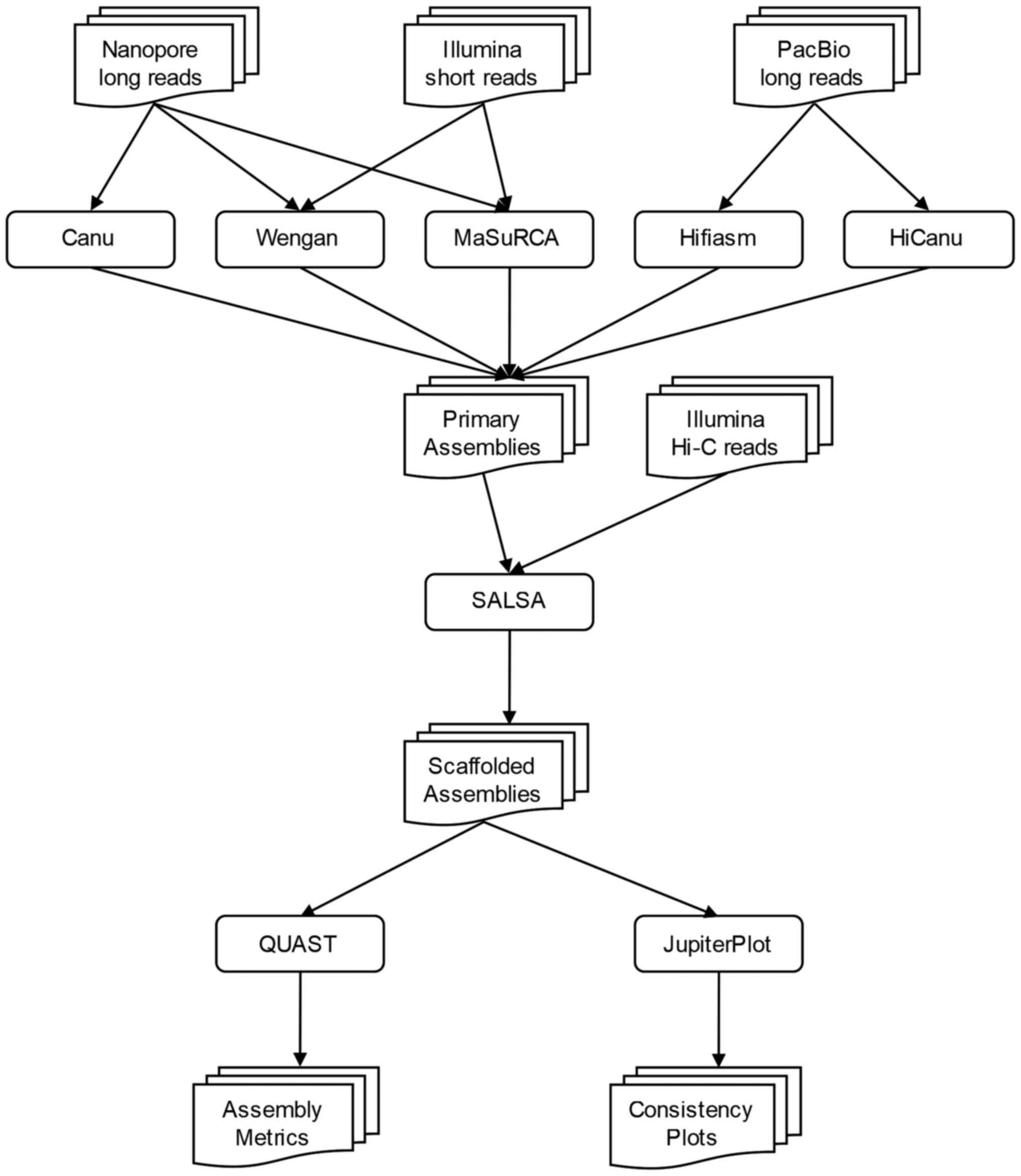
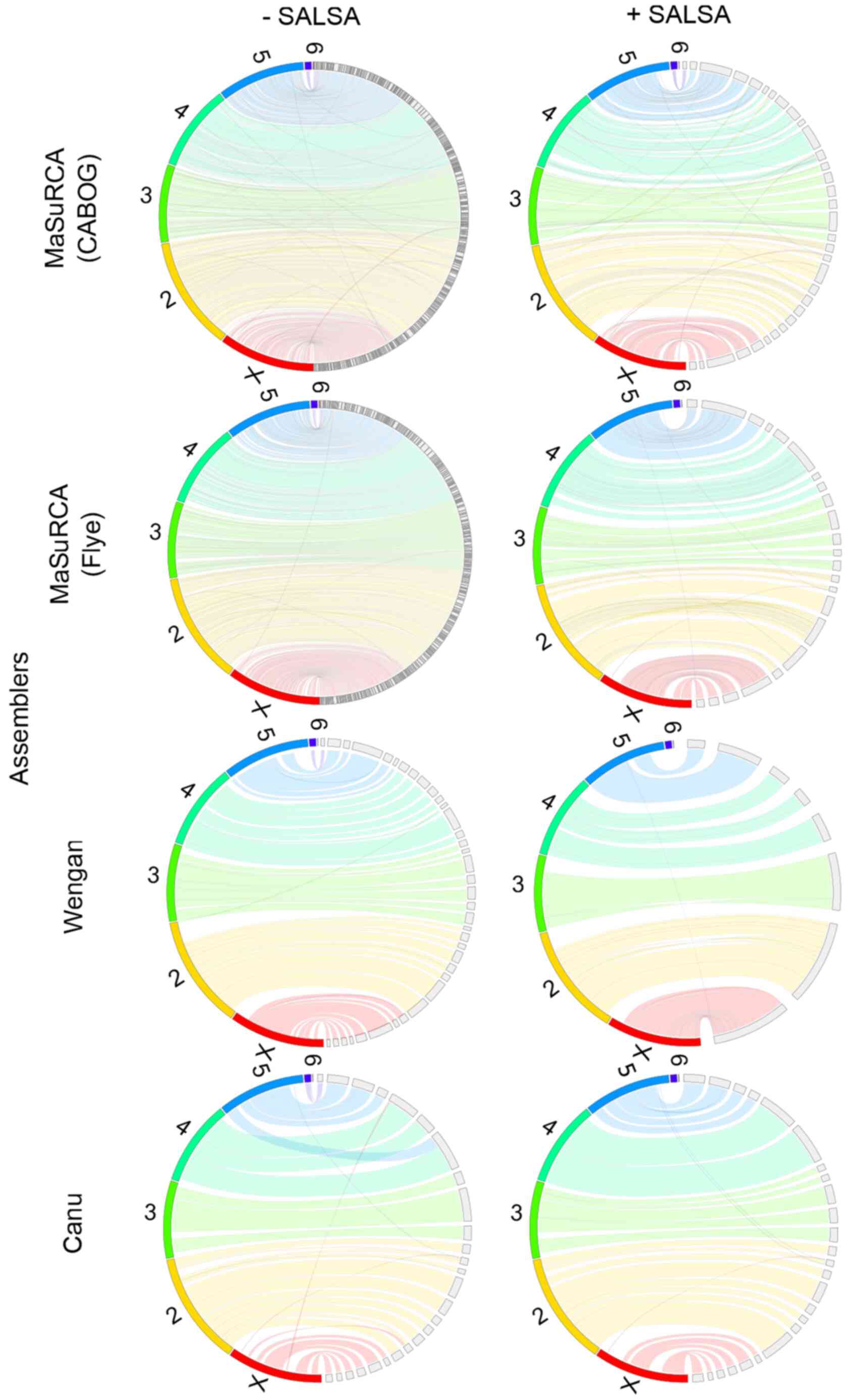
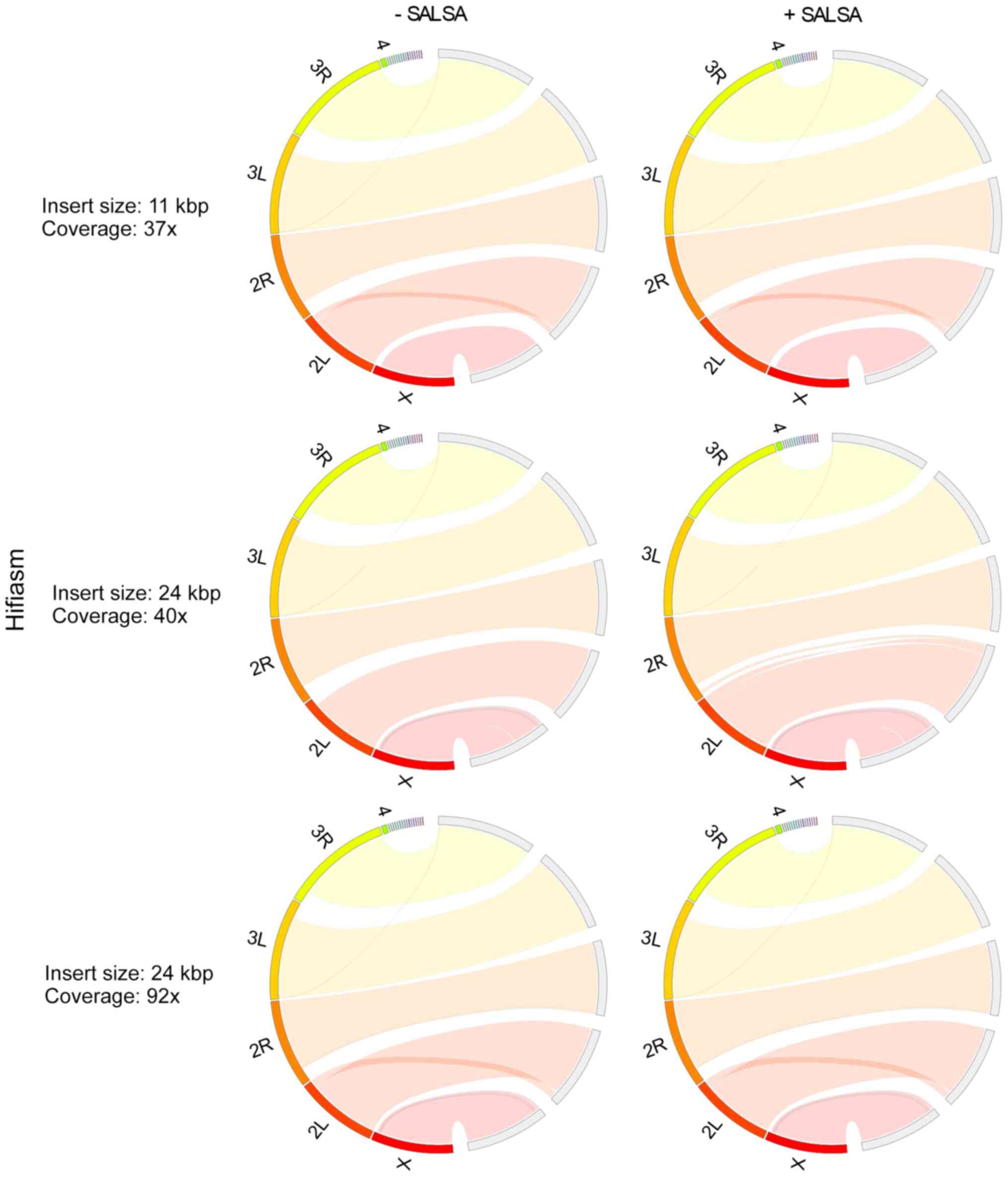
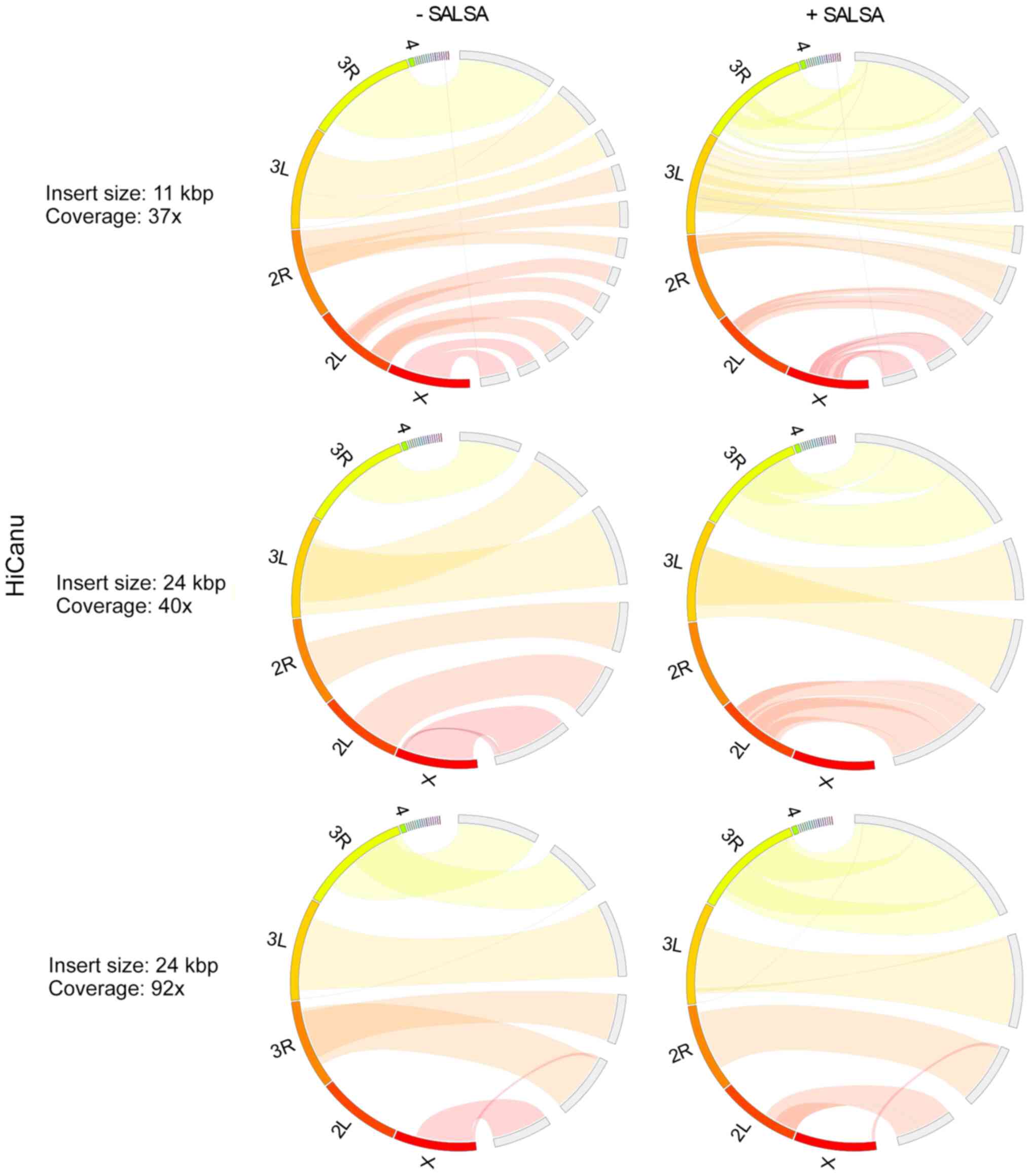
|
1
|
Lander ES, Linton LM, Birren B, Nusbaum C,
Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al
International Human Genome Sequencing Consortium, : Initial
sequencing and analysis of the human genome. Nature. 409:860–921.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sanger F, Nicklen S and Coulson AR: DNA
sequencing with chain-terminating inhibitors. Proc Natl Acad Sci
USA. 74:5463–5467. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kent WJ and Haussler D: Assembly of the
working draft of the human genome with GigAssembler. Genome Res.
11:1541–1548. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shendure J, Balasubramanian S, Church GM,
Gilbert W, Rogers J, Schloss JA and Waterston RH: DNA sequencing at
40: Past, present and future. Nature. 550:345–353. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Salzberg SL and Yorke JA: Beware of
mis-assembled genomes. Bioinformatics. 21:4320–4321. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chaisson MJ, Wilson RK and Eichler EE:
Genetic variation and the de novo assembly of human genomes. Nat
Rev Genet. 16:627–640. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
van Dijk EL, Jaszczyszyn Y, Naquin D and
Thermes C: The Third Revolution in sequencing technology. Trends
Genet. 34:666–681. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kasianowicz JJ, Brandin E, Branton D and
Deamer DW: Characterization of individual polynucleotide molecules
using a membrane channel. Proc Natl Acad Sci USA. 93:13770–13773.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Haque F, Li J, Wu HC, Liang XJ and Guo P:
Solid-state and biological nanopore for real-time sensing of single
chemical and sequencing of DNA. Nano Today. 8:56–74. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Eid J, Fehr A, Gray J, Luong K, Lyle J,
Otto G, Peluso P, Rank D, Baybayan P, Bettman B, et al: Real-time
DNA sequencing from single polymerase molecules. Science.
323:133–138. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Aird D, Ross MG, Chen W-S, Danielsson M,
Fennell T, Russ C, Jaffe DB, Nusbaum C and Gnirke A: Analyzing and
minimizing PCR amplification bias in Illumina sequencing libraries.
Genome Biol. 12:R182011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jain M, Koren S, Miga KH, Quick J, Rand
AC, Sasani TA, Tyson JR, Beggs AD, Dilthey AT, Fiddes IT, et al:
Nanopore sequencing and assembly of a human genome with ultra-long
reads. Nat Biotechnol. 36:338–345. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lin Y, Yuan J, Kolmogorov M, Shen MW,
Chaisson M and Pevzner PA: Assembly of long error-prone reads using
de Bruijn graphs. Proc Natl Acad Sci USA. 113:E8396–E8405. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tan MH, Austin CM, Hammer MP, Lee YP,
Croft LJ and Gan HM: Finding Nemo: Hybrid assembly with Oxford
Nanopore and Illumina reads greatly improves the clownfish
(Amphiprion ocellaris) genome assembly. Gigascience. 7:1–6.
2018. View Article : Google Scholar
|
|
15
|
Nowak RM, Jastrzębski JP, Kuśmirek W,
Sałamatin R, Rydzanicz M, Sobczyk-Kopcioł A, Sulima-Celińska A,
Paukszto Ł, Makowczenko KG, Płoski R, et al: Hybrid de novo
whole-genome assembly and annotation of the model tapeworm
Hymenolepis diminuta. Sci Data. 6:3022019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Korlach J and Turner SW: Zero-Mode
Waveguides. Encyclopedia of Biophysics. Roberts GC: Springer;
Heidelberg: pp. 2793–2795. 2013, View Article : Google Scholar
|
|
17
|
Wenger AM, Peluso P, Rowell WJ, Chang PC,
Hall RJ, Concepcion GT, Ebler J, Fungtammasan A, Kolesnikov A,
Olson ND, et al: Accurate circular consensus long-read sequencing
improves variant detection and assembly of a human genome. Nat
Biotechnol. 37:1155–1162. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Silvestre-Ryan J and Holmes I: Pair
consensus decoding improves accuracy of neural network basecallers
for nanopore sequencing. Genome Biol. 22:382021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ghurye J and Pop M: Modern technologies
and algorithms for scaffolding assembled genomes. PLoS Comput Biol.
15:e10069942019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lieberman-Aiden E, van Berkum NL, Williams
L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ,
Dorschner MO, et al: Comprehensive mapping of long-range
interactions reveals folding principles of the human genome.
Science. 326:289–293. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Auton A, Brooks LD, Durbin RM, Garrison
EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA and
Abecasis GR; 1000 Genomes Project Consortium, : A global reference
for human genetic variation. Nature. 526:68–74. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Koepfli KP, Paten B and O'Brien SJ; Genome
10K Community of Scientists, : The Genome 10K Project: A way
forward. Annu Rev Anim Biosci. 3:57–111. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
ICGC/TCGA Pan-Cancer Analysis of Whole
Genomes Consortium: Pan-cancer analysis of whole genomes. Nature.
578:82–93. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Logsdon GA, Vollger MR, Hsieh P, Mao Y,
Liskovykh MA, Koren S, Nurk S, Mercuri L, Dishuck PC and Rhie A:
The structure, function, and evolution of a complete human
chromosome 8. bioRxiv. Sep 8–2020.(Epub ahead of print).
https://doi.org/10.1101/2020.09.08.285395.
|
|
25
|
Miga KH, Koren S, Rhie A, Vollger MR,
Gershman A, Bzikadze A, Brooks S, Howe E, Porubsky D, Logsdon GA,
et al: Telomere-to-telomere assembly of a complete human X
chromosome. Nature. 585:79–84. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Amid C, Alako BTF, Balavenkataraman
Kadhirvelu V, Burdett T, Burgin J, Fan J, Harrison PW, Holt S,
Hussein A, Ivanov E, et al: The European nucleotide archive in
2019. Nucleic Acids Res. 48:D70–D76. 2020.PubMed/NCBI
|
|
27
|
Zimin AV, Marçais G, Puiu D, Roberts M,
Salzberg SL and Yorke JA: The MaSuRCA genome assembler.
Bioinformatics. 29:2669–2677. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zimin AV, Puiu D, Luo MC, Zhu T, Koren S,
Marçais G, Yorke JA, Dvořák J and Salzberg SL: Hybrid assembly of
the large and highly repetitive genome of Aegilops tauschii,
a progenitor of bread wheat, with the MaSuRCA mega-reads algorithm.
Genome Res. 27:787–792. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Di Genova A, Buena-Atienza E, Ossowski S
and Sagot MF: Wengan: Efficient and high quality hybrid de novo
assembly of human genomes. bioRxiv. Nov 25–2019.(Epub ahead of
print). doi: https://doi.org/10.1101/840447.
|
|
30
|
Miller JR, Delcher AL, Koren S, Venter E,
Walenz BP, Brownley A, Johnson J, Li K, Mobarry C and Sutton G:
Aggressive assembly of pyrosequencing reads with mates.
Bioinformatics. 24:2818–2824. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan
J, He G, Chen Y, Pan Q, Liu Y, et al: SOAPdenovo2: An empirically
improved memory-efficient short-read de novo assembler.
Gigascience. 1:182012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kolmogorov M, Yuan J, Lin Y and Pevzner
PA: Assembly of long, error-prone reads using repeat graphs. Nat
Biotechnol. 37:540–546. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Weisenfeld NI, Yin S, Sharpe T, Lau B,
Hegarty R, Holmes L, Sogoloff B, Tabbaa D, Williams L, Russ C, et
al: Comprehensive variation discovery in single human genomes. Nat
Genet. 46:1350–1355. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Koren S, Walenz BP, Berlin K, Miller JR,
Bergman NH and Phillippy AM: Canu: Scalable and accurate long-read
assembly via adaptive k-mer weighting and repeat separation. Genome
Res. 27:722–736. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Myers EW, Sutton GG, Delcher AL, Dew IM,
Fasulo DP, Flanigan MJ, Kravitz SA, Mobarry CM, Reinert KH,
Remington KA, et al: A whole-genome assembly of Drosophila.
Science. 287:2196–2204. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cheng H, Concepcion GT, Feng X, Zhang H
and Li H: Haplotype-resolved de novo assembly with phased assembly
graphs. arXiv. Aug 3–2020.(Epub ahead of print).
|
|
37
|
Nurk S, Walenz BP, Rhie A, Vollger MR,
Logsdon GA, Grothe R, Miga KH, Eichler EE, Phillippy AM and Koren
S: HiCanu: Accurate assembly of segmental duplications, satellites,
and allelic variants from high-fidelity long reads. Genome Res.
30:1291–1305. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chin C-S, Peluso P, Sedlazeck FJ,
Nattestad M, Concepcion GT, Clum A, Dunn C, O'Malley R,
Figueroa-Balderas R, Morales-Cruz A, et al: Phased diploid genome
assembly with single-molecule real-time sequencing. Nat Methods.
13:1050–1054. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Arima Genomics, Inc., . Arima-HiC Mapping
Pipeline. San Diego. 2019, GitHub. https://github.com/ArimaGenomics/mapping_pipeline/blob/master/Arima_Mapping_UserGuide_A160156_v02.pdf
|
|
40
|
Ghurye J, Rhie A, Walenz BP, Schmitt A,
Selvaraj S, Pop M, Phillippy AM and Koren S: Integrating Hi-C links
with assembly graphs for chromosome-scale assembly. PLoS Comput
Biol. 15:e10072732019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gurevich A, Saveliev V, Vyahhi N and
Tesler G: QUAST: Quality assessment tool for genome assemblies.
Bioinformatics. 29:1072–1075. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Earl D, Bradnam K, St John J, Darling A,
Lin D, Fass J, Yu HO, Buffalo V, Zerbino DR, Diekhans M, et al:
Assemblathon 1: A competitive assessment of de novo short read
assembly methods. Genome Res. 21:2224–2241. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Seppey M, Manni M and Zdobnov EM: BUSCO:
Assessing genome assembly and annotation completeness. Methods Mol
Biol. 1962:227–245. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chu J: Jupiter Plot: A circos-based tool
to visualize genome assembly consistency. 2018, GitHub. https://github.com/JustinChu/JupiterPlot/find/master
|
|
45
|
Krzywinski M, Schein J, Birol I, Connors
J, Gascoyne R, Horsman D, Jones SJ and Marra MA: Circos: An
information aesthetic for comparative genomics. Genome Res.
19:1639–1645. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kyriakidis K, Charalampidou A, Natsiavas
P, Vizirianakis IS and Malousi A: Linking exome sequencing data
with drug response aberrations. Stud Health Technol Inform.
264:1845–1846. 2019.PubMed/NCBI
|
|
47
|
Wei X, Ju X, Yi X, Zhu Q, Qu N, Liu T,
Chen Y, Jiang H, Yang G, Zhen R, et al: Identification of sequence
variants in genetic disease-causing genes using targeted
next-generation sequencing. PLoS One. 6:e295002011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kanakoglou DS, Michalettou TD, Vasileiou
C, Gioukakis E, Maneta D, Kyriakidis KV, Georgakilas AG and
Michalopoulos I: Effects of high-dose ionizing radiation in human
gene expression: A meta-analysis. Int J Mol Sci. 21:212020.
View Article : Google Scholar
|
|
49
|
DePristo MA, Banks E, Poplin R, Garimella
KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA,
Hanna M, et al: A framework for variation discovery and genotyping
using next-generation DNA sequencing data. Nat Genet. 43:491–498.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
50
|
Vollger MR, Logsdon GA, Audano PA,
Sulovari A, Porubsky D, Peluso P, Wenger AM, Concepcion GT,
Kronenberg ZN, Munson KM, et al: Improved assembly and variant
detection of a haploid human genome using single-molecule,
high-fidelity long reads. Ann Hum Genet. 84:125–140. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chen Z, Erickson DL and Meng J:
Benchmarking hybrid assembly approaches for genomic analyses of
bacterial pathogens using Illumina and Oxford Nanopore sequencing.
BMC Genomics. 21:6312020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Johnson LK, Sahasrabudhe R, Gill JA, Roach
JL, Froenicke L, Brown CT and Whitehead A: Draft genome assemblies
using sequencing reads from Oxford Nanopore Technology and Illumina
platforms for four species of North American Fundulus killifish.
Gigascience. 9:92020. View Article : Google Scholar
|
|
53
|
Coombe L, Zhang J, Vandervalk BP, Chu J,
Jackman SD, Birol I and Warren RL: ARKS: Chromosome-scale
scaffolding of human genome drafts with linked read kmers. BMC
Bioinformatics. 19:2342018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yeo S, Coombe L, Warren RL, Chu J and
Birol I: ARCS: Scaffolding genome drafts with linked reads.
Bioinformatics. 34:725–731. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lam ET, Hastie A, Lin C, Ehrlich D, Das
SK, Austin MD, Deshpande P, Cao H, Nagarajan N, Xiao M, et al:
Genome mapping on nanochannel arrays for structural variation
analysis and sequence assembly. Nat Biotechnol. 30:771–776. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wallberg A, Bunikis I, Pettersson OV,
Mosbech MB, Childers AK, Evans JD, Mikheyev AS, Robertson HM,
Robinson GE and Webster MT: A hybrid de novo genome assembly of the
honeybee, Apis mellifera, with chromosome-length scaffolds.
BMC Genomics. 20:2752019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Crosswell LC and Thornton JM: ELIXIR: A
distributed infrastructure for European biological data. Trends
Biotechnol. 30:241–242. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kodama Y, Shumway M and Leinonen R;
International Nucleotide Sequence Database Collaboration, : The
sequence read archive: Explosive growth of sequencing data. Nucleic
Acids Res. 40:D54–D56. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Leinonen R, Sugawara H and Shumway M;
International Nucleotide Sequence Database Collaboration, : The
sequence read archive. Nucleic Acids Res. 39:D19–D21. 2011.
View Article : Google Scholar : PubMed/NCBI
|