Introduction
Cardiac arrest (CA) poses a significant public
health burden. Although modern cardiopulmonary resuscitation (CPR)
improves the return of spontaneous circulation (ROSC) rate in
patients who have suffered from CA, the survival to hospital
discharge rate remains poor (1,2).
Significant left ventricular systolic and diastolic dysfunction
early after ROSC is termed post-cardiac arrest myocardial
dysfunction (PAMD) and is a common symptom noted in these patients
(3). Although PAMD is temporary
following resuscitation, approximately two-thirds of the patients
who achieve ROSC do not survive due to PAMD within the first 72 h
(4–6). Currently, pharmacological treatments
that attenuate PAMD are not available (6,7).
Mitochondria-mediated cardiomyocyte apoptosis is one
of the primary mechanisms underlying PAMD due to CA (6,8,9).
Global myocardial ischemia associated with CA induces mitochondrial
permeability transition pore (mPTP) opening, which results in
cytochrome c leakage from the mitochondrial matrix to the
cytoplasm, leading to caspase-3 activation and induction of
apoptosis (10–12). Inhibition of mPTP opening and
apoptosis induction may represent a potential therapeutic approach
for PAMD (13,14).
Prostaglandin E1 (PGE1) is an essential member of
the prostaglandin family that demonstrates several physiological
and pharmacological activities. PGE1 is widely used in the clinic
for the treatment of ischemic injury (15–17).
PGE1 decreases mPTP opening and apoptosis in animal models of
myocardial infarction and coronary microembolization (17–19).
However, protective effects of PGE1 on PAMD have not been reported
to date.
The present study evaluated whether PGE1 treatment
could attenuate PAMD and improve survival rates in a rat model of
CA. In addition, whether PGE1 might exert cardioprotective effects
by inhibiting mitochondria-mediated cardiomyocyte apoptosis was
also assessed.
Materials and methods
Animals
Animal experiments were conducted following the
guide for the Care and Use of Laboratory Animals of the National
Institutes of Health. The present study was approved by the Animal
Use and Care Committee of Shandong University. Male Wistar rats
weighing 380–430 g, aged ~14 weeks were purchased from the
Department of Experimental Animals of Shandong University. The rats
were housed in independent ventilation cages at an ambient
temperature of 23±1°C and a humidity of 55±5% under a 12-h
light/dark cycle. They were allowed access to food and tap water
ad libitum.
Asphyxia-induced CA model
establishment and animal treatments
A previously published rat model of CA was used in
the present study. Briefly, rats were anesthetized with 5%
isoflurane in room air (21% oxygen) in a plastic induction box. The
trachea was orally intubated once the rats were fully anesthetized
(no response to pain absence of corneal reflex). The rats were
mechanically ventilated and maintained under anesthesia with 2%
isoflurane. A PE-50 catheter was advanced through the left femoral
artery into the aorta for measurement of the mean aortic pressure
(MAP). A microcatheter was inserted into the left femoral vein for
drug administration. A conventional lead II electrocardiogram was
continuously recorded. Pancuronium bromide (2 mg/kg) was
intravenously administered for complete muscle relaxation.
Following pancuronium bromide administration, the rats were
administered 0.5% isoflurane in room air (21% oxygen) for 5
min.
The rats were randomly allocated to three groups as
follows (Fig. 1): i) In the CA+PGE1
group, the rats underwent 8 min asphyxia followed by CPR, and 1
µg/kg Lipo-PGE1 (Qilu Pharmaceutical Co., Ltd.) was intravenously
administered at the onset of ROSC (19,20);
ii) in the CA group, the rats underwent 8 min of asphyxia and CPR;
and iii) in the sham group, the rats underwent the same surgical
procedure without PGE1 or asphyxia/CPR. The rat model of
asphyxia-induced CA was established as described previously
(21,22). Following pancuronium-bromide
administration, the rats were administered 0.5% isoflurane in room
air (21% oxygen) for 5 min. CA was induced by asphyxia, which
occurred after turning off the ventilator and clamping the
endotracheal tube. The hypotension and heart rate dropped quickly.
CA was defined as a MAP of <20 mmHg. Following 8 min of
asphyxia, chest compression (23,24)
was initiated at a rate of 200 beats/min with a pneumatically
driven mechanical chest compressor (Fig. S1). The depth of compressions (1–1.5
cm) was adjusted to maintain the MAP >25 mmHg during
resuscitation. Mechanical ventilation with 100% oxygen and a bolus
injection of 10 µg/kg epinephrine via a venous line were initiated
30 sec prior to chest compression. Chest compressions were
continued and epinephrine was injected every 5 min until ROSC was
achieved. ROSC was defined as the return of supraventricular rhythm
with an increase in MAP >50 mmHg for 5 min. Only rats that
achieved ROSC within 10 min were used for further studies (25–27).
This criterion was to avoid non-homogeneous CA periods leading to
pathophysiological differences and subsequently to the need for
larger numbers of animals. In this CA model, most of rats could get
back to supraventricular rhythm within 5 min after performing CPR
and maintain MAP more than 50 mmHg. At 4 h post-ROSC, 21 rats
(n=7/group) were euthanized with Euthasol (390 mg/kg pentobarbital
sodium and 50 mg/kg phenytoin sodium; intraperitoneal injection)
(28). Death was confirmed by the
cessation of the heartbeat and respiration. The remaining 30 rats
(n=10/group) were monitored for 72-h survival analysis.
Cardiac function monitoring
Cardiac function was measured using an ultra-high
frequency ultrasound system for small animals (Vevo 2100;
VisualSonics, Inc.) at baseline and 1, 2, 3 and 4 h post-ROSC. Left
ventricular end-diastolic diameter (LVEDD), left ventricular
end-systolic diameter (LVESD), left ventricular end-diastolic
volume (LVEDV), left ventricular end-systolic volume (LVESV) and
heart rate (HR) were recorded from M-mode images. Ejection fraction
(EF) was calculated as EF=[(LVEDV-LVESV)/LVEDV] ×100. Cardiac
output was calculated as CO=(LVEDV-LVESV) × HR. All measurements
were performed and reviewed by two investigators blinded to group
assignment.
Survival analysis
For the 72-h survival study, all catheters were
removed and wounds were surgically closed at 4 h following ROSC.
Rats received a subcutaneous injection of 1 mg/kg buprenorphine to
relieve pain when they were returned to their cages. Rats were
observed every 2 h within first 24 h after ROSC. Then rats were
observed at 48 and 72 h. During the 72-h observation period,
gasping or respiratory rate under 5 breaths/min required immediate
euthanasia (29). At 72 h, all
surviving rats were re-anesthetized with 5% isoflurane and had a
vein catheter placed for blood sampling. At the end of the
experiment rats were euthanized.
Hypoxia-reoxygenation (H/R) cell model
establishment
The H9c2 rat cardiomyoblast cell line was obtained
from the Cell Bank of the Chinese Academy of Sciences. H9c2 cells
were grown in high-glucose DMEM (Gibco; Thermo Fisher Scientific,
Inc.) supplemented with 10% fetal bovine serum (Gibco; Thermo
Fisher Scientific, Inc.) and 1% penicillin/streptomycin at 37°C in
a humidified atmosphere of 5% CO2.
In the present study, H9C2 cells were digested and
divided into three groups. The control group was incubated in
normal culture medium. The H/R group was treated as described
previously (30,31). Briefly, H9c2 cells in glucose-free
DMEM (Gibco; Thermo Fisher Scientific, Inc.) were exposed to 1%
O2, 94% N2 and 5% CO2 in an
anaerobic chamber (Don Whitley Scientific, Ltd.) for 12 h to mimic
ischemia. Subsequently, the medium was replaced with high-glucose
DMEM and the cells were transferred to a regular incubator at 37°C
with 5% CO2 for 12 h to mimic reperfusion. To study the
effects of PGE1 on H/R model, the H/R+PGE1 group was treated with
0.5 µM PGE1 at the start of reoxygenation.
TUNEL assay
Cardiomyocyte apoptosis was detected in rats using a
TUNEL assay using with the in situ Apoptosis Detection Kit
(EMD Millipore) according to the manufacturer's instructions.
Briefly, the heart tissue was separated and fixed in 4%
paraformaldehyde overnight at room temperature. Fixed heart tissue
was embedded in paraffin and cut transversely into 5-µm sections.
TUNEL reaction mixture (100 µl) was added to cover the tissue
section and incubated in a humidified chamber for 1 h at 37°C. The
nuclei were counterstained with 0.5% methyl green for 5 min at room
temperature. The tissue sections were washed with xylene (three
washes; each wash, 2 min) and covered with coverslips using neutral
balsam mounting medium. The sections were examined under a BX41
light microscope (Olympus Corporation) and images of selected areas
were captured.
H9c2 cell apoptosis was detected using a TUNEL assay
with an Apoptosis Assay Kit (Roche Diagnostics), according to the
manufacturer's instructions. Briefly, the H9c2 cells were fixed in
4% paraformaldehyde for 1 h at room temperature. The samples were
incubated with the TUNEL reaction mixture (50 µl) at 37°C for 60
min. The nuclei were counterstained with DAPI for 5 min at room
temperature. The samples were washed in PBS (three washes; each
wash, 5 min) and coverslipped in fluorescence mounting medium. The
cells were visualized under an IX73 fluorescence microscope
(Olympus Corporation) using excitation and emission wavelengths of
540 and 580 nm, respectively. The cells exhibiting red fluorescence
were defined as TUNEL-positive apoptotic cells.
The levels of apoptosis in tissue and H9c2 cells
were calculated by counting the TUNEL-positive cardiomyocyte nuclei
in three randomly selected fields in each slide (magnification,
×400). The levels were reported as a percentage of total
cardiomyocyte nuclei.
Measurement of mPTP opening
The mitochondria derived from rat myocardium and/or
from H9c2 cells were isolated by differential centrifugation using
a Tissue/Cell Mitochondria Isolation Kit (Beyotime Institute of
Biotechnology) according to the manufacturer's instructions. Fresh
mitochondria were used for the measurement of mPTP opening, while
the mitochondria-free cytoplasmic protein extract was used for the
measurement of cytochrome c expression.
mPTP opening was measured using a Purified
Mitochondrial Membrane Pore Channel Colorimetric Assay kit
(Genmed). mPTP opening was induced by 200 µM CaCl2 and
presented as mitochondrial swelling, which results in a reduction
of the absorbance at 520 nm (A520). The changes in
A520 at various time points were measured for each
sample. The value at −1 min was normalized to the value at 10 min
and was used for statistical analysis.
An additional immunofluorescence method was used to
measure mPTP opening in H9c2 cells. Briefly, cells were washed with
PBS and subsequently stained with 1 µmol/l calcein-AM (Invitrogen;
Thermo Fisher Scientific, Inc.) in the presence of 8 mmol/l
CoCl2 (Sigma-Aldrich; Merck KGaA) at room temperature
for 20 min in the dark. CoCl2 was added to quench the
cytoplasmic signal so that only fluorescence in the mitochondria
was captured. A change in fluorescence intensity corresponded to
the level of mPTP opening.
Western blot analysis
Total protein was extracted from heart tissues and
H9c2 cells using 1X RIPA buffer (Sigma-Aldrich; Merck KGaA) with a
Protease Inhibitor Cocktail (Sigma-Aldrich; Merck KGaA). The
protein concentration was determined using the Pierce BCA Protein
Assay (Pierce; Thermo Fisher Scientific, Inc.). Total protein was
used to measure the expression levels of GSK3β and caspase-3.
Cytoplasmic protein without mitochondria was used to measure
cytochrome c expression. The samples with equal amounts of
protein (50 µg) were loaded onto 10% sodium dodecyl
sulfate-polyacrylamide gels, subjected to electrophoresis and
subsequently transferred onto 0.22-µM PVDF membranes (EMD
Millipore). Following blocking with 5% nonfat milk for 1 h at room
temperature, the membranes were incubated with primary antibodies
overnight at 4°C: Specific for total GSK3β (1:1,000; cat. no.
12456; Cell Signaling Technology, Inc.), phosphorylated (p)-GSK3β
(1:1,000; Ser9; cat. no. 55558; Cell Signaling Technology, Inc.),
caspase-3 (1:1,000; cat. no. 9662; Cell Signaling Technology,
Inc.), cleaved-caspase-3 (1:1,000; cat. no. 9664; Cell Signaling
Technology, Inc.), cytochrome c (1:1,000; cat. no. 11940;
Cell Signaling Technology, Inc.). β-actin (1:1,000; cat. no.
60008-1; ProteinTech Group, Inc.) was used to detect reference
protein expression. After incubation with an HRP-conjugated
anti-rabbit (1:20,000; cat. no. ab205718; Abcam) or anti-mouse
secondary antibody (1:20,000; cat. no. ab205719; Abcam) for 1 h at
room temperature, the labeled proteins were visualized using the
Pierce ECL Western Blotting Substrate (Pierce; Thermo Fisher
Scientific, Inc.). Signals from immunoblots were scanned using
Amersham Imager 600 imagers (GE Healthcare). The relative band
intensities were quantified using the ImageJ software (version
1.52a; National Institutes of Health).
Statistical analysis
All data are presented as the mean ± SD of three
independent experiments. Multi-group comparisons were performed
using one-way ANOVA followed by Tukey's post hoc test. Student's
t-test was used to compare two groups. The comparisons between
time-based measurements within each group were performed using
repeated-measures ANOVA. Survival was analyzed using Kaplan-Meier
curves and log-rank tests. Two-sided P<0.05 was considered to
indicate a statistically significant difference. Statistical
analysis was carried out using GraphPad Prism 7.0 (GraphPad
Software, Inc.).
Results
Baseline characteristics of rats and
resuscitation characteristics
In total, 58 rats were used in the present study, of
which 51 rats achieved ROSC within 10 min and were included in
further studies. Two rats died for the anesthesia induction before
CA, and 5 rats were euthanized because that they did not get back
to supraventricular rhythm after 5 min CPR or could not maintain
MAP more than 50 mmHg for 5 min within 10 min. The mortality rate
during model establishment in this study was consistent with the
previous report (27).
No significant differences were noted in the
parameters body weight, baseline MAP and HR between the treatment
groups (Table I). No significant
differences were noted in the duration of CA, epinephrine dose,
duration of CPR, MAP and HR at 1 h following ROSC between the CA
and the CA+PGE1 groups (Table
I).
 | Table I.Baseline characteristics of rats and
resuscitation characteristics. |
Table I.
Baseline characteristics of rats and
resuscitation characteristics.
| Variable | CA | CA+PGE1 | Sham |
|---|
| Body weight,
(g) | 400.7±17.8 | 411.4±14.7 | 413.5±10.1 |
| Heart rate before
asphyxia, bpm | 357.2±33.4 | 363.5±45.6 | 340.8±29.9 |
| MAP before
asphyxia, mmHg | 133.7±15.6 | 131.8±18.9 | 137.3±10.1 |
| MAP at 1 h after
ROSC, mmHg | 96.2±15.2 | 94±17.83 | – |
| Cardiac arrest
time, sec | 274.9±37.2 | 271.1±29.8 | – |
| CPR duration,
sec | 43.0±12.1 | 43.7±16.5 | – |
| Adrenaline dose,
µg | 4.1±0.2 | 4.1±0.2 | – |
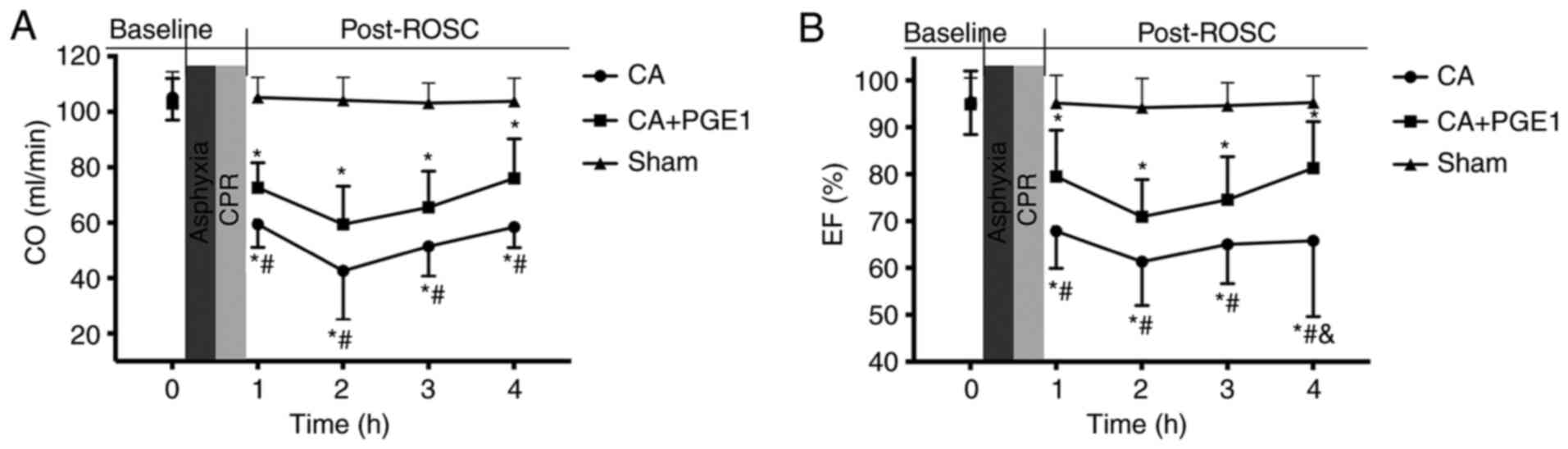
PGE1 ameliorates cardiac
dysfunction
CO and EF were measured in the three treatment
groups at baseline and at 1, 2, 3 and 4 h following ROSC (Fig. 2). No significant differences were
noted in baseline cardiac function between the groups. CO and EF
were significantly decreased within 4 h following ROSC in the CA
group compared with the sham group. However, CO was significantly
increased following PGE1 treatment, compared with the CA group
(P<0.05). A highly significant difference was also noted at 4 h
following ROSC when comparing the mean EF values between the CA and
CA+PGE1 groups (65.8±16.2 vs. 81.2±9.91%; P<0.01).
PGE1 improves the survival rate
The survival rates were monitored for 72 h.
Kaplan-Meier survival curves indicated a rapid decline in the
survival rate of rats in the CA group within 24 h following ROSC
(Fig. 3). The survival rate in the
CA+PGE1 group was significantly higher than that in the CA group
(P<0.05; Fig. 3).
PGE1 suppresses cardiomyocyte
apoptosis
Myocardial apoptosis was detected using TUNEL
staining. In vivo, the levels of apoptosis in the CA group
were significantly increased compared with those in the sham group
(12.11±4.37 vs. 48.33±19.07%, P<0.05, Fig. 4A). In contrast to these findings,
the levels of cardiac apoptosis decreased following treatment with
PGE1, compared with the CA group (31.02±13.51 vs. 48.33±19.07,
respectively; P<0.05; Fig. 4A).
In vitro, the fraction of apoptotic H9c2 cells was
significantly higher in the in H/R group than that in the control
group (52.13±7.86 vs. 6.38±2.02%, respectively; P<0.01; Fig. 4B). The fraction of apoptotic H9c2
cells in the H/R+PGE1 group significantly decreased, compared with
the H/R group (31.20±12.62 vs. 52.13±7.86%, respectively;
P<0.01, Fig. 4B).
PGE1 blocks mPTP opening
The mitochondrial swelling assay results
demonstrated that the change of OD value A520 in the CA
group was lower compared with the sham group (0.61±0.11 vs.
0.82±0.13, respectively; P<0.05; Fig. 5A and B), which indicated the level
of mPTP opening was increased in the CA group. PGE1 treatment
reduced mPTP opening, compared with the CA group (0.76±0.11 vs.
0.61±0.12, respectively; P<0.05; Fig. 5A and B). Moreover, mPTP opening was
decreased by PGE1 treatment in vitro, compared with the H/R
group (0.77±0.13 vs. 0.58±0.10, respectively; P<0.05; Fig. 5C and D). Consistent with the
mitochondrial swelling assay results, the relative fluorescence
intensity of calcein-AM was significantly increased in the H/R+PGE1
group, compared with the H/R group (0.72±0.24 vs. 0.41±0.12,
respectively; P<0.01; Fig.
5E-H).
PGE1 regulates GSK3β, cleaved
caspase-3 and cytochrome c expression
GSK3β protein phosphorylation (Ser9) was also
analyzed. In vivo, GSK3β phosphorylation decreased in the CA
group compared with the sham group (P<0.01; Fig. 6A). GSK3β phosphorylation was higher
in the CA+PGE1 group than in the CA group (P<0.01; Fig. 6A). In vitro, GSK3β
phosphorylation was decreased in the H/R group (P<0.01),
compared with the control group. However, it was partly restored
following PGE1 treatment (P<0.01; Fig. 6B).
The cytoplasmic protein levels of cleaved caspase-3
and cytochrome c were significantly increased in the CA
group in vivo, compared with the sham group (P<0.01;
Fig. 6A). PGE1 treatment partially
suppressed these effects (P<0.01; Fig. 6B). The results obtained in
vitro were consistent with the in vivo data. Indeed,
PGE1 treatment inhibited the increase in the expression levels of
cleaved caspase-3 and cytochrome c induced by H/R
(P<0.01; Fig. 6B).
Discussion
PAMD is a common condition that can reduce survival
in resuscitated patients following CA. No effective treatment
strategies have been reported for PAMD (6,32). In
the present study, it was demonstrated that PGE1 treatment improved
cardiac function and survival outcome following ROSC in a rat model
of asphyxia-induced CA. Moreover, in vitro experiments
demonstrated that inhibition of mitochondria-mediated cardiomyocyte
apoptosis was involved in the mechanism underlying the protective
effect of PGE1.
The rat model of asphyxia-induced CA used in the
present study is commonly used (33). The EF and CO parameters were
measured in the experimental animals, which confirmed a significant
impairment of cardiac function following CA. It is interesting to
note that PGE1 treatment attenuated cardiac dysfunction at 1 h
following ROSC. Furthermore, PGE1 significantly improved EF at 4 h
following ROSC. These results indicated that PGE1 exhibited
protective effects against PAMD.
The underlying mechanism was investigated in the
in vivo and in vitro models. The primary causes of
PAMD are myocardial apoptosis and stunning following
ischemia-reperfusion injury (6,34).
Inhibition of apoptosis can effectively alleviate organ disorders
(35). Gu et al (36) reported that inhibition of
cardiomyocyte apoptosis could improve cardiac function following
CA. In the present study, the induction of cardiomyocyte apoptosis
was investigated following CA. The results indicated that PGE1
treatment significantly ameliorated cardiomyocyte apoptosis
following CA.
Since mitochondrial disorder plays a vital role in
myocardial apoptosis (34,37–39),
mPTP opening was assessed. mPTP opening in the inner mitochondrial
membrane results in collapse of the membrane potential, matrix
swelling and the release of cytochrome c into the cytoplasm,
leading to activation of caspase-3 and induction of apoptosis
(11,37). Growing evidence indicates that
inhibition of mPTP opening can reduce apoptosis in several
pathological conditions (14,40,41).
An increase in mPTP was noted following CA and H/R. However, PGE1
treatment effectively prevented excessive mPTP opening. Similarly,
Cour et al (42) reported
that cyclosporine A (an inhibitor of mPTP opening) attenuated
post-CA syndrome and improved short-term survival in a rabbit
model. Moreover, Zhu et al (19) reported that PGE1 pretreatment
prevented mPTP opening in a rat model of coronary
microembolization. These findings support the hypothesis that PGE1
inhibits mPTP opening.
Cytochrome c and caspase-3 are downstream
proteins in mitochondria-mediated cardiomyocyte apoptosis. When a
cell is stimulated by particular pathological factors,
macromolecules, including cytochrome c, procaspase-2, and
procaspase-9, are released through the mPTP. Cytochrome c
plays a central role in apoptosis. Once released into the cytosol,
it forms a complex known as the apoptosome (43–45).
These events allow for the activation of caspase-3, which
eventually mediates apoptosis (10–12).
To identify the mechanism underlying the antiapoptotic effects of
PGE1, the cytosolic protein levels of cytochrome c and
cleaved caspase-3 were examined. CA increased cytochrome c
and cleaved caspase-3 expression in the heart, consistent with the
findings reported by Garcia et al (46). PGE1 treatment significantly
attenuated the pathological increase in cytochrome c and
cleaved caspase-3 that were induced by CA.
The GSK3β pathway is a primary upstream mPTP
regulator (47–49). A recent study (50) indicated that a prostaglandin E
receptor subtype 4 agonist inhibited mPTP opening following
ischemia-reperfusion in hepatocytes through the GSK3β pathway. To
evaluate the mechanism by which PGE1 treatment inhibits mPTP
opening, GSK3β activation was measured. GSK3β (Ser9) inhibitory
phosphorylation was decreased after CA, whereas PGE1 treatment
caused inhibition of (Ser9) phosphorylation of GSK3β. Therefore, it
may be hypothesized that PGE1 inhibited mPTP opening by
inactivating GSK3β.
The present study exhibits certain limitations. The
CA model used was induced by asphyxia. It remains unknown whether
PGE1 also exerts cardioprotective effects in CA induced by
ventricular fibrillation. Moreover, primary adult rat cardiac
myocytes isolated from the rats would have provided more reliable
results than a cardiomyocyte cell line. However, the use H9c2 cells
undergoing H/R model remains a reasonable model for the purpose of
this study. H9c2 cell line was originally derived from embryonic
rat ventricular tissue. Although H9c2 cells are no longer able to
beat, they share several characteristics with primary
cardiomyocytes (51). The H/R model
is also widely used to simulate circulatory arrest (23,52–54)
and as a cellular model of cardiac arrest (23,53,55).
In addition, inhibitors were not used to further evaluate the role
of the GSK3β pathway in the cardioprotective mechanism of PGE1.
Lastly, the side effects of PGE1 on hemodynamics were not fully
assessed. For example, PGE1 may increase the risk of decreasing
blood pressure when used for clinical treatment. The recording of
MAP was terminated at 1 h following ROSC. Although the MAP at 1 h
following ROSC was not significantly different between the CA and
CA+PGE1 groups, further studies should be conducted to assess the
safety of PGE1 before its cardioprotective effects are investigated
in clinical studies.
In conclusion, the present study indicated that PGE1
treatment attenuated PAMD at the onset of ROSC and improved the
survival rate following CA. Its benefits were partially attributed
to inhibition of mitochondria-mediated cardiomyocyte apoptosis,
which possibly involves the GSK3β pathway. These findings offer
significant insight into developing new strategies for the
treatment of PAMD.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Key
R&D Program of China (grant no. 2017YFC0908700,
2017YFC0908703), National Natural Science Foundation of China
(grant nos. 81772036, 81671952, 81873950, 81873953, 81570401 and
81571934), National S&T Fundamental Resources Investigation
Project (2 grant nos. 018FY100600 and 2018FY100602), Taishan
Pandeng Scholar Program of Shandong Province (grant no.
tspd20181220), Taishan Young Scholar Program of Shandong Province
(grant nos. tsqn20161065 and tsqn201812129), Key R&D Program of
Shandong Province (grant nos. 2016ZDJS07A14 and 2018GSF118003) and
the Fundamental Research Funds of Shandong University (grant no.
2018JC011).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YC and JW designed the experiments. CS and XF
performed the experiments. CS and FX collected and analyzed data.
The manuscript was written by CS. FX revised the manuscript
critically for important intellectual content. All authors read and
approved the manuscript, and agree to be accountable for all
aspects of the research in ensuring that the accuracy or integrity
of any part of the work are appropriately investigated and
resolved. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Animal Use and
Care Committee of Shandong University (approval no.
KYLL-2018KS-219).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Neumar RW: Doubling cardiac arrest
survival by 2020: Achieving the American heart association impact
goal. Circulation. 134:2037–2039. 2016. View Article : Google Scholar
|
|
2
|
Andersen LW, Holmberg MJ, Berg KM, Donnino
MW and Granfeldt A: In-hospital cardiac arrest: A review. JAMA.
321:1200–1210. 2019. View Article : Google Scholar
|
|
3
|
Jentzer JC, Chonde MD, Shafton A, Abu-Daya
H, Chalhoub D, Althouse AD and Rittenberger JC: Echocardiographic
left ventricular systolic dysfunction early after resuscitation
from cardiac arrest does not predict mortality or vasopressor
requirements. Resuscitation. 106:58–64. 2016. View Article : Google Scholar
|
|
4
|
Laurent I, Monchi M, Chiche JD, Joly LM,
Spaulding C, Bourgeois B, Cariou A, Rozenberg A, Carli P, Weber S
and Dhainaut JF: Reversible myocardial dysfunction in survivors of
out-of-hospital cardiac arrest. J Am Coll Cardiol. 40:2110–2116.
2002. View Article : Google Scholar
|
|
5
|
Ruiz-Bailen M, Aguayo de Hoyos E,
Ruiz-Navarro S, Díaz-Castellanos MA, Rucabado-Aguilar L,
Gómez-Jiménez FJ, Martínez-Escobar S, Moreno RM and Fierro-Rosón J:
Reversible myocardial dysfunction after cardiopulmonary
resuscitation. Resuscitation. 66:175–181. 2005. View Article : Google Scholar
|
|
6
|
Jentzer JC, Chonde MD and Dezfulian C:
Myocardial dysfunction and shock after cardiac arrest. Biomed Res
Int. 2015:3147962015. View Article : Google Scholar
|
|
7
|
Chonde M, Flickinger KL, Sundermann ML,
Koller AC, Salcido DD, Dezfulian C, Menegazzi JJ and Elmer J:
Intra-Arrest administration of cyclosporine and methylprednisolone
does not reduce postarrest myocardial dysfunction. Biomed Res Int.
2019:65390502019. View Article : Google Scholar
|
|
8
|
Liu S, Xu J, Gao Y, Shen P, Xia S, Li Z
and Zhang M: Multi-organ protection of ulinastatin in traumatic
cardiac arrest model. World J Emerg Surg. 13:512018. View Article : Google Scholar
|
|
9
|
Heusch G, Boengler K and Schulz R:
Inhibition of mitochondrial permeability transition pore opening:
The Holy Grail of cardioprotection. Basic Res Cardiol. 105:151–154.
2010. View Article : Google Scholar
|
|
10
|
Wu HY, Huang CH, Lin YH, Wang CC and Jan
TR: Cannabidiol induced apoptosis in human monocytes through
mitochondrial permeability transition pore-mediated ROS production.
Free Radic Biol Med. 124:311–318. 2018. View Article : Google Scholar
|
|
11
|
Sileikyte J and Forte M: The mitochondrial
permeability transition in mitochondrial disorders. Oxid Med Cell
Longev. 2019:34030752019. View Article : Google Scholar
|
|
12
|
Morciano G, Bonora M, Campo G, Aquila G,
Rizzo P, Giorgi C, Wieckowski MR and Pinton P: Mechanistic role of
mPTP in ischemia-reperfusion injury. Adv Exp Med Biol. 982:169–189.
2017. View Article : Google Scholar
|
|
13
|
Huang CH, Tsai MS, Hsu CY, Su YJ, Wang TD,
Chang WT and Chen WJ: Post-cardiac arrest myocardial dysfunction is
improved with cyclosporine treatment at onset of resuscitation but
not in the reperfusion phase. Resuscitation. 82 (Suppl 2):S41–S47.
2011. View Article : Google Scholar
|
|
14
|
Cour M, Loufouat J, Paillard M, Augeul L,
Goudable J, Ovize M and Argaud L: Inhibition of mitochondrial
permeability transition to prevent the post-cardiac arrest
syndrome: A pre-clinical study. Eur Heart J. 32:226–235. 2011.
View Article : Google Scholar
|
|
15
|
Hew MR and Gerriets V: Prostaglandin E1.
StatPearls; Treasure Island, FL: 2019
|
|
16
|
Weiss T, Fischer D, Hausmann D and Weiss
C: Endothelial function in patients with peripheral vascular
disease: Influence of prostaglandin E1. Prostaglandins Leukot
Essent Fatty Acids. 67:277–281. 2002. View Article : Google Scholar
|
|
17
|
Schutte H, Lockinger A, Seeger W and
Grimminger F: Aerosolized PGE1, PGI2 and nitroprusside protect
against vascular leakage in lung ischaemia-reperfusion. Eur Respir
J. 18:15–22. 2001. View Article : Google Scholar
|
|
18
|
Johnson RG: Prostaglandin E1 and
myocardial reperfusion injury. Crit Care Med. 28:2649–2650. 2000.
View Article : Google Scholar
|
|
19
|
Zhu H, Ding Y, Xu X, Li M, Fang Y, Gao B,
Mao H, Tong G, Zhou L and Huang J: Prostaglandin E1 protects
coronary microvascular function via the glycogen synthase kinase
3β-mitochondrial permeability transition pore pathway in rat hearts
subjected to sodium laurate-induced coronary microembolization. Am
J Transl Res. 9:2520–2534. 2017.
|
|
20
|
Fang WT, Li HJ and Zhou LS: Protective
effects of prostaglandin E1 on human umbilical vein endothelial
cell injury induced by hydrogen peroxide. Acta Pharmacol Sin.
31:485–492. 2010. View Article : Google Scholar
|
|
21
|
Wei L, Zhao W, Hu Y, Wang X, Liu X, Zhang
P and Han F: Exploration of the optimal dose of HOE-642 for the
protection of neuronal mitochondrial function after cardiac arrest
in rats. Biomed Pharmacother. 110:818–824. 2019. View Article : Google Scholar
|
|
22
|
Kim T, Paine MG, Meng H, Xiaodan R, Cohen
J, Jinka T, Zheng H, Cranford JA and Neumar RW: Combined intra- and
post-cardiac arrest hypothermic-targeted temperature management in
a rat model of asphyxial cardiac arrest improves survival and
neurologic outcome compared to either strategy alone.
Resuscitation. 107:94–101. 2016. View Article : Google Scholar
|
|
23
|
Huang CH, Tsai MS, Chiang CY, Su YJ, Wang
TD, Chang WT, Chen HW and Chen WJ: Activation of mitochondrial
STAT-3 and reduced mitochondria damage during hypothermia treatment
for post-cardiac arrest myocardial dysfunction. Basic Res Cardiol.
110:592015. View Article : Google Scholar
|
|
24
|
Yin L, Yang Z, Yu H, Qian J, Zhao S, Wang
J, Wu X, Cahoon J and Tang W: Changes in sublingual
microcirculation is closely related with that of bulbar
conjunctival microcirculation in a rat model of cardiac arrest.
Shock. 45:428–433. 2016. View Article : Google Scholar
|
|
25
|
Keilhoff G, Esser T, Titze M, Ebmeyer U
and Schild L: High-potential defense mechanisms of neocortex in a
rat model of transient asphyxia induced cardiac arrest. Brain Res.
1674:42–54. 2017. View Article : Google Scholar
|
|
26
|
Uray T, Empey PE, Drabek T, Stezoski JP,
Janesko-Feldman K, Jackson T, Garman RH, Kim F, Kochanek PM and
Dezfulian C: Nitrite pharmacokinetics, safety and efficacy after
experimental ventricular fibrillation cardiac arrest. Nitric Oxide.
93:71–77. 2019. View Article : Google Scholar
|
|
27
|
Incagnoli P, Ramond A, Joyeux-Faure M,
Pepin JL, Levy P and Ribuot C: Erythropoietin improved initial
resuscitation and increased survival after cardiac arrest in rats.
Resuscitation. 80:696–700. 2009. View Article : Google Scholar
|
|
28
|
McAdams RM, McPherson RJ, Dabestani NM,
Gleason CA and Juul SE: Left ventricular hypertrophy is prevalent
in sprague-dawley rats. Comp Med. 60:357–363. 2010.
|
|
29
|
Magnet IAM, Ettl F, Schober A, Warenits
AM, Grassmann D, Wagne M, Schriefl C, Clodi C, Teubenbacher U,
Högler S, et al: Extracorporeal life support increases survival
after prolonged ventricular fibrillation cardiac arrest in the rat.
Shock. 48:674–680. 2017. View Article : Google Scholar
|
|
30
|
Huang X, Zuo L, Lv Y, Chen C, Yang Y, Xin
H, Li Y and Qian Y: Asiatic acid attenuates myocardial
Ischemia/Reperfusion injury via Akt/GSK-3β/HIF-1α signaling in rat
H9c2 Cardiomyocytes. Molecules. 21:12482016. View Article : Google Scholar
|
|
31
|
Pu Y, Wu D, Lu X and Yang L: Effects of
GCN2/eIF2α on myocardial ischemia/hypoxia reperfusion and
myocardial cells injury. Am J Transl Res. 11:5586–5598. 2019.
|
|
32
|
Kang Y: Management of post-cardiac arrest
syndrome. Acute Crit Care. 34:173–178. 2019. View Article : Google Scholar
|
|
33
|
Vognsen M, Fabian-Jessing BK, Secher N,
Løfgren B, Dezfulian C, Andersen LW and Granfeldt A: Contemporary
animal models of cardiac arrest: A systematic review.
Resuscitation. 113:115–123. 2017. View Article : Google Scholar
|
|
34
|
Piao L, Fang YH, Hamanaka RB, Mutlu GM,
Dezfulian C, Archer SL and Sharp WW: Suppression of
superoxide-hydrogen peroxide production at Site IQ of mitochondrial
Complex I attenuates myocardial stunning and improves postcardiac
arrest outcomes. Crit Care Med. 48:e133–e140. 2020. View Article : Google Scholar
|
|
35
|
Zhao Z, Qu F, Liu R and Xia Y:
Differential expression of miR-142-3p protects cardiomyocytes from
myocardial ischemia-reperfusion via TLR4/NFκB axis. J Cell Biochem.
Nov 20–2019.(Epub ahead of print). doi: 10.1002/jcb.29506.
|
|
36
|
Gu W, Li C, Yin W, Guo Z, Hou X and Zhang
D: Shen-fu injection reduces postresuscitation myocardial
dysfunction in a porcine model of cardiac arrest by modulating
apoptosis. Shock. 38:301–306. 2012. View Article : Google Scholar
|
|
37
|
Kuznetsov AV, Javadov S, Margreiter R,
Grimm M, Hagenbuchner J and Ausserlechner MJ: The role of
mitochondria in the mechanisms of Cardiac Ischemia-Reperfusion
injury. Antioxidants (Basel). 8:4542019. View Article : Google Scholar
|
|
38
|
Zhou X, Qu Y, Gan G, Zhu S, Huang Y, Liu
Y, Zhu J, Xie B and Tan Z: Cyclosporine a plus ischemic
postconditioning improves neurological function in rats after
cardiac resuscitation. Neurocrit Care. 32:812–821. 2020. View Article : Google Scholar
|
|
39
|
Zheng JH, Xie L, Li N, Fu ZY, Tan XF, Tao
R, Qin T and Chen MH: PD98059 protects the brain against
mitochondrial-mediated apoptosis and autophagy in a cardiac arrest
rat model. Life Sci. 232:1166182019. View Article : Google Scholar
|
|
40
|
Wang H, Chen S, Zhang Y, Xu H and Sun H:
Electroacupuncture ameliorates neuronal injury by
Pink1/Parkin-mediated mitophagy clearance in cerebral
ischemia-reperfusion. Nitric Oxide. 91:23–34. 2019. View Article : Google Scholar
|
|
41
|
Sun T, Ding W, Xu T, Ao X, Yu T, Li M, Liu
Y, Zhang X, Hou L and Wang J: Parkin regulates programmed necrosis
and myocardial ischemia/reperfusion injury by targeting
cyclophilin-D. Antioxid Redox Signal. 31:1177–1193. 2019.
View Article : Google Scholar
|
|
42
|
Cour M, Abrial M, Jahandiez V, Loufouat J,
Belaïdi E, Gharib A, Varennes A, Monneret G, Thibault H, Ovize M
and Argaud L: Ubiquitous protective effects of cyclosporine A in
preventing cardiac arrest-induced multiple organ failure. J Appl
Physiol (1985). 117:930–936. 2014. View Article : Google Scholar
|
|
43
|
Chalkias A, Kuzovlev A, Noto A, d'Aloja E
and Xanthos T: Identifying the role of cytochrome c in
post-resuscitation pathophysiology. Am J Emerg Med. 33:1826–1830.
2015. View Article : Google Scholar
|
|
44
|
Sun L, Jia H, Ma L, Yu M, Yang Y, Liu Y,
Zhang H and Zou Z: Metabolic profiling of hypoxia/reoxygenation
injury in H9c2 cells reveals the accumulation of phytosphingosine
and the vital role of Dan-Shen in Xin-Ke-Shu. Phytomedicine.
49:83–94. 2018. View Article : Google Scholar
|
|
45
|
Borutaite V and Brown GC: Mitochondria in
apoptosis of ischemic heart. FEBS Lett. 541:1–5. 2003. View Article : Google Scholar
|
|
46
|
Garcia NA, Moncayo-Arlandi J, Vazquez A,
Genovés P, Calvo CJ, Millet J, Martí N, Aguado C, Knecht E,
Valiente-Alandi I, et al: Hydrogen sulfide improves cardiomyocyte
function in a cardiac arrest model. Ann Transplant. 22:285–295.
2017. View Article : Google Scholar
|
|
47
|
Yang K, Chen Z, Gao J, Shi W, Li L, Jiang
S, Hu H, Liu Z, Xu D and Wu L: The key roles of GSK-3β in
regulating mitochondrial activity. Cell Physiol Biochem.
44:1445–1459. 2017. View Article : Google Scholar
|
|
48
|
Nikolaou PE, Boengler K, Efentakis P,
Vouvogiannopoulou K, Zoga A, Gaboriaud-Kolar N, Myrianthopoulos V,
Alexakos P, Kostomitsopoulos N, Rerras I, et al: Investigating and
re-evaluating the role of glycogen synthase kinase 3 beta kinase as
a molecular target for cardioprotection by using novel
pharmacological inhibitors. Cardiovasc Res. 115:1228–1243. 2019.
View Article : Google Scholar
|
|
49
|
Tanaka T, Saotome M, Katoh H, Satoh T,
Hasan P, Ohtani H, Satoh H, Hayashi H and Maekawa Y: Glycogen
synthase kinase-3β opens mitochondrial permeability transition pore
through mitochondrial hexokinase II dissociation. J Physiol Sci.
68:865–871. 2018. View Article : Google Scholar
|
|
50
|
Cai LL, Xu HT, Wang QL, Zhang YQ, Chen W,
Zheng DY, Liu F, Yuan HB, Li YH and Fu HL: EP4 activation
ameliorates liver ischemia/reperfusion injury via
ERK1/2GSK3β-dependent MPTP inhibition. Int J Mol Med. 45:1825–1837.
2020.
|
|
51
|
Kimes BW and Brandt BL: Properties of a
clonal muscle cell line from rat heart. Exp Cell Res. 98:367–381.
1976. View Article : Google Scholar
|
|
52
|
Kwon WY, Suh GJ, Kim KS, Jung YS, Kim SH,
Lee AR, You KM and Park MJ: Niacin and selenium attenuate brain
injury after cardiac arrest in rats by up-regulating DJ-1-akt
signaling. Crit Care Med. 46:e788–e796. 2018. View Article : Google Scholar
|
|
53
|
Zhang R, Liu B, Fan X, Wang W, Xu T, Wei
S, Zheng W, Yuan Q, Gao L, Yin X, et al: Aldehyde dehydrogenase 2
protects against post-cardiac arrest myocardial dysfunction through
a novel mechanism of suppressing mitochondrial reactive oxygen
species production. Front Pharmacol. 11:3732020. View Article : Google Scholar
|
|
54
|
Zhou T, Lin H, Jiang L, Yu T, Zeng C, Liu
J and Yang Z: Mild hypothermia protects hippocampal neurons from
oxygen-glucose deprivation injury through inhibiting caspase-3
activation. Cryobiology. 80:55–61. 2018. View Article : Google Scholar
|
|
55
|
Yeh CH, Chen TP, Wang YC, Lin YM and Fang
SW: MicroRNA-27a regulates cardiomyocytic apoptosis during
cardioplegia-induced cardiac arrest by targeting interleukin
10-related pathways. Shock. 38:607–614. 2012. View Article : Google Scholar
|