Introduction
At present, high-throughput reverse
transcription-quantitative PCR (RT-qPCR) allows for the detection
and quantification of small amounts of DNA, even individual
molecules, in an accurate and quantitative manner (1). However, limited sample sizes of rare
tissues, liquid biopsies, fine-needle aspirates, and single cells
have been the bottleneck of research studies and clinical
assessments based on DNA and RNA analyses (2–4). The
amplification reaction fails for limited samples and poor cDNA
templates. Thus, researchers urgently require an easy and
reproducible method to prepare available cDNA for high-throughput
qPCR screening.
Pre-amplification is the most common strategy for
the enrichment of target cDNA templates (5). Pre-amplification, multiplex PCR with
specific primer pairs (6), can
target all DNA in an unselective manner (7) and specifically target only genes of
interest (8–12). The formation of non-specific PCR
products and the competition of reagents between the parallel
reactions limit the application of pre-amplification during
template enrichment (13). Fewer
cycles and lower primer concentration will reduce the limitation of
pre-amplification. However, despite its wide application, targeted
pre-amplification during DNA template quantification, particularly
its properties and characteristics, such as templates and dNTP mix
concentrations, is poorly understood (14). The process is still a
time-consuming and it is expensive to amplify specific primer pairs
during multiplex PCR. Furthermore, the whole process is poorly
repeatable (15).
Heterogeneity of all types of cancer leads to
differences in the sensitivity of patients to chemotherapy drugs
(16). As the cost decreases, to
achieve precision medicine, RNA-sequencing in individual patients
is possible in the future. The results of sequencing require
further verification using conventional PCR. However, it is
impossible to verify thousands of differentially expressed genes in
individual patients using traditional PCR. Therefore,
high-throughput PCR, with enough cDNA template, may provide a
suitable method to be used for precision medicine of tumors.
In the present study, a commercially available
RNeasy Micro kit (Qiagen GmbH) was used to improve the quality of
total RNA extracted from cultured cells. Saturated
phenol-chloroform extraction was also used to remove PCR inhibitors
in the samples. The high-throughput qPCR was performed using the
BioMark™ HD system. Notably, the aforementioned workflow was used
to verify peripheral blood mononuclear cells (PBMC) separated from
blood cells in patients infected with Hepatitis B virus. Using the
novel method, an easy and reproducible strategy was developed to
prepare cDNA templates for high-throughput qPCR screening using the
BioMark™ HD system.
Materials and methods
Cell lines
All cell lines, which were purchased from the China
Center for Type Culture Collection, were cultured at 37°C in a
humidified incubator with 5% CO2. Liver cancer cell
lines, HepG2 and Hep3B, were authenticated using STR profiling and
cultured in minimum essential medium supplemented with 10% fetal
bovine serum, 100 U/ml penicillin, and 100 µg/ml streptomycin (all
from Thermo Fisher Scientific, Inc.).
Total RNA extraction using
TRIzol®
Cells were washed three times with cold PBS. To
avoid fragmenting DNA, harvested cells were directly lysed with 800
µl TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.)
and homogenized gently using a pipette. The lysate was added to 160
µl chloroform and mixed thoroughly. After incubation at room
temperature for 15 min, the mixture was centrifuged at 12,000 × g
at 4°C for 15 min. The RNA was transferred to a fresh RNase-free
centrifuge tube and mixed with 200 µl isopropanol at room
temperature for 10 min. The total RNA was collected and centrifuged
at 12,000 × g for 10 min at room temperature. The RNA precipitate
was washed with 800 µl 70% ethanol, and re-precipitated and
centrifuged at 8,000 × g for 5 min at 4°C. After diluting the
sample in 10 µl RNase-free water, the total RNA (~600 ng/µl) was
stored at 80°C until further experimentation.
Total RNA extraction using RNeasy
Micro kit
Total RNA was extracted from cultured (HepG2 and
Hep3B, 80% confluence in 6-well tissue culture plate) cells and
PBMCs (1×106) using a RNeasy Micro kit (Qiagen GmbH)
according to the manufacturer's protocol. Briefly, harvested cells
were directly lysed with 350 µl RLT buffer containing 1%
β-mercaptoethanol (Sigma-Aldrich; Merck KGaA) and homogenized using
a pipette. To precipitate mRNA, 70% ethanol was added to the cell
lysates, and mixed by pipetting. Then, the sample was transferred
to a RNeasy MinElute spin column in a 2 ml collection tube and
centrifuged immediately for 15 sec at 8,000 × g at 4°C. After the
flow-through was discarded, the collected mRNA was washed with 350
µl Buffer RW1 and centrifuged for 15 sec at 8,000 × g at 4°C. The
DNA in the sample was digested with 80 µl DNaseI solution for 15
min at room temperature. After washing with Buffer RW1 and
centrifuging for 15 sec at 8,000 × g at 4°C, 500 µl 80% ethanol was
added to completely wash the sample. The RNeasy MinElute spin
column was centrifuged in a new 2 ml collection tube at 13,000 × g
for 5 min at 4°C to dry the membrane. RNase-free water (14 µl) was
added directly to the center of the spin column membrane to elute
the total RNA, which was stored at −80°C until further use.
cDNA synthesis
RT was performed using the SuperScript®
III First-Strand Synthesis kit for RT-qPCR (Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol.
Briefly, each component was mixed and centrifuged at 2,000 × g for
15 sec at 4°C before use. Random hexamer primers (5 ng/µl), dNTP
mix (1 mM), total RNA (≤2.5 µg), and RNase-free water were added to
a final volume of 5 µl. Samples were denatured at 65°C for 5 min
and subsequently cooled on ice at least for 2 min. The following
reagents were added to a total volume of 10 µl:
SuperScript® III (10 U), RNaseOUT (2 U),
MgCl2 (5 mM), DL-Dithiothreitol (10 mM) and RT buffer.
The following temperature protocol were used: 25°C for 10 min, 50°C
for 60 min, 85°C for 5 min and 4°C to infinity. cDNA was diluted 1,
5, 10 and 20 times in diethyl pyrocarbonate (DEPC)-treated water
and stored at −20°C, until further use.
Removal of PCR inhibitors and cDNA
template enrichment
Saturated phenol was used to remove proteins in the
diluted cDNA, and chloroform was used to remove the phenol
dissolved in water. Briefly, 200 µl cDNA was added with an equal
volume of the saturated phenol-chloroform mixture (ratio, 25:24),
incubated on ice for 10 min and centrifuged at 12,000 × g for 10
mins at 4°C to separate the cDNA and protein. The upper aqueous
phase was transferred to a fresh 1.5 ml centrifuge tube. To
precipitate the cDNA from the aqueous phase, 2 µg glycogen and 500
µl ethanol was added to the aqueous phase and the solution was
stored at −80°C for 8 h. The sample was centrifuged at 14,000 × g
for 30 min at room temperature to separate the cDNA precipitate.
After washing with 1 ml 70% ethanol, the cDNA was centrifuged for a
final time at 10,000 × g for 5 min at room temperature. The
enriched cDNA was then diluted in 10 µl DEPC-treated water and
stored at −20°C.
High-throughput qPCR
High-throughput qPCR was performed using the
BioMark™ HD system and the 48.48 or 96.96 Dynamic Array™ integrated
fluidic circuit (IFC) for gene expression according to the
manufacturer's protocol (Fluidigm Corporation). Briefly, control
line fluid was injected into each accumulator of the IFC. After the
blue film was removed from the bottom of the IFC, the primer script
was run in the instrument. For 10X assay preparation, 1.5 µl primer
(10 µM; Shanghai Sangon Pharmaceutical Co., Ltd.), 1.5 µl probe (10
µM; Shanghai Sangon Pharmaceutical Co., Ltd.), and 2X assay loading
reagent (Fluidigm, Corporation) were mixed together. All primers
used are listed in Table SI. For
the pre-mix preparation, 3 µl TaqMan Universal PCR master mix (2X;
Thermo Fisher Scientific, Inc.), 0.3 µl 20X GE sample loading
reagent (Fluidigm Corporation), and 2.7 µl enriched cDNA were mixed
together. The primed IFC was removed from the instrument and 5 µl
2X assay and pre-mixed sample was pipetted into the assay and
sample inlets, respectively. The following thermocycling conditions
were used: 50° for 2 min, pre-denaturation at 95°C for 1 min,
denaturation at 95°C for 15 sec, annealing at 56°C for 30 sec,
elongation at 72°C for 50 sec, for 50 cycles, then the samples were
held at 4°C forever. Amplification data were analyzed using the
Biomark Real-Time PCR analysis software version 1.3 (Fluidigm
Corporation). The housekeeping genes, GAPDH and ACTB, served as
internal controls. For quality control, in each test, a positive
and a negative control was used, which was provided by the supplier
(Fluidigm Corporation). If none of the 96 samples detected the
result, then it was sufficient evidence that there was a problem
with the detector. On the contrary, if none of the 96 genes
detected the result, then there was have sufficient evidence that
there was an issue with the sample.
Conventional qPCR
As performed in our previous study (17,18),
the primer and probe [ABL proto-oncogene 1 (ABL1), cyclin dependent
kinase inhibitor 1B (CDKN1B), CyclinA2, tissue inhibitor of matrix
metalloproteinase (TIMP-1) and cyclin dependent kinase 7 (CDK7)]
mixture solutions were prepared by adding 10 µM forward primer (2
µl), 10 µM reverse primer (2 µl), 10 µM probe (2 µl), and
double-distilled (dd)H2O (14 µl). The qPCR reaction
solution was prepared with 2X TaqMan Universal PCR Master Mix (4
µl), cDNA diluted in DEPC-treated water (1 µl), ddH2O (1 µl), and
primer and probe mixture solution (2 µl). The qPCR was run using a
384-well system using the aforementioned conditions. All primer and
probes used are listed in Table
SI.
Patients
A total of 21 residual whole blood samples (2 ml),
collected from 4 patients infected with Hepatitis B virus, were
obtained from the Clinical Laboratory of Beijing YouAn Hospital
(Beijing, China). The Ethical Committee of Beijing YouAn Hospital,
Capital Medical University, approved all studies (approval no.
2018011) and written informed consent was provided from all
patients prior to the start of the study. The study methodologies
conformed to the standards set by the Declaration of Helsinki.
There were a total of four patients, three male and one female,
which were between the ages of 43 and 67 years old. All samples
were collected in May 2019.
PBMCs separation
Peripheral blood, 2 ml, was collected into heparin
anticoagulation tubes and centrifuged at 500 × g for 10 min at room
temperature. The cell pellet was diluted with an equal volume (~0.8
ml) of 1X PBS and mix gently with a disposable plastic dropper. A
total of 4 ml lymphocyte isolate (Beijing Solarbio Science and
Technology Co., Ltd.), was added to a fresh 15 ml centrifuge tube,
following which the blood cells were added, gently down the side of
the tube, on top of the lymphocyte separating fluid. After
centrifugation at 500 × g for 10 min at 4°C, PBMCs were removed
into a fresh centrifuge tube and washed with 1X PBS, twice. The
cell pellet was centrifuged in between each wash with PBS. The
collected cell pellet was resuspended in 3 ml red blood cell lysis
buffer to lyse red blood cells. PBMCs were washed with 1X PBS twice
and centrifuged at 500 × g for 5 min at 4°C to obtain the cell
pellet.
Statistical analysis
Differences between groups were analyzed using
Pearson's χ2 test in SPSS v17.0 (SPSS, Inc.) for
Windows. All experiments were repeated three times, the relative
gene expression is presented as the Cq value and the positive rate
is presented as a percentage. P<0.05 was considered to indicate
a statistically significant difference. Heat maps were constructed
using HemI v1.0 (19).
Results
Limited cDNA template without
pre-amplification
To prepare cDNA for high-throughput qPCR, the total
RNA was extracted from HepG2 and Hep3B cells, followed by RT using
SuperScript® III First-Strand Synthesis kit. The
synthesized cDNA was diluted 20X DEPC-treated water. The standard
temperature profile was performed using the BioMark™ HD system for
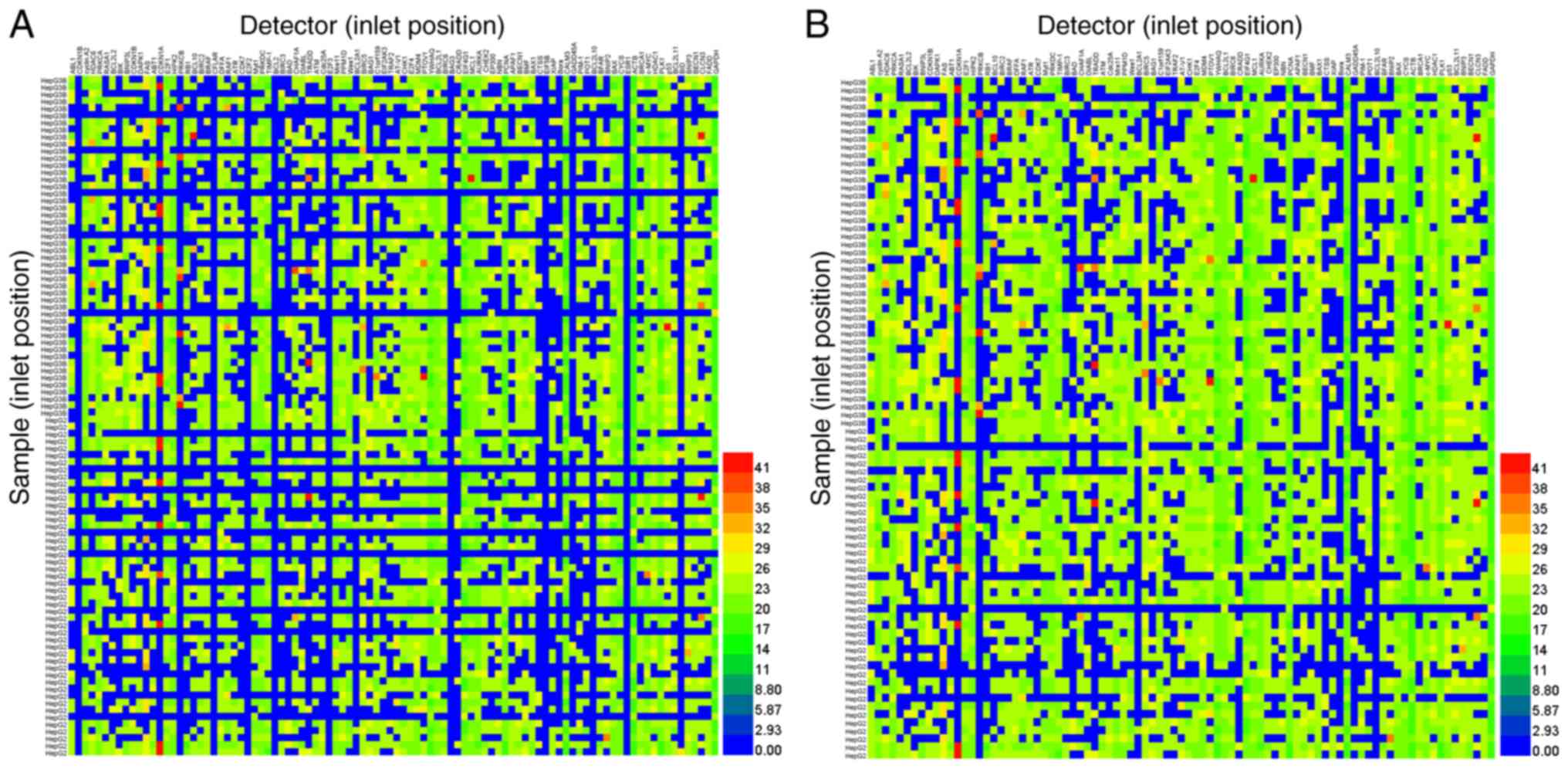
the Cq value of target gene expression. However, only 56% of the
9,216 tests were detected using the Biomark™ HD (Fig. 1A). After the samples and detectors
(primers and probes), which had failed completely were removed from
the total number of samples, 70.18% of 7,329 tests were detectable
(Fig. 1B). Notably, target genes
failed to be detected in samples when the Cq values of housekeeping
genes were >20 (Fig. 1A). Taken
together, the low positive detection rate in the tested samples
suggested that cDNA without pre-amplification was limited due to
its limited template with high-throughput qPCR in the BioMark™ HD
system. Thus, the aim of the present study was to identify an easy
and reproducible strategy that enriches the cDNA template for
high-throughput screening.
BioMark™ HD system fails to detect the
target by directly reducing the dilution factor
To prepare a high concentration of cDNA template for
high-throughput qPCR, the synthesized cDNA from HepG2 and Hep3B
cells was diluted 1, 5, 10 and 20 times in DEPC-treated water for a
serial gradient of cDNA. The standard temperature profile was
performed as aforementioned for 48.48 Dynamic Array™ IFC.
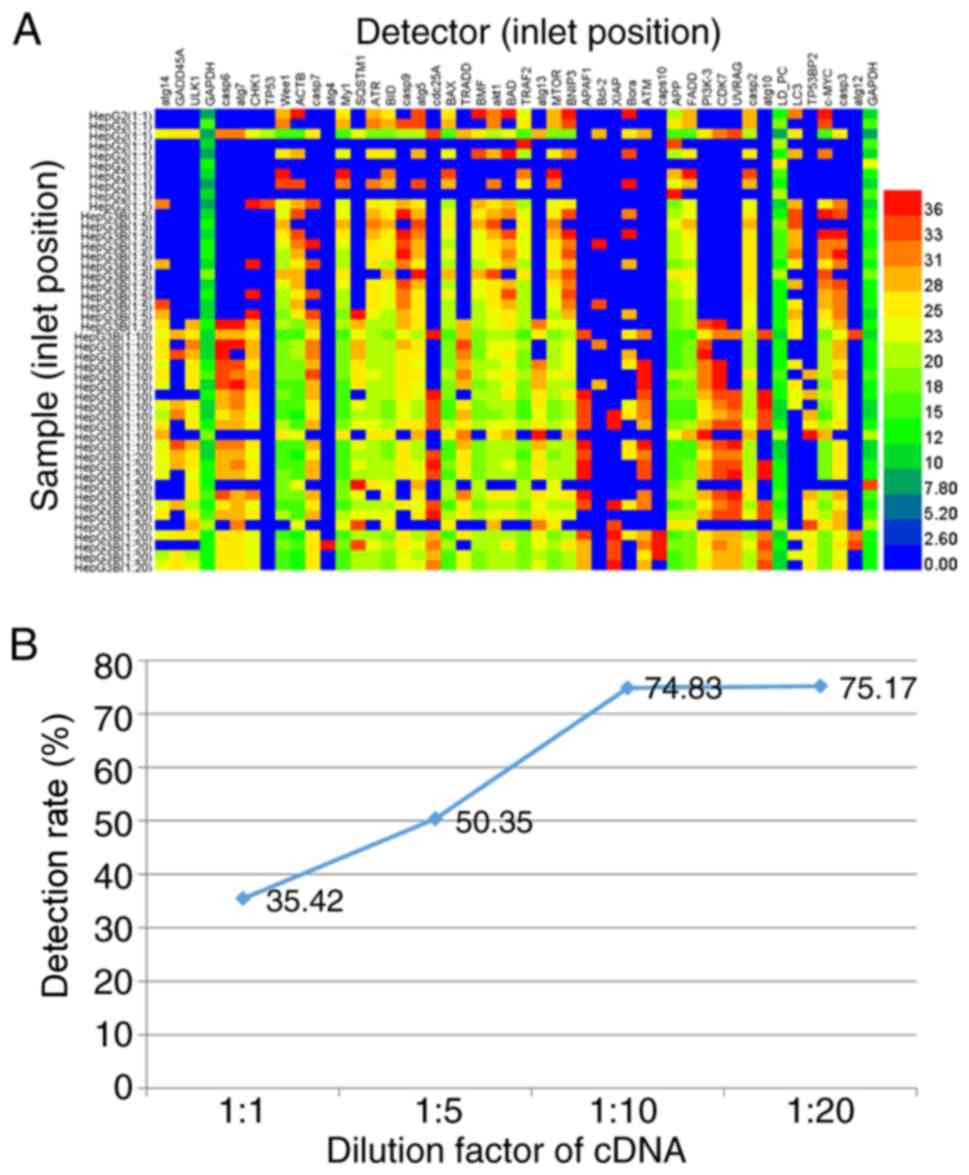
Consistently, in the 20-fold diluted cDNA, only 75.17% of 576
target samples were analyzed using the BioMark™ HD system (left,
lines 35–46; Fig. 2A). By
contrast, the 10-fold diluted cDNA exhibited a positive detection
rate (74.83% of 576 tests), which did not change markedly (left,
lines 23–34; Fig. 2A). However, in
the 1- and 5-fold diluted cDNA samples, the positive detection rate
decreased to 35.42% in 480 tests (left, lines 1–10; Fig. 2A) and 50.35% in 576 samples (left,
lines 11–22; Fig. 2A),
respectively. Taken together, it indicates that increasing the
dilution factor increased the positive detection rate; however, PCR
inhibitors (proteins and soluble salt ions) in the sample inhibited
the subsequent PCR amplification. The PCR amplification requires a
relatively high concentration of template but lower levels of PCR
inhibitors (20).
Removal of PCR inhibitors using
phenol-chloroform extraction
To remove proteins in the samples, an equal volume
(~200 µl) of saturated phenol-chloroform mixture was mixed with the
aforementioned 20× diluted cDNA. The high concentration of soluble
salt ion was removed using centrifugation after being stored on ice
for 10 min. The cDNA pellet was diluted in 10 µl DNase-free water.
The standard temperature profile was performed as aforementioned
using the 96.96 Dynamic Array™ IFC. Notably, 70.11% of 9,216 target
samples were analyzed using the Biomark™ HD system (Fig. 3A). After samples and detectors
which had failed were removed, the positive detection rate
increased to 90.28% in 7,138 samples analyzed (Fig. 3B). Thus, following removal of PCR
inhibitors (proteins and soluble ions), a higher number of cDNA
samples were analyzed using high-throughput qPCR screening and the
BioMark™ HD system.
High quality total RNA prepared using
RNeasy Micro kit
The aforementioned results revealed that saturated
phenol-chloroform extraction markedly improved the cDNA templates
for qPCR. However, as shown in Fig.
3A, the sample concentration (left, lines 24, 25, 49, 58–60,
70, 72, 82, and 84; Fig. 3A) was
too small to be detected using the BioMark™ HD system. Factors from
total RNA extraction using TRIzol® (i.e., protein
pollution in RNA separation and precipitation of soluble salts in
RNA centrifugation) limited the subsequent qPCR reaction. A
commercially available RNA extraction kit (RNeasy Micro kit; Qiagen
GmbH) was used for an easy and reproducible RNA extraction. After
dilution in 10 µl DEPC-treated water, the standard temperature
profile was performed aforementioned for 96.96 Dynamic Array™ IFC.
Notably, 86.09% of 5,148 tests were analyzed using the BioMark™ HD
system (Fig. 4A). After detectors
which had failed were removed (top, line numbers 12, 17, 22, 24,
36, 48, 57, 60, 71, 72, and 78; Fig.
4A), the positive detection rate increased to 97.04% in 4,590
samples analyzed (Fig. 4B). To
compare the results of BioMark™ qPCR and conventional RT-qPCR, the
same samples and 5 detectors (ABL1, CDKN1B, CyclinA2, TIMP-1 and
CDK7) were added to a 384-well plate for conventional qPCR. As
shown in Fig. 4C, the positive
detection rate increased to 96.6% in 270 tests. Thus, there was no
difference between BioMark™ HD system and conventional qPCR (97.04
vs. 96.6%); however, the gene expression detected by the new method
was higher compared with that in conventional PCR. Taken together,
the results showed that using a combination of the commercially
available RNA extraction kit from Qiagen GmbH and saturated
phenol-chloroform extraction, cDNA sample preparation was easy and
reproducible for high-throughput qPCR screening using the BioMark™
HD system.
Preparation of cDNA template from
PBMCs
As aforementioned, the cDNA template preparation was
easy and reproducible. Furthermore, the assay was performed in
cultured cell lines, therefore the same method was used with cDNA
prepared from a limited PBMC sample to determine its suitability
with high-throughput qPCR screening using the BioMark™ HD system.
The residual blood samples were obtained from patients with
Hepatitis B virus, recruited at the clinical laboratory of Beijing
YouAn Hospital (Beijing, China). Following extraction from the
blood cells, cDNA templates from PBMCs were prepared as
aforementioned. After dilution in 10 µl DEPC-treated water, the
standard temperature profile was performed as aforementioned for
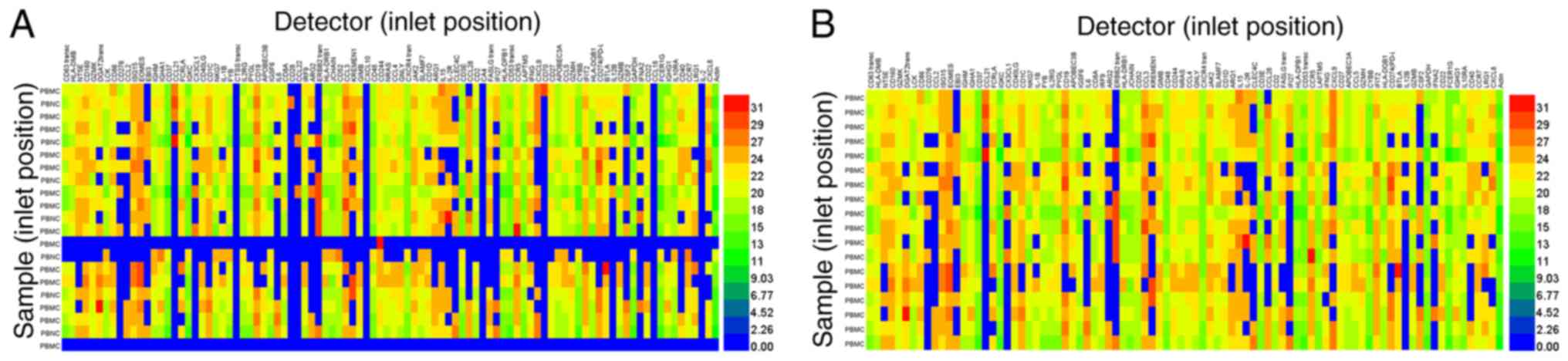
96.96 Dynamic Array™ IFC. A total of 70.4% of 2,016 tests were
analyzed using the BioMark™ HD system (Fig. 5A). After the detectors, which had
failed detectors (top, line numbers 34, 45, 62, 71, 87, and 94) and
samples (left, line numbers 13, 14, and 21) were removed, the
positive detection rate increased to 81.55% of 1,729 samples
(Fig. 5B). Taken together, in
addition to cultured cells, PBMCs were suitable for cDNA sample
preparation for high-throughput qPCR screening using the BioMark™
HD system, using a combination of a commercially available RNA
extraction kit (Qiagen GmbH) and saturated phenol-chloroform
extraction.
Discussion
Limited sample amounts are increasingly used in
laboratory research and in clinical laboratories. At present,
various analytes, such as protein, RNA and DNA, can be accurately
analyzed and quantified, even from an individual single cell
(21–23). Next-generation sequencing and qPCR
are emerging as the two most commonly used techniques to analyze
mRNA sequence and expression levels, respectively (24). However, pre-amplification is
typically required to increase the template of limited samples
(25). The pre-amplification step
is not necessary when few genes (≤10), intermediately or highly
expressed, are to be accurately analyzed (26). When analyzing only one gene,
pre-amplification should be avoided as the conventional qPCR method
is sufficient (27). In the
present study, in the cultured cell lines, HepG2 and Hep3B,
individual gene expression failed to be detected using
high-throughput q-PCR in Biomark™ HD. Target gene expression of a
sample with a Cq value >20 for the housekeeping genes had poor
detectability, which may be due to the limited amount of the
template. Therefore, template concentrations were increased by
reducing the dilution factor. The cDNA prepared using the novel
method showed no difference in the positive detection rate compared
with that using the Biomark™ HD system; however, the Cq value was
higher. The possible reason may be that some cDNA templates were
lost. In addition, when the PCR inhibitor was removed, success of
PCR depends on the concentration of the cDNA template.
However, the presence of PCR inhibitors (e.g.,
blood, aqueous and vitreous humors, heparin,
ethylenediaminetetraacetic acid, urine, polyamines, and plant
polysaccharides) are common limiting products in PCR-based methods
and can lead to failed amplification (28–31).
By reducing the dilution factor, it was found that the cDNA
template concentration increases. However, the PCR amplification
reaction was still inhibited due to the presence of PCR
inhibitors.
There are 4 common methods for removing PCR
inhibitors in samples, including the Power Clean® DNA
clean-up kit (MO BIO Laboratories, Inc.; Qiagen, Inc.), DNAIQ™
System (Promega Corporation), Chelex 1–100 method (Sigma-Aldrich;
Merck KGaA), and phenol-chloroform extraction (Tiangen Biotech Co.,
Ltd.) (32–35). To remove PCR inhibitors and
increase the concentration of the cDNA template, it was found that
secondary extraction using saturated phenol-chloroform for the
library preparation of cDNA could be used to analyze mRNA
quantification for high-throughput qPCR screening using the
BioMark™ HD system. Although saturated phenol chloroform extraction
was added here, pre-amplification was avoided in high-throughput
qPCR, which makes the widespread use of high-throughput qPCR
screening using the BioMark™ HD system possible. Importantly, the
positive detection rate of individual target gene expression was
increased to 90.28% (Fig. 3B).
Total mRNA extraction is an important process that
influences the RT-qPCR reaction. In addition to the amount of mRNA
in cells, the efficiency of RNA extraction may also have a
significant impact on the PCR template. Some common methods to
extract mRNA from samples include phenol (Tiangen Biotech Co.,
Ltd.), anionic detergent, LiCl-urea (LiCI, 3 M; urea, 6 M; NaOAc,
10 mM), modified Gomez, bismuth isothiocyanate (Amresco, LLC),
cetyl trimethylammonium bromide (Amresco, LLC), modified or
conventional hot boric acid (Chemical Book), and TRIzol®
reagent rapid extraction (36–40).
The results in the present study revealed (Figs. 1 and 3) that although TRIzol®
reagent rapid extraction is currently widely used in laboratories,
in order to avoid contamination of phenol and protein this method
requires an experienced experimenter. Thus, the reproducibility of
the results are unpredictable, which is why in the present study
commercially available kits were used for RNA extraction. RNase in
cells and the mRNA extraction process degrade mRNA, and therefore
protein, DNA, and soluble salts can have a notable negative impact
on the subsequent qPCR reaction (33,41).
Finally, Trizol® prepares poor quality cDNA. Using the
commercially available RNeasy Micro kit (Qiagen GmbH) good quality
cDNA was prepared. As a result, almost none samples were below the
detection limit (Cq values of housekeeping genes were >20), and
the positive detection rate increased to 97.04%. Notably, in
addition to the cultured cell lines, high-quality and
high-throughput PCR cDNA samples were prepared using the novel
strategy, and the positive detection rate of samples from PBMCs
extracted from patients with Hepatitis B virus infection was
notably increased using this protocol (Fig. 5).
The novel method described in the present study
produced an easy and reproducible method for template preparation
and high-throughput qPCR, however, the quality and quantity of the
sample were essential factors that could influence the final
result. RNase is commonly found in the environment (42). Thus, once the permeability of the
cell membrane changes, RNase in the environment can enter the cell
and degrade mRNA rapidly (43).
Therefore, fresh or preserved samples at −80°C are required. Unlike
traditional pre-amplification, the concentration of the template
was increased by reducing the dilution factor. Thus, the right
number of samples, 80% confluent HepG2 and Hep3B cells in 6-well
tissue culture plate or 1×106 PBMCs, was required.
Normally, 2 days are required to perform all the
experimental procedures. When cDNA was precipitated overnight at
−80°C, a total of 3 days was required. The traditional techniques
could be completed in <2 days; however, the design of the
pre-amplification primers can be a time-consuming and complicated
process (44). Notably, the novel
method may overcome the pre-amplification bias in cDNA template
preparation, which was the primary reason for the development of a
cDNA enrichment method. However, automatic procedures of for this
method were not developed. At present, litter cDNA in the sample
was not sufficient to recycle using the commercial kit, therefore
glycogen was added to promote the precipitation of cDNA. However,
using the principles in the present study, commercial kits could be
developed in the future to achieve automatic cDNA preparation.
In summary, high-quality total RNA was repeatedly
extracted using a commercially available RNeasy Micro kit (Qiagen
GmbH). PCR inhibitors in samples were removed using saturated
phenol-chloroform extraction. By decreasing the dilution factor,
the positive detection rate for high-throughput qPCR screening in
the BioMark™ HD system was increased to 97.04%. Notably, the easy
and reproducible novel method is suitable for both cultured cell
lines and PBMCs separated from blood cells. Therefore, large sample
preparation would be possible for high-throughput qPCR screening
using the BioMark™ HD system.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Mr. Rifeng Jin
(Oregon State University, Chemical Biological Environmental
Engineering College, Oregon State University, Corvallis, USA) for
his help drafting and editing this manuscript.
Funding
This study was supported by the Capital's Funds for
Health Improvement and Research (grant no. 2018-1-1151), the
National Natural Science Foundation of China (grant no. 81672026),
the National Science and Technology Major Project of China (grant
no. 2018ZX10302205-005) and the Clinical Medical Research Project
(grant no. 2017Z21).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DC and YZ designed the study and wrote the
manuscript. TY drafted the manuscript and prepared the cDNA. YO
helped to operate the BioMark™ HD system and analyzed data. YG
performed cell cultures. DL helped collect blood and performed PBMC
separation. All authors read and approved the final manuscript. All
authors approved the final version of the manuscript.
Ethics approval and consent to
participate
The Ethics Committee of Beijing YouAn Hospital,
Capital Medical University, (Beijing, China) approved all studies
involving patients and informed consent was provided from all the
patients prior to the start of the study (approval no.
2018011).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Moore MD, Panjwani S, Gray KD, Finnerty
BM, Zarnegar R and Fahey TJ III: The role of molecular diagnostic
testing in the management of thyroid nodules. Expert Rev Mol Diagn.
17:3541–576. 2017. View Article : Google Scholar
|
|
2
|
Scher HI, Heller G, Molina A, Attard G,
Danila DC, Jia X, Peng W, Sandhu SK, Olmos D, Riisnaes R, et al:
Circulating tumor cell biomarker panel as an individual-level
surrogate for survival in metastatic castration-resistant prostate
cancer. J Clin Oncol. 33:1348–1355. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Labourier E, Shifrin A, Busseniers AE,
Lupo MA, Manganelli ML, Andruss B, Wylie D and Beaudenon-Huibregtse
S: Molecular testing for miRNA, mRNA, and DNA on fine-needle
aspiration improves the preoperative diagnosis of thyroid nodules
with indeterminate cytology. J Clin Endocrinol Metab.
100:2743–2750. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Patel AP, Tirosh I, Trombetta JJ, Shalek
AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT,
Martuza RL, et al: Single-cell RNA-seq highlights intratumoral
heterogeneity in primary glioblastoma. Science. 344:1396–1401.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Muthukumar T, Lee JR, Dadhania DM, Ding R,
Sharma VK, Schwartz JE and Suthanthiran M: Allograft rejection and
tubulointerstitial fibrosis in human kidney allografts:
interrogation by urinary cell mRNA profiling. Transplant Rev
(Orlando). 28:145–154. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ståhlberg A and Kubista M: The workflow of
single-cell expression profiling using quantitative real-time PCR.
Expert Rev Mol Diagn. 14:323–331. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Eberwine J, Yeh H, Miyashiro K, Cao Y,
Nair S, Finnell R, Zettel M and Coleman P: Analysis of gene
expression in single live neurons. Proc Natl Acad Sci USA.
89:3010–3014. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lao K, Xu NL, Sun YA, Livak KJ and Straus
NA: Real time PCR profiling of 330 human micro-RNAs. Biotechnol J.
2:33–35. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lao K, Xu NL, Yeung V, Chen C, Livak KJ
and Straus NA: Multiplexing RT-PCR for the detection of multiple
miRNA species in small samples. Biochem Biophys Res Commun.
343:85–89. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Morrison JA, Box AC, Mckinney MC, Mclennan
R and Kulesa PM: Quantitative single cell gene expression profiling
in the avian embryo. Dev Dyn. 244:774–784. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rusnakova V, Honsa P, Dzamba D, Ståhlberg
A, Kubista M and Anderova M: Heterogeneity of astrocytes: From
development to injury-single cell gene expression. PLoS One.
8:e697342013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tang F, Hajkova P, Barton SC, Lao K and
Surani MA: MicroRNA expression profiling of single whole embryonic
stem cells. Nucleic Acids Res. 34:e92006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Walder RY, Hayes JR and Walder JA: Use of
PCR primers containing a 3′-terminal ribose residue to prevent
cross-contamination of amplified sequences. Nucleic Acids Res.
21:4339–4343. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Moghaddaszadeh-Ahrabi S, Farajnia S,
Rahimi-Mianji G and Nejati-Javaremi A: A short and simple
improved-primer extension preamplification (I-PEP) procedure for
whole genome amplification (WGA) of bovine cells. Anim Biotechnol.
23:24–42. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xia P, Radpour R, Kohler C, Dang CX, Fan
AX, Holzgreve W and Zhong XY: A selected pre-amplification strategy
for genetic analysis using limited DNA targets. Clin Chem Lab Med.
47:288–293. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vessoni AT, Filippi-Chiela EC, Lenz G and
Batista LFZ: Tumor propagating cells: Drivers of tumor plasticity,
heterogeneity, and recurrence. Oncogene. 39:2055–2068. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang T, Wu T, Lv L, Zhang Z, Liu D, Xu J,
Chen D and Wu G: Ceria oxide nanoparticles an ideal carrier given
little stress to cells and rats. J Nanosci Nanotechnol.
18:3865–3869. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang T, Gao Y, Liu D, Wang Y, Wu J, Liu X,
Shi Y and Chen D: ASPP2 enhances chemotherapeutic sensitivity
through the down-regulation of XIAP expression in a p53 independent
manner in hepatocellular carcinoma. Biochem Biophys Res Commun.
508:769–774. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Deng W, Wang Y, Liu Z, Cheng H and Xue Y:
HemI: A toolkit for illustrating heatmaps. PLoS One. 9:e1119882014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schrader C, Schielke A, Ellerbroek L and
Johne R: PCR inhibitors-occurrence, properties and removal. J Appl
Microbiol. 113:1014–1026. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Picelli S, Faridani OR, Björklund AK,
Winberg G, Sagasser S and Sandberg R: Full-length RNA-seq from
single cells using Smart-seq2. Nat Protoc. 9:171–181. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ståhlberg A, Thomsen C, Ruff D and Åman P:
Quantitative PCR analysis of DNA, RNAs, and proteins in the same
single cell. Clin Chem. 58:1682–1691. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kroneis T, Geigl JB, El-Heliebi A, Auer M,
Ulz P, Schwarzbraun T, Dohr G and Sedlmayr P: Combined molecular
genetic and cytogenetic analysis from single cells after isothermal
whole-genome amplification. Clin Chem. 57:1032–1041. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Devonshire AS, Sanders R, Wilkes TM,
Taylor MS, Foy CA and Huggett JF: Application of next generation
qPCR and sequencing platforms to mRNA biomarker analysis. Methods.
59:89–100. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vermeulen J, Derveaux S, Lefever S, De
Smet E, De Preter K, Yigit N, De Paepe A, Pattyn F, Speleman F and
Vandesompele J: RNA pre-amplification enables large-scale RT-qPCR
gene-expression studies on limiting sample amounts. BMC Res Notes.
2:2352009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Okino ST, Kong M, Sarras H and Wang Y:
Evaluation of bias associated with high-multiplex, target-specific
pre-amplification. Biomol Detect Quantif. 6:13–21. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ståhlberg A, Kubista M and Aman P:
Single-cell gene-expression profiling and its potential diagnostic
applications. Expert Rev Mol Diagn. 11:735–740. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ahokas H and Erkkilä M: Interference of
PCR amplification by the polyamines, spermine and spermidine. PCR
Methods Appl. 3:65–68. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Holodniy M, Kim S, Katzenstein D, Konrad
M, Groves E and Merigan TC: Inhibition of human immunodeficiency
virus gene amplification by heparin. J Clin Microbiol. 29:676–679.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Khan G, Kangro HO, Coates PJ and Heath RB:
Inhibitory effects of urine on the polymerase chain reaction for
cytomegalovirus DNA. J Clin Pathol. 44:360–365. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wiedbrauk DL, Werner JC and Drevon AM:
Inhibition of PCR by aqueous and vitreous fluids. J Clin Microbiol.
33:2643–2646. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hu Q, Liu Y, Yi S and Huang D: A
comparison of four methods for PCR inhibitor removal. Forensic Sci
Int Genet. 16:94–97. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Faber KL, Person EC and Hudlow WR: PCR
inhibitor removal using the NucleoSpin® DNA Clean-Up XS
kit. Forensic Sci Int Genet. 7:209–213. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hudlow WR, Krieger R, Meusel M, Sehhat JC,
Timken MD and Buoncristiani MR: The NucleoSpin® DNA
Clean-up XS kit for the concentration and purification of genomic
DNA extracts: An alternative to microdialysis filtration. Forensic
Sci Int Genet. 5:226–230. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Thompson RE, Duncan G and McCord BR: An
investigation of PCR inhibition using Plexor(®)-based
quantitative PCR and short tandem repeat amplification. J Forensic
Sci. 59:1517–1529. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gómez JC, Reátegui Adel C, Flores JT,
Saavedra RR, Ruiz MC and Correa SA: Isolation of high-quality total
RNA from leaves of myrciaria dubia ‘CAMU CAMU’. Prep Biochem
Biotechnol. 43:527–538. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen Q, Yu HW, Wang XR, Xie XL, Yue XY and
Tang HR: An alternative cetyltrimethylammonium bromide-based
protocol for RNA isolation from blackberry (Rubus L.). Genet Mol
Res. 11:1773–1782. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhao L, Ding Q, Zeng J, Wang FR, Zhang J,
Fan SJ and He XQ: An improved CTAB-ammonium acetate method for
total RNA isolation from cotton. Phytochem Anal. 23:647–650. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Christou A, Georgiadou EC, Filippou P,
Manganaris GA and Fotopoulos V: Establishment of a rapid,
inexpensive protocol for extraction of high quality RNA from small
amounts of strawberry plant tissues and other recalcitrant fruit
crops. Gene. 537:169–173. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gambino G, Perrone I and Gribaudo I: A
rapid and effective method for RNA extraction from different
tissues of grapevine and other woody plants. Phytochem Anal.
19:520–525. 2010. View Article : Google Scholar
|
|
41
|
Romsos EL and Vallone PM: Rapid PCR of STR
markers: Applications to human identification. Forensic Sci Int
Genet. 18:90–99. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bisbal C: RNase L: Effector nuclease of an
activatable RNA degradation system in mammals. Prog Mol Subcell
Biol. 18:19–34. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kaplan R and Apirion D: The fate of
ribosomes in Escherichia coli cells starved for a carbon source. J
Biol Chem. 250:1854–1863. 1975.PubMed/NCBI
|
|
44
|
Korenková V, Scott J, Novosadová V,
Jindřichová M, Langerová L, Švec D, Šídová M and Sjöback R:
Pre-amplification in the context of high-throughput qPCR gene
expression experiment. BMC Mol Biol. 16:52015. View Article : Google Scholar : PubMed/NCBI
|