Introduction
Prompt reperfusion is essential for recovery
following acute myocardial infarction; however, reperfusion can
also lead to ischemia/reperfusion (I/R) injury (1). Although myocardial I/R injury
involves complex pathophysiological mechanisms that have not yet
been fully elucidated, oxidative stress and intracellular
Ca2+ overload are considered to be two of the main
mechanisms contributing to myocardial I/R injury (2–4).
Increases in reactive oxygen species (ROS) and intracellular
Ca2+ levels are described as mutually causative, forming
a vicious cycle (5). Intracellular
Ca2+ homeostasis and ROS production are closely related
to the endoplasmic reticulum (ER) and mitochondria. When faced with
oxidative stress, ischemia, hypoxia, Ca2+ imbalance and
other conditions, unfolded proteins accumulate in the (ER), and
upon exceeding its capacity to deal with unfolded proteins, ER
homeostasis is lost and the ER stress (ERS) response is activated
(6). Accumulating studies have
reported that ERS serves an important role in myocardial I/R injury
(7,8).
The ER is associated with the mitochondria at
multiple levels through mitochondrial-associated membranes (MAMs),
which are specific protein-rich regions of the ER located in close
proximity to the mitochondria. MAMs regulate several functions,
including the synthesis and transport of phospholipids,
Ca2+ transfer between organelles and cell signaling
pathways (9–11). ER oxidase 1 (ERO1) is a
glycosylated flavonase located at MAMs, of which there are two
mammalian isoforms; the ERO1α isotype, which is found distributed
throughout the body, and the ERO1β isotype, which is most abundant
in pancreatic β cells and lymphocytes (12,13).
Both isoforms respond to ERS, but only ERO1α is induced by hypoxia
(14,15). ERO1α serves an important role in
catalyzing the formation of protein disulfide bonds, ER redox and
Ca2+ homeostasis (16),
and the ERO1α-dependent ER-mitochondrial calcium flux has been
observed to contribute to ERS (17). However, the exact role of ERO1α in
myocardial I/R injury remains unclear.
In the present study, myocardial I/R injury was
simulated using myocardial hypoxia/reoxygenation (H/R). The effects
of ERO1α on myocardial H/R were observed by genetically knocking
down the expression of ERO1α with short hairpin RNA (shRNA), and
treatment with the ERS activator, TM or the ERS inhibitor,
4-Phenylbutyric acid (4-PBA).
Materials and methods
Lentiviral cell transfection
Using the ERO1α gene mRNA sequence (GenBank
accession no. NM_138528.1), three shRNA candidate target sequences
(1832, 1833 and 1834) were designed and synthesized by Shanghai
GeneChem Co., Ltd. The above target sequences were cloned into the
lentiviral vector pMAGic4.1 and scrambled shRNA was cloned into the
pMAGic4.1 vector as the negative control. H9C2 cells (American Type
Culture Collection) were plated in 6-well plates at a density of
1×106 cells/well, and upon reaching 70% confluence, the
cells were transfected with either recombinant lentivirus
ERO1α-shRNA (Table I) or scrambled
shRNA with titers of 5×106 TU/ml. Following transfection
for 48 h, the efficiency of ERO1α silencing was assessed using
reverse transcription-quantitative PCR and western blotting. The
shRNA with the best silencing effect was selected for subsequent
experiments.
 | Table I.Short hairpin RNA primer
sequences. |
Table I.
Short hairpin RNA primer
sequences.
| ID | Primer sequence
(5′→3′) |
|---|
| 1832 | F:
ccggGACCATCGATAAGTTTAATAActcgagATTAAACTTATCGATGGTCTCttttttg |
|
| R:
aattcaaaaaaGAGACCATCGATAAGTTTAATctcgagTTATTAAACTTATCGATGGTC |
| 1833 | F:
ccggGAGCATTCTACAGGCTTATATctcgagATAAGCCTGTAGAATGCTCTCttttttg |
|
| R:
aattcaaaaaaGAGAGCATTCTACAGGCTTATctcgagATATAAGCCTGTAGAATGCTC |
| 1834 | F:
ccggGTGGACGAAACACGATGATTCctcgagATCATCGTGTTTCGTCCACTGttttttg |
|
| R:
aattcaaaaaaCAGTGGACGAAACACGATGATctcgagGAATCATCGTGTTTCGTCCAC |
Exposure of H9C2 cardiomyocytes to H/R
and treatments
H9C2 cardiomyocytes were purchased from the American
Type Culture Collection and the H/R model was established using the
AnaeroPack® method (18). Briefly, a hypoxic atmosphere was
created by incubating an AnaeroPack® (Mitsubishi Gas
Chemical Company, Inc.), which absorbs oxygen, and H9C2
cardiomyocytes in a sealed airtight container together at 37°C.
Following incubation for 3 h, the AnaeroPack® container
was opened to terminate the hypoxic conditions. The cells in the
culture plates were removed and subsequently placed in a
CO2 incubator at 37°C for 6 h. The morphology was
observed under an inverted light microscope at ×200 magnification.
In the 4-PBA + H/R group, H9C2 cardiomyocytes were treated with 0.5
mM 4-PBA (Sigma-Aldrich; Merck KGaA), a selective ERS inhibitor, 2
h prior to H/R induction. In the TM + ERO1α-shRNA + H/R,
cardiomyocytes were pretreated with 2 µg/l TM (Cayman Chemical), an
ERS activator, 24 h prior to H/R exposure to counteract the
reduction in ERS following ERO1α knockdown.
Morphological analysis following
Hoechst 33258 staining
A total of 1×105 H9C2 cardiomyocytes/well
were incubated in 24-well plates. Cells were fixed by 4%
paraformaldehyde at 4°C for 1 h, washed twice with PBS and stained
with Hoechst 33258 (Beyotime Institute of Biotechnology) at room
temperature for 5 min. Hoechst 33258 staining solution was
subsequently aspirated and washed twice with PBS for 5 min each.
Stained cell nuclei were visualized using an IX70 fluorescence
microscope (Olympus Corporation) (magnification, ×200). A total of
five fields were randomly selected from each well, and the
proportion of apoptotic cells was calculated as the ratio of
nuclear pyknosis cells to the total cells.
Flow cytometric analysis of
apoptosis
The Annexin V-FITC Apoptosis Assay kit (Beyotime
Institute of Biotechnology) was used to detect cell apoptosis.
Briefly, following treatment, cells were transferred to individual
tubes and a solution containing 195 µl binding buffer, 5 µl Annexin
V-FITC and 10 µl propidium iodide was added to each test tube. The
tubes were subsequently mixed and incubated at room temperature in
the dark for 15 min. Apoptotic cells were detected using a FACSScan
flow cytometer (FACSVerse; BD Biosciences). The data analysis was
conducted using CellQuest software version 3.3 (BD
Biosciences).
Detection of caspase-3 activity
Caspase-3 activity was measured using a caspase-3
activity kit (Beyotime Institute of Biotechnology). Briefly, cells
were suspended in lysis buffer on ice for 15 min and subsequently
centrifuged at 16,000 × g for 10 min. The supernatants were then
incubated with 20 ng Ac-DEVD-pNA in a 96-well plate for 2 h at 37°C
(19). The absorbance of pNA was
measured at 405 nm using an Infinite® 200 PRO microplate
reader (Tecan Group, Ltd.).
Measurement of intracellular
Ca2+ levels
Cells were washed twice with PBS buffer and then
incubated with Fluo-3/AM working fluid for 30 min in the dark at
37°C. The fluorescence intensity was determined using a confocal
laser scanning microscope (TCS SP5; Leica Microsystems GmbH), with
an excitation wavelength of 488 nm and an emission wavelength of
525 nm (magnification, ×400). A total of five fields were randomly
selected from each dish. Semi-quantitative analysis was conducted
using ImageJ 1.49 software (National Institutes of Health).
Measurement of intracellular ROS
levels
Intracellular ROS levels were detected by ROS
fluorescent probe dichlorodihydrofluorescein diacetate (DCFH-DA).
Briefly, 1×106 H9C2 cardiomyocytes/ml were incubated
with 10 µM DCFH-DA at 37°C for 30 min and then washed three times
with PBS buffer. The fluorescent intensity was detected with an
excitation wavelength of 488 nm and an emission wavelength of 525
nm using an Infinite® 200 PRO microplate reader (Tecan
Group, Ltd.). The results represent the percentage variation
relative to the untreated control.
Extraction of the cytoplasmic and
mitochondrial components
The cytoplasmic and mitochondrial proteins were
extracted using a cell mitochondria isolation kit (Beyotime
Institute of Biotechnology). Briefly, cells were incubated in lysis
buffer at 4°C and centrifuged at 3,000 × g for 10 min.
Subsequently, the supernatants were centrifuged for a second time
at 4°C for 10 min at 13,000 × g. The cytoplasmic components were
present in the supernatant, whereas the cell pellet contained the
mitochondrial components.
Measurement of mitochondrial membrane
potential (Δψm)
A total of 100 µl (1 mg/ml) purified mitochondria
was added to 900 µl JC-1 staining solution and subsequently
incubated at 37°C for 30 min. To detect the JC-1 monomer, the
excitation and emission wavelength was set to 490 and 530 nm,
respectively. To detect the JC-1 polymer, the excitation and
emission wavelength was set to 525 and 590 nm, respectively
(20). The fluorescent intensity
was determined using an Infinite® 200 PRO microplate
reader (Tecan Group, Ltd.).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted using TRIzol®
reagent (Takara Bio, Inc.). Total RNA was reverse transcribed into
cDNA using the PrimeScript RT reagent kit (Takara Bio, Inc.). qPCR
was subsequently performed using the SYBR® Premix Ex
Taq™ kit (Takara Bio, Inc.), according to the manufacturer's
protocol, and a qTower2.2 quantitative PCR instrument (21). Primer sequences of the genes used
for the qPCR are presented in Table
II. Relative expression of genes was calculated using the
comparative cycle threshold (Ct) (2−ΔΔCt) method with
18s RNA as the internal control (22).
| Table II.Primer sequences used for reverse
transcription-quantitative PCR. |
Table II.
Primer sequences used for reverse
transcription-quantitative PCR.
| Gene | Primer sequence
(5′→3′) | PCR conditions |
|---|
| ERO1α | F:
TGTGCTGTCAAACCCTGCCA | Denaturation, 95°C,
30 sec; annealing, 57°C, 20 sec; extension, |
|
| R:
CAGCCTGCTCACACTCCTCA | 72°C, 1 min; 35
cycles |
| GRP78 | F:
AAGGAAACTGCCGAGGCGTA | Denaturation: 95°C,
30 sec; annealing, 56°C, 20 sec; extension, |
|
| R:
AAGGAAACTGCCGAGGCGTA | 72°C, 1 min; 35
cycles |
| CHOP | F:
TCCTGAGTGGCGGACTGTTC | Denaturation, 95°C,
30 sec; annealing, 57°C, 20 sec; extension, |
|
| R:
GGCAGAGACTCAGCTGCCAT | 72°C, 1 min; 35
cycles |
| Cytochrome c | F:
TGGTCTGTTTGGGCGGAAGA | Denaturation, 95°C,
30 sec; annealing, 57°C, 20 sec; extension, |
|
| R:
TGGTCTGTTTGGGCGGAAGA | 72°C, 1 min; 35
cycles |
| Caspase-12 | F:
TGGAGAAGGAAGGCCGAACC | Denaturation, 95°C,
30 sec; annealing, 57°C, 20 sec; extension, |
|
| R:
TGGACGGCCAGCAAACTTCA | 72°C, 1 min; 35
cycles |
| 18s | F:
CGGCTACCACATCCAAGGAA | Denaturation, 95°C,
30 sec; annealing, 61.5°C, 20 sec; extension, |
|
| R:
GCTGGAATTACCGCGGCT | 72°C, 1 min; 35
cycles |
Western blotting
Total cell proteins were extracted using RIPA Lysis
Buffer (Beyotime Institute of Biotechnology). Mitochondria proteins
were extracted using the Cell Mitochondria Isolation kit (Beyotime
Institute of Biotechnology) according to the manufacturer's
protocol. Protein concentration was determined via BCA protein
assay kit (Beyotime Institute of Biotechnology). A total of 50 µg
of extracted protein were electroblotted onto a PVDF membrane
following separation on a 10% SDS-PAGE. The membranes were blocked
with 5% non-fat milk for 1 h at room temperature prior to being
incubated with the following primary antibodies overnight at 4°C:
Anti-ERO1α (1:1,000; cat. no. sc-365526) purchased from Santa Cruz
Technology, and anti-78 kDa glucose-regulated protein (GRP78;
1:2,000; cat. no. ab108615), anti-C/EBP homologous protein (CHOP;
1:2,000; cat. no. ab179823), anti-caspase-12 (1:2,000; cat. no.
ab62484), anti-cytochrome c (1:2,000; cat. no. ab133504)
anti-cytochrome c oxidase subunit IV (COX IV; 1:2,000; cat. no.
ab153709) and anti-GAPDH (1:5,000; cat. no. ab181602) purchased
from Abcam. Following the primary antibody incubation, membranes
were washed three times with wash buffer and incubated with
horseradish peroxidase-conjugated IgG secondary antibodies
(1:2,000; cat. nos. A0208 and A0216, respectively; Beyotime
Institute of Biotechnology) for 2 h at room temperature. Protein
bands were visualized using an enhanced chemiluminescence system
(Beyotime Institute of Biotechnology), with GAPDH as the loading
control (COX IV was used as a mitochondrial loading control).
Semi-quantitative analysis was conducted using ImageJ 1.49 software
(National Institutes of Health).
Statistical analysis
Statistical analysis was performed using SPSS
version 19.0 software (IBM Corp.) and data are presented as the
mean ± SD. To determine statistical differences amongst >2
groups, one-way ANOVA was used, followed by Tukey's post hoc test
for multiple group comparisons. P<0.05 was considered to
indicate a statistically significant difference.
Results
Expression levels of ERO1α protein
increase following H/R induction
H9C2 cardiomyocytes were exposed to hypoxia for 3 h
and reoxygenation for 1, 3, 6 and 12 h, and ERO1α protein
expression levels were subsequently assessed using western
blotting. In the control (CON) group, H9C2 cardiomyocytes did not
undergo hypoxia and reoxygenation. The cells in the CON group grew
well and most of them were fusiform. Compared with the CON group,
hypoxia-reoxygenation caused some cells to appear irregular or
round in shape, and floating cells and cell debris increased
significantly (Fig. 1A). ERO1α
protein expression levels significantly increased following H/R
induction compared with the CON group (Fig. 1B); and ERO1α expression levels
reached their highest in H9C2 cardiomyocytes following 3 h of
hypoxia and 6 h of reoxygenation. In addition, ERS marker GRP78
protein expression levels peaked following 3 h of reoxygenation.
Therefore, subsequent experiments in the present study were
performed in cells following 3 h of hypoxia and 6 h of
reoxygenation.
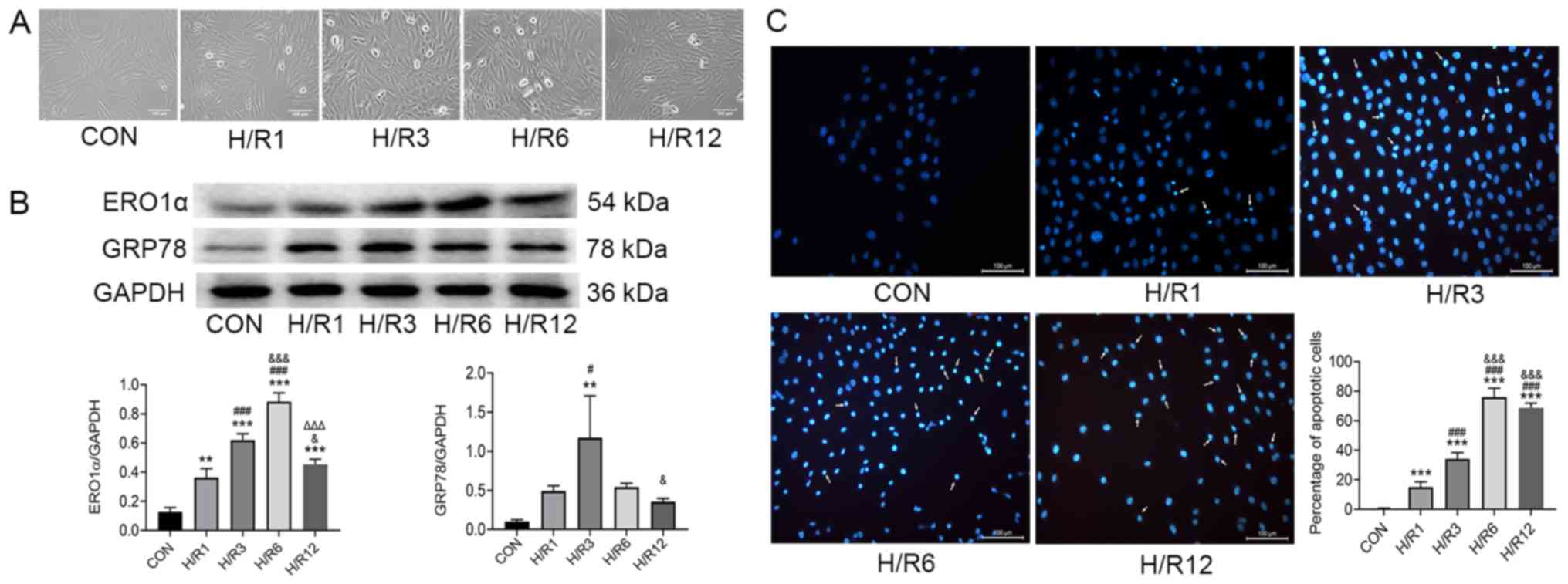 | Figure 1.H9C2 cardiomyocytes exposed to
hypoxia for 3 h and subsequently 1, 3, 6 and 12 h of reoxygenation.
(A) Optical images of H9C2 cardiomyocytes. (B) Protein expression
levels of ERO1α and GRP78 as determined by western blot analysis.
(C) Apoptosis detected by Hoechst 33258 staining. Scale bar, 100
µm. Data are presented as the mean ± SD from three independent
experiments. **P<0.01, ***P<0.001 vs. CON group;
#P<0.05, ###P<0.001 vs. H/R1 group;
&P<0.05, &&&P<0.001 vs.
H/R3 group; ΔΔΔP<0.001 vs. H/R6 group. ERO1α,
endoplasmic reticulum oxidase 1α; GRP78, 78 kDa glucose-regulated
protein; CON, control; H/R, hypoxia/reoxygenation. |
Consistent with ERO1α expression, H/R-stimulated
H9C2 cardiomyocytes demonstrated increased the proportion of
apoptotic cells. The nuclei of H9C2 cardiomyocytes in the CON group
were round and homogeneously stained, whereas those in the H/R
group were observed to have significant apoptotic characteristics,
such as cell shrinkage and chromatin condensation (Fig. 1C). Counting the cells with
apoptotic characteristics found that the percentage of apoptotic
cells in the H/R group was significantly increased compared with
the CON group.
Transfection with specific
ERO1α-shRNAs decreases ERO1α expression levels in H9C2
cardiomyocytes
The role of ERO1α following H/R was chosen for
further investigation because previous studies had reported that
ERS served an important role in myocardial I/R injuries (23,24).
ERO1α-shRNA carrier vectors were observed to contain the expected
sequence (Table SI), indicating
their successful construction. Following transfection of H9C2
cardiomyocytes with three different ERO1α-shRNAs (1832, 1833 and
1834), the mRNA expression levels of ERO1α were significantly
decreased in the H9C2 cardiomyocytes (Fig. 2A). Although all three lentiviral
shRNAs significantly inhibited ERO1α protein expression (Fig. 2B), the knockdown efficiency of
shRNA 1832 was more effective compared with shRNA 1833 and shRNA
1834. Therefore, the lentivirus shRNA 1832 was selected for use in
subsequent experiments.
ERO1α knockdown decreases H/R-induced
ERS
The mRNA and protein expression levels of
ERS-related molecules, including ERO1α, GRP78 and CHOP were
detected using RT-qPCR and western blotting. The expression levels
of ERO1α, GRP78 and CHOP mRNA and protein significantly increased
following H/R compared with the CON group, whereas 4-PBA inhibited
the expression levels of ERO1α, GRP78 and CHOP protein (Fig. 3A). Furthermore, ERO1α knockdown
reversed the H/R-induced increase in mRNA and protein expression
levels of GRP78 and CHOP (Fig. 3B and
C). Together, these data strongly suggested that ERS may serve
an important role in myocardial H/R injury, and that ERO1α may have
a crucial role in H/R-induced ERS in H9C2 cardiomyocytes.
 | Figure 3.ERO1α knockdown decreases H/R-induced
ER stress. (A) Protein expression levels of ERO1α, GRP78 and CHOP
in H9C2 cardiomyocytes following pretreatment with 4-PBA for 2 h
prior to H/R induction. (B) Protein expression levels of ERO1α,
GRP78 and CHOP in ERO1α-shRNA-transfected H9C2 cardiomyocytes. (C)
mRNA expression levels of ERO1α, GRP78 and CHOP in H9C2
cardiomyocytes following transfection with ERO1α-shRNA. Data are
presented as the mean ± SD from three independent experiments.
*P<0.05, **P<0.01, ***P<0.001 vs. CON group;
#P<0.05, ##P<0.01,
###P<0.001 vs. H/R group. ERO1α, endoplasmic
reticulum oxidase 1α; GRP78, 78 kDa glucose-regulated protein;
CHOP, C/EBP homologous protein; CON, control; H/R,
hypoxia/reoxygenation; shRNA, short hairpin RNA; ns, not
significant; 4-PBA, 4-Phenylbutyric acid. |
ERO1α knockdown inhibits H/R-induced
cell apoptosis
The percentage of apoptotic cells, caspase-3
activity and the expression levels of caspase-12 and
cleaved-caspase-12 (c-caspase-12) were investigated to determine
the role of ERO1α in H/R injury. H9C2 cardiomyocytes underwent
hypoxia for 3 h, then reoxygenation for 6 h, and the percentage of
apoptotic cells was evaluated using flow cytometry. The percentage
of apoptotic cells in the H/R group was significantly increased
compared with the CON group (Fig.
4A); however, the percentage of apoptotic cells of the
ERO1α-shRNA + H/R group was decreased compared with the H/R
group.
Apoptosis requires the activation of cysteine
protease, which both promotes and executes cell death (25). Excessive ERS was reported to induce
apoptotic signaling in I/R injury (26). Thus, the expression levels of
caspase-12 and c-caspase-12 proteins, and caspase-3 activity were
subsequently investigated (Fig. 4B and
C). The expression levels of c-caspase-12 in the H/R group were
significantly increased compared with the CON group; however, ERO1α
knockdown reduced c-caspase-12 expression compared with the H/R
group. These data suggested that ERO1α knockdown may inhibit
H/R-induced apoptosis in H9C2 cardiomyocytes.
ERO1α knockdown decreases H/R-induced
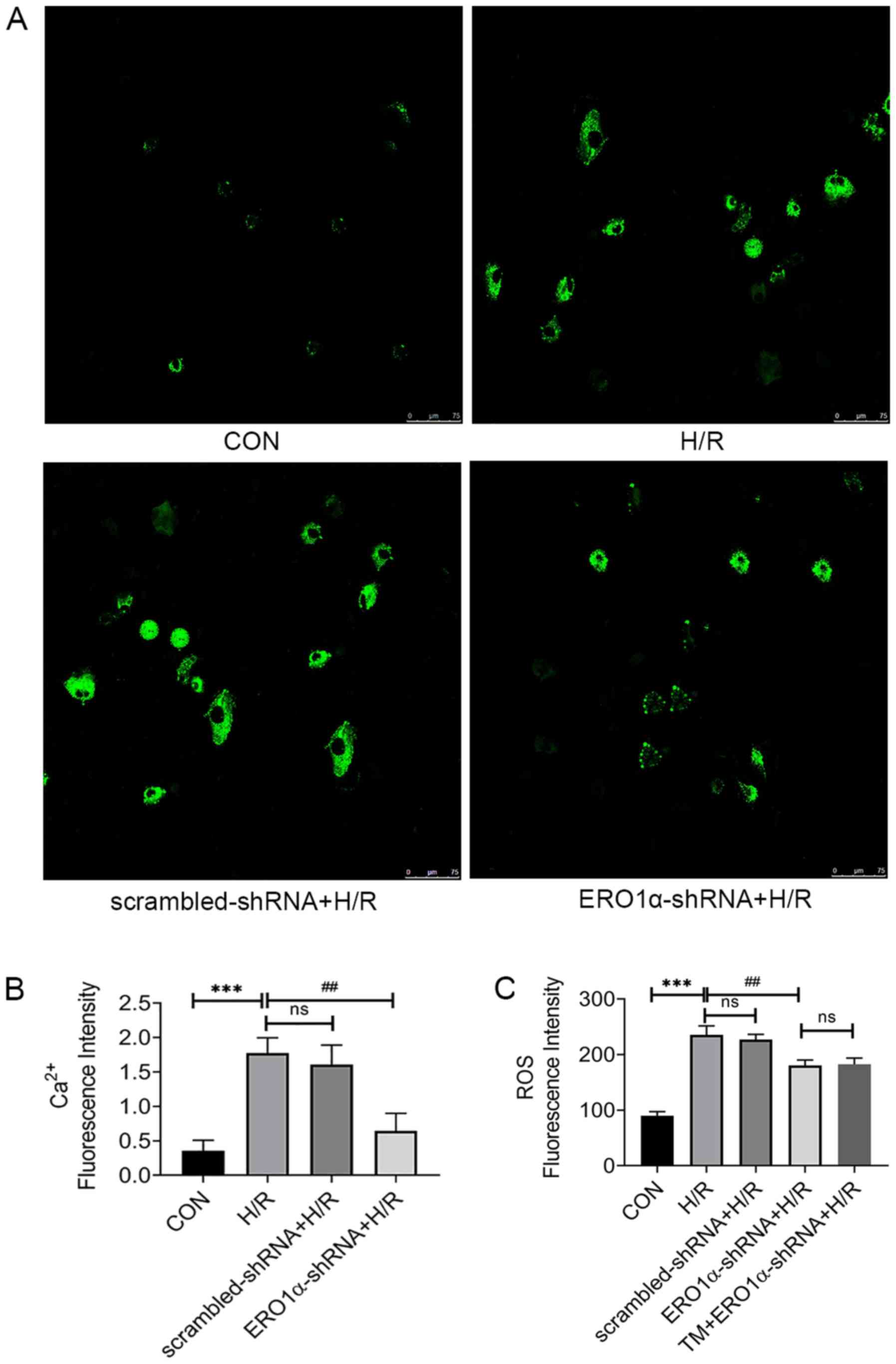
increases in intracellular ROS and Ca2+ levels
Under stress conditions, increases in intracellular
Ca2+ levels promote apoptosis (27), thus the intracellular
Ca2+ influx in response to H/R was measured.
Intracellular Ca2+ levels in H9C2 cardiomyocytes were
markedly increased in the H/R group, whereas ERO1α knockdown
decreased the intracellular Ca2+ levels (Fig. 5A and B). Furthermore, oxidative
stress is related to apoptosis-induced H/R injury (28), thus intracellular ROS levels were
analyzed to investigate the association between ERO1α and oxidative
stress. ERO1α knockdown significantly decreased intracellular ROS
levels in the H/R group (Fig.
5C).
ERO1α knockdown alleviates H/R-induced
mitochondrial dysfunction through reducing ERS
To investigate whether ERO1α knockdown protected
mitochondrial function in H/R injury, cardiomyocytes were
pretreated with 2 µg/l TM, an ERS activator, 24 h prior to H/R
exposure to counteract the reduction in ERS following ERO1α
knockdown. Intracellular ROS levels, Δψm, and expression levels of
cytochrome c in mitochondrial and cytosolic fractions were
evaluated. TM pretreatment of ERO1α-shRNA-transfected H9C2
cardiomyocytes undergoing H/R injury did not reverse the reduction
in intracellular ROS levels observed following ERO1α knockdown
(Fig. 5C). Compared with the H/R
group, decreased expression levels of cytochrome c were observed in
the cytoplasm in the ERO1α-shRNA + H/R group, whereas increased
levels were found in the mitochondria. However, no significant
differences were observed between the TM + ERO1α-shRNA + H/R and
the ERO1α-shRNA + H/R groups (Fig.
6A). After H/R, the Δψm significantly decreased, while ERO1α
knockdown had a trend towards an increase in Δψm in the ERO1α-shRNA
+ H/R group; however, there was no significant difference when
comparing the H/R and ERO1α-shRNA + H/R groups (Fig. 6B). Taken together, these findings
suggested that ERO1α may be responsible for the H/R-induced
mitochondrial damage of H9C2 cardiomyocytes, which is associated
with ERS.
 | Figure 6.ERO1α knockdown alleviates
mitochondrial dysfunction by reducing endoplasmic reticulum stress
following H/R injury. (A) Cyt c expression levels in the cytoplasm
and mitochondria were analyzed using western blotting. (B) Δψm was
detected using a microplate reader following staining with JC-1.
Data are presented as the mean ± SD from three independent
experiments. ***P<0.001 vs. CON group; ##P<0.01
vs. H/R group. ERO1α, endoplasmic reticulum oxidase 1α; CON,
control; Cyt c, cytochrome c; H/R, hypoxia/reoxygenation; Cyto,
cytoplasmic; Mito, mitochondria; COX IV, cyt c oxidase subunit IV;
Δψm, mitochondrial membrane potential; shRNA, short hairpin RNA;
ns, not significant; TM, tunicamycin. |
Discussion
In mammalian cells, the ER has an important role in
the proper folding and assembly of polypeptide chains,
Ca2+ storage and post-translational modifications
(29). ERS occurs following
I/R-induced increases in ROS, which results in the accumulation of
misfolded or unfolded proteins in the ER lumen (30). Thus, there is increasing evidence
to suggest that ERS is associated with I/R injury (31–33).
ERO1α is a conserved glycoprotein, which has been
demonstrated to accept electrons from reduced protein disulfide
isomerase and transfer them to oxygen molecules, catalyze the
formation of oxygen-mediated protein disulfide bonds and contribute
to ERS (34,35). ERO1α activity may be an important
factor contributing to the large production of ROS in cells, as it
has been previously found that ERO1α dysfunction may result in a
rapid decrease in ER-derived oxidative stress (16). However, in homocysteine-induced
ERS, ERO1α demonstrated a negative regulatory effect (36). In the present study, ERO1α
expression levels in H9C2 cardiomyocytes were confirmed using
western blotting, and H/R induction was subsequently used to
simulate I/R in these cells to further verify whether ERO1α
expression was altered as a result of oxidative stress. ERO1α
expression levels in H9C2 cardiomyocytes were markedly increased
following H/R, reaching their highest levels following 6 h of
reoxygenation, whereas 4-PBA decreased the expression levels. In
addition, the number of apoptotic cells was significantly increased
following H/R induction. Therefore, it was hypothesized that ERO1α
may serve an important role in H/R development.
To further understand ERO1α function in H9C2
cardiomyocytes following H/R, lentiviral shRNA was used to reduce
ERO1α expression levels. In previous studies, it has been reported
that following myocardial I/R, ERS promoted apoptosis in
cardiomyocytes through the CHOP and caspase-12 signaling pathways
(37); that c-caspase-12
expression levels, and caspase-3 activity were increased following
H/R (38,39); and that caspase-12, which
indirectly activates cytoplasmic caspase-3, was considered to be a
crucial mediator of ERS-induced apoptosis (40). Consistent with these studies, the
data from the present study revealed that the expression levels of
GRP78, CHOP and c-caspase-12, as well as caspase-3 activity, were
significantly increased. However, following the transfection of
H9C2 cardiomyocytes with ERO1α-shRNA, the expression levels and
activity significantly decreased. These results indicated that
ERO1α may have an important role in ERS, and that the
downregulation of ERO1α may decrease ERS and apoptosis in
myocardial cells following H/R injury.
The ER is the main storage location of
Ca2+ in cells and participates in dynamic
Ca2+ exchange with the mitochondria (41); Ca2+ is transferred to
the mitochondrial matrix to stimulate mitochondrial ATP synthesis
by activating the tricarboxylic acid cycle. During I/R, the
increase in intracellular ROS levels and oxidative stress from
multiple different sources leads to a large amount of
Ca2+ dissociating from the ER to the mitochondria
(42). This subsequently promotes
mitochondrial Ca2+ overload, which triggers cell
apoptosis by opening the mitochondrial permeability transition pore
(43). Oxidative stress promotes
ERS, and persistent ERS has been observed to promote mitochondrial
dysfunction, which in turn induces oxidative stress (44). As previously mentioned, ERO1α is
strongly associated with the generation of ROS, and it has been
demonstrated that due to excessive oxidation, ERO1α enhances
inositol triphosphate receptor (IP3R) activity, and promotes
IP3R-mediated Ca2+ release and transfer to the
mitochondria, facilitating apoptosis (45,46).
Thus, further investigation into the relationship between ERO1α and
mitochondrial function following H/R is required.
The Δψm reflects the functional status of the
mitochondria. In the present study, it was observed that oxidative
stress led to a decrease in Δψm, accompanied by significantly
increased ERO1α expression levels. Compared with the H/R group,
following ERO1α-shRNA transfection, the concentration of
Ca2+ decreased, while ERO1α knockdown had a trend
towards an increase in Δψm. ERO1α knockdown reduced the release of
cytochrome c from the mitochondria to the cytoplasm. However, the
pretreatment of ERO1α-transfected H9C2 cardiomyocytes with TM
following H/R injury did not reverse the reduced intracellular ROS
levels, the ratio of cytoplasmic/mitochondrial cytochrome c
achieved by ERO1α knockdown. These results suggested that the
downregulation of ERO1α may attenuate oxidative stress, and
decrease the intracellular Ca2+ concentration and the
percentage of apoptotic cells in H9C2 cardiomyocytes following
H/R.
In conclusion, the findings in the present study
indicated that ERO1α may serve as a positive mediator of the
apoptotic pathway during H/R; however, the exact mechanism by which
ERO1α achieves this function remains to be investigated. In
addition, the effects of ERO1α on the H9C2 cardiomyocytes in
vitro may not reflect the in vivo scenario; thus, future
studies should encompass animal models for further validation of
these results.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported by the Science and
Technology Innovation Team Project of Changzhi Medical College
(grant no. CX201409).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LNL and XJZ conceived and designed the experiments;
YL, YYL, SLC and NZ performed the experiments; and DW and LNL were
involved in analyzing the data and drafting the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Braunwald E and Kloner RA: Myocardial
reperfusion: A double-edged sword? J Clin Invest. 76:1713–1719.
1985. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hausenloy DJ and Yellon DM: Ischaemic
conditioning and reperfusion injury. Nat Rev Cardiol. 13:193–209.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hausenloy DJ and Yellon DM: Myocardial
ischemia-reperfusion injury: A neglected therapeutic target. J Clin
Invest. 123:92–100. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dhalla NS, Elmoselhi AB, Hata T and Makino
N: Status of myocardial antioxidants in ischemia-reperfusion
injury. Cardiovasc Res. 47:446–456. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chang JC, Lien CF, Lee WS, Chang HR, Hsu
YC, Luo YP, Jeng JR, Hsieh JC and Yang KT: Intermittent hypoxia
prevents myocardial mitochondrial Ca2+ overload and cell
death during ischemia/reperfusion: The role of reactive oxygen
species. Cells. 8:E5642019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Farrukh MR, Nissar UA, Afnan Q, Rafiq RA,
Sharma L, Amin S, Kaiser P, Sharma PR and Tasduq SA: Oxidative
stress mediated Ca (2+) release manifests endoplasmic reticulum
stress leading to unfolded protein response in UV-B irradiated
human skin cells. J Dermatol Sci. 75:24–35. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Martindale JJ, Fernandez R, Thuerauf D,
Whittaker R, Gude N, Sussman MA and Glembotski CC: Endoplasmic
reticulum stress gene induction and protection from
ischemia/reperfusion injury in the hearts of transgenic mice with a
tamoxifen-regulated form of ATF6. Circ Res. 98:1186–1193. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jian L, Lu Y, Lu S and Lu C: Chemical
chaperone 4-phenylbutyric acid reduces cardiac ischemia/reperfusion
injury by alleviating endoplasmic reticulum stress and oxidative
stress. Med Sci Monit. 22:5218–5227. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kerkhofs M, Giorgi C, Marchi S, Seitaj B,
Parys JB, Pinton P, Bultynck G and Bittremieux M: Alterations in
Ca2+ signalling via ER-mitochondria contact site
remodelling in cancer. Adv Exp Med Biol. 997:225–254. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kerkhofs M, Bittremieux M, Morciano G,
Giorgi C, Pinton P, Parys JB and Bultynck G: Emerging molecular
mechanisms in chemotherapy: Ca2+ signaling at the
mitochondria-associated endoplasmic reticulum membranes. Cell Death
Dis. 9:3342018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Morciano G, Marchi S, Morganti C, Sbano L,
Bittremieux M, Kerkhofs M, Corricelli M, Danese A,
Karkucinska-Wieckowska A, Wieckowski MR, et al: Role of
mitochondria-associated ER membranes in calcium regulation in
cancer-specific settings. Neoplasia. 20:510–523. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dias-Gunasekara S, Gubbens J, van Lith M,
Dunne C, Williams JA, Kataky R, Scoones D, Lapthorn A, Bulleid NJ
and Benham AM: Tissue specific expression and dimerization of the
endoplasmic reticulum oxidoreductase Ero1beta. J Biol Chem.
280:33066–33075. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zito E, Chin KT, Blais J, Harding HP and
Ron D: ERO1-beta, a pancreas-specific disulfide oxidase, promotes
insulin biogenesis and glucose homeostasis. J Cell Biol.
188:821–832. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pagani M, Fabbri M, Benedetti C, Fassio A,
Pilati S, Bulleid NJ, Cabibbo A and Sitia R: Endoplasmic reticulum
oxidoreductin 1-Lbeta (ERO1-Lbeta), a human gene induced in the
course of the unfolded protein response. J Biol Chem.
275:23685–23692. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gess B, Hofbauer KH, Wenger RH, Lohaus C,
Meyer HE and Kurtz A: The cellular oxygen tension regulates
expression of the endoplasmic oxidoreductase ERO1-Lalpha. Eur J
Biochem. 270:2228–2235. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sevier CS and Kaiser CA: Ero1 and redox
homeostasis in the endoplasmic reticulum. Biochim Biophys Acta.
1783:549–556. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Seervi M, Sobhan PK, Joseph J, Ann Mathew
K and Santhoshkumar TR: ERO1α-dependent endoplasmic
reticulum-mitochondrial calcium flux contributes to ER stress and
mitochondrial permeabilization by procaspase-activating compound-1
(PAC-1). Cell Death Dis. 4:e9682013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nakamura K, Tamaki H, Kang MS, Mochimaru
H, Lee ST, Nakamura K and Kamagata Y: A six-well plate method: Less
laborious and effective method for cultivation of obligate
anaerobic microorganisms. Microbes Environ. 26:301–306. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li J, Xia X, Ke Y, Nie H, Smith MA and Zhu
X: Trichosanthin induced apoptosis in HL-60 cells via mitochondrial
and endoplasmic reticulum stress signaling pathways. Biochim
Biophys Acta. 1770:1169–1180. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lai LN, Zhang XJ, Zhang XY, Song LH, Guo
CH, Lei JW and Song XL: Lazaroid U83836E protects the heart against
ischemia reperfusion injury via inhibition of oxidative stress and
activation of PKC. Mol Med Rep. 13:3993–4000. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lai L, Chen Y, Tian X, Li X, Zhang X, Lei
J, Bi Y, Fang B and Song X: Artesunate alleviates hepatic fibrosis
induced by multiple pathogenic Factors and inflammation through the
inhibition of LPS/TLR4/NF-κB signaling pathway in rats. Eur J
Pharmacol. 765:234–241. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu H, Ye M, Yang J and Ding J: Modulating
endoplasmic reticulum stress to alleviate myocardial ischemia and
reperfusion injury from basic research to clinical practice: A long
way to go. Int J Cardiol. 223:630–631. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yu L, Li S, Tang X, Li Z, Zhang J, Xue X,
Han J, Liu Y, Zhang Y, Zhang Y, et al: Diallyl trisulfide
ameliorates myocardial ischemia-reperfusion injury by reducing
oxidative stress and endoplasmic reticulum stress-mediated
apoptosis in type 1 diabetic rats: Role of SIRT1 activation.
Apoptosis. 22:942–954. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yoon S, Park SJ, Han JH, Kang JH, Kim JH,
Lee J, Park S, Shin HJ, Kim K, Yun M and Chwae YJ:
Caspase-dependent cell death-associated release of nucleosome and
damage-associated molecular patterns. Cell Death Dis. 5:e14942014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Groenendyk J, Sreenivasaiah PK, Kim DH,
Agellon LB and Michalak M: Biology of endoplasmic reticulum stress
in the heart. Circ Res. 107:1185–1197. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Weiss JN, Korge P, Honda HM and Ping P:
Role of the mitochondrial permeability transition in myocardial
disease. Circ Res. 93:292–301. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kalogeris T, Baines CP, Krenz M and
Korthuis RJ: Cell biology of ischemia/reperfusion injury. Int Rev
Cell Mol Biol. 298:229–317. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gelebart P, Opas M and Michalak M:
Calreticulin, a Ca2+-binding chaperone of the
endoplasmic reticulum. Int J Biochem Cell Biol. 37:260–266. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Doroudgar S and Glembotski CC: New
concepts of endoplasmic reticulum function in the heart: Programmed
to conserve. J Mol Cell Cardiol. 55:85–91. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guan G, Zhang J, Liu S, Huang W, Gong Y
and Gu X: Glucagon-like peptide-1 attenuates endoplasmic reticulum
stress-induced apoptosis in H9c2 cardiomyocytes during
hypoxia/reoxygenation through the GLP-1R/PI3K/Akt pathways. Naunyn
Schmiedebergs Arch Pharmacol. 392:715–722. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang XB, Huang XM, Ochs T, Li XY, Jin HF,
Tang CS and Du JB: Effect of sulfur dioxide preconditioning on rat
myocardial ischemia/reperfusion injury by inducing endoplasmic
reticulum stress. Basic Res Cardiol. 106:865–878. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xu J, Hu H, Chen B, Yue R, Zhou Z, Liu Y,
Zhang S, Xu L, Wang H and Yu Z: Lycopene protects against
hypoxia/reoxygenation injury by alleviating ER stress induced
apoptosis in neonatal mouse cardiomyocytes. PLoS One.
10:e01364432015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zito E: ERO1: A protein disulfide oxidase
and H2O2 producer. Free Radic Biol Med. 83:299–304. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zeeshan HM, Lee GH, Kim HR and Chae HJ:
Endoplasmic reticulum stress and associated ROS. Int J Mol Sci.
17:3272016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang X, Xu H, Hao Y, Zhao L, Cai X, Tian
J, Zhang M, Han X, Ma S, Cao J and Jiang Y: Endoplasmic reticulum
oxidoreductin 1α mediates hepatic endoplasmic reticulum stress in
homocysteine-induced atherosclerosis. Acta Biochim Biophys Sin
(Shanghai). 46:902–910. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Neuber C, Uebeler J, Schulze T, Sotoud H,
El-Armouche A and Eschenhagen T: Guanabenz interferes with ER
stress and exerts protective effects in cardiac myocytes. PLoS One.
9:e988932014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhu X, Zhang ZL, Li P, Liang WY, Feng XR
and Liu ML: Shenyuan, an extract of American ginseng and corydalis
tuber formula, attenuates cardiomyocyte apoptosis via inhibition of
endoplasmic reticulum stress and oxidative stress in a porcine
model of myocardial infarction. J Ethnopharmacol. 150:672–681.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mei Y, Thompson MD, Shiraishi Y, Cohen RA
and Tong X: Sarcoplasmic/endoplasmic reticulum Ca2+
ATPase C674 promotes ischemia- and hypoxia-induced angiogenesis via
coordinated endothelial cell and macrophage function. J Mol Cell
Cardiol. 76:275–282. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Szegezdi E, Fitzgerald U and Samali A:
Caspase-12 and ER-stress-mediated apoptosis: The story so far. Ann
NY Acad Sci. 1010:186–194. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Giorgi C, De Stefani D, Bononi A, Rizzuto
R and Pinton P: Structural and functional link between the
mitochondrial network and the endoplasmic reticulum. Int J Biochem
Cell Biol. 41:1817–1827. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Park SJ, Lee SB, Suh Y, Kim SJ, Lee N,
Hong JH, Park C, Woo Y, Ishizuka K, Kim JH, et al: DISC1 modulates
neuronal stress responses by gate-keeping ER-mitochondria
Ca2+ transfer through the MAM. Cell Rep. 21:2748–2759.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Marchi S, Bittremieux M, Missiroli S,
Morganti C, Patergnani S, Sbano L, Rimessi A, Kerkhofs M, Parys JB,
Bultynck G, et al: Endoplasmic reticulum-mitochondria communication
through Ca2+ signaling: The importance of
mitochondria-associated membranes (MAMs). Adv Exp Med Biol.
997:49–67. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yang L, Guan G, Lei L, Liu J, Cao L and
Wang X: Oxidative and endoplasmic reticulum stresses are involved
in palmitic acid-induced H9c2 cell apoptosis. Biosci Rep.
39:BSR201902252019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kang S, Kang J, Kwon H, Frueh D, Yoo SH,
Wagner G and Park S: Effects of redox potential and Ca2+
on the inositol 1,4,5-trisphosphate receptor L3-1 loop region:
Implications for receptor regulation. J Biol Chem. 283:25567–25575.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Li G, Mongillo M, Chin KT, Harding H, Ron
D, Marks AR and Tabas I: Role of ERO1-alpha-mediated stimulation of
inositol 1,4,5-triphosphate receptor activity in endoplasmic
reticulum stress-induced apoptosis. J Cell Biol. 186:783–792. 2009.
View Article : Google Scholar : PubMed/NCBI
|