–1050. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang H, Zheng Z, Cai H, Li H, Ye X, Zhang
X, Wang Z and Fu Q: Three novel missense mutations in the filamin B
gene are associated with isolated congenital talipes equinovarus.
Hum Genet. 135:1181–1189. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kuleshov MV, Jones MR, Rouillard AD,
Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM,
Lachmann A, et al: Enrichr: A comprehensive gene set enrichment
analysis web server 2016 update. Nucleic Acids Res. 44:W90–W97.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Utami KH, Winata CL, Hillmer AM, Aksoy I,
Long HT, Liany H, Chew EG, Mathavan S, Tay SK, Korzh V, et al:
Impaired development of neural-crest cell-derived organs and
intellectual disability caused by MED13L haploinsufficiency. Hum
Mutat. 35:1311–1320. 2014.PubMed/NCBI
|
|
31
|
Muncke N, Jung C, Rüdiger H, Ulmer H,
Roeth R, Hubert A, Goldmuntz E, Driscoll D, Goodship J, Schön K, et
al: Missense mutations and gene interruption in PROSIT240, a novel
TRAP240-like gene, in patients with congenital heart defect
(transposition of the great arteries). Circulation. 108:2843–2850.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Asadollahi R, Oneda B, Sheth F,
Azzarello-Burri S, Baldinger R, Joset P, Latal B, Knirsch W, Desai
S, Baumer A, et al: Dosage changes of MED13L further delineate its
role in congenital heart defects and intellectual disability. Eur J
Hum Genet. 21:1100–1104. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hamdan FF, Srour M, Capo-Chichi JM, Daoud
H, Nassif C, Patry L, Massicotte C, Ambalavanan A, Spiegelman D,
Diallo O, et al: De novo mutations in moderate or severe
intellectual disability. PLoS Genet. 10:e10047722014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
van Haelst MM, Monroe GR, Duran K, van
Binsbergen E, Breur JM, Giltay JC and van Haaften G: Further
confirmation of the MED13L haploinsufficiency syndrome. Eur J Hum
Genet. 23:135–138. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Adegbola A, Musante L, Callewaert B,
Maciel P, Hu H, Isidor B, Picker-Minh S, Le Caignec C, Delle Chiaie
B, Vanakker O, et al: Redefining the MED13L syndrome. Eur J Hum
Genet. 23:1308–1317. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lin HY, Wang XF, Ng-Eaton E, Weinberg RA
and Lodish HF: Expression cloning of the TGF-β type II receptor, a
functional transmembrane serine/threonine kinase. Cell. 68:775–785.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mizuguchi T, Collod-Beroud G, Akiyama T,
Abifadel M, Harada N, Morisaki T, Allard D, Varret M, Claustres M,
Morisaki H, et al: Heterozygous TGFBR2 mutations in Marfan
syndrome. Nat Genet. 36:855–860. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Loeys BL, Chen J, Neptune ER, Judge DP,
Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, et
al: A syndrome of altered cardiovascular, craniofacial,
neurocognitive and skeletal development caused by mutations in
TGFBR1 or TGFBR2. Nat Genet. 37:275–281. 2005. View Article : Google Scholar : PubMed/NCBI
|