Introduction
Preeclampsia (PE) is a systemic disorder of
pregnancy characterized by the onset of high blood pressure and
proteinuria. The clinical manifestations of PE include maternal
placental hypoxia, endothelial dysfunction, and imbalance of
angiogenic factors (1). PE is a
complication in over 5% of human pregnancies and a leading cause of
feto-maternal morbidity and mortality (2).
Although PE ranks as one of the most critical
problems in obstetrics, its etiology remains unknown. It is widely
accepted that the dysfunction of the placenta due to placental
ischemia and hypoxia result in PE (3). Poor placental perfusion caused by
failure of the trophoblast cells to replace the spiral artery and
imbalanced angiogenic factors increase placental oxidative stress
(4). Vascular endothelial growth
factor (VEGF) and its receptors play crucial roles in placental
angiogenesis. VEGF activates two high-affinity receptor tyrosine
kinases, fms-like tyrosine kinase-1 (Flt-1) and fetal liver kinase
1 (Flk-1), to mediate its angiogenic functions. However, binding of
the soluble form of Flt-1 (sFlt-1) with VEGF prevents its
interaction with endogenous receptors and antagonizes the
VEGF-mediated proangiogenic activity. In PE, circulating levels of
sFlt-1 levels are high, and thus, the levels of free VEGF are
reduced in maternal circulation (5). These conditions lead to fetal growth
restriction and low placental weight (6).
The imbalance in the levels of placental steroid
hormones and steroidogenic enzymes is associated with PE
pathogenesis. The placenta is the major endocrine organ during
pregnancy and secretes several steroid hormones. The multiple
steroid hormones are produced by a process called steroidogenesis,
which is mediated by steroidogenic enzymes (7). Pregnenolone (P5) is synthesized by
cholesterol side-chain cleavage enzyme (CYP11A1) and converted into
progesterone (P4) or dehydroepiandrosterone (DHEA) by
3β-hydroxysteroid dehydrogenase/δ5 4-isomerase type 1 (HSD3B1) or
17α-hydroxylase/17,20-lyase (CYP17A1), respectively. The enzymes
including 17β-dehydrogenase 3 (HSD17B3) and HSD3B1 catalyze the
formation of androgens, such as testosterone (T), from DHEA and P5.
The final step of steroidogenesis, estrone (E1) and estradiol (E2)
biosynthesis, is mediated by the aromatase cytochrome P450
(CYP19A1) and HSD17B. A previous study has shown that the levels of
P4 and E2 were downregulated in women with PE, and their levels
were modulated by the expression of steroidogenic enzymes in the PE
placenta (8).
To study the various aspects of PE, several in
vitro and in vivo models have been established (4). One of the well-known models is
established by reducing or depleting oxygen in the trophoblast
cells and the placenta. Hypoxic conditions tend to inhibit
placental invasion and trigger symptoms of PE (9). Inhibition of nitric oxide synthase
with N-nitro-L-arginine methyl ester hydrochloride (L-NAME) is also
known to mimic PE (10). The
combination of a deficiency of nitric oxide (NO) and an increase in
peroxynitrite (ONOO−) results in the majority of
pathological changes associated with PE, such as high blood
pressure, proteinuria, and increased glomerular filtration rate
(11). Catechol-o-methyltransferase
(COMT) metabolizes 2-hydroxyestradiol to 2-methoxyoestradiol
(2-ME), which is involved in the biosynthesis of E2. COMT
expression increases with the progression of pregnancy, generating
higher levels 2-ME. Pregnant animals deficient in COMT develop
PE-like conditions, including proteinuria and placental hypoxia
(12). Since PE is caused by a
reduction in uterine blood flow due to abnormal trophoblast
invasion of the spiral arteries, the reduced uterine perfusion
pressure (RUPP) operation generates a suitable PE animal model
(1). The RUPP rat model mimics the
physiological features of PE, including hypertension and
proteinuria, and exhibits increased plasma and placental sFlt-1 and
decreased plasma VEGF and placental growth factor (13,14).
Although these models have some features of PE, they are not
representative of the full spectrum of the human disease because
the etiology of PE is most likely multifactorial and may have
several forms (15).
Due to the life-threatening risk of PE and the lack
of effective treatment options, there have been many attempts to
understand the pathogenesis of PE. Recently, many studies
addressing the relationship between human PE and steroid hormones
have been conducted (8,10,16).
However, only steroid hormones in human PE have been studied, and
the in vitro and in vivo PE conditions have not been
ascertained. To understand the entire steroidogenic machinery and
its underlying mechanisms in PE, we established various cellular
and animal PE models and compared the levels of steroid hormones
during steroidogenesis. Additionally, the levels of steroid
hormones in early-stage pregnant women were investigated, who
ultimately showed PE symptoms at full term.
Materials and methods
Cell lines and culture
The BeWo human choriocarcinoma-derived cell line
(Korean Cell Line Bank, Seoul, Republic of Korea) were seeded on
6-well plates (7×105 cells/well) in Dulbecco's modified
Eagle medium (DMEM; Hyclone) containing 10% fetal bovine plasma
(FBS; Hyclone), 100 IU/ml of penicillin, and 100 µg/ml of
streptomycin and grown at 5% CO2 at 37°C. After 24 h
(~70%), the cells were placed in a hypoxic chamber (Modular
Incubator Chamber; Billups-Rothenberg) to establish a hypoxic model
witsh inflow and outflow connectors, and the hypoxic gas consisted
of 2% O2, 5% CO2, and 93% N2.
Experiments were conducted in an environment at 37°C by placing the
chambers in an incubator for 24 h. For other in vitro PE
models, the cells were treated with L-NAME (100 µM; Sigma-Aldrich),
Ro 41-0960 (COMT-I; 10 µM; Sigma-Aldrich), or EtOH as a vehicle
control for 24 h in a 37°C incubator. The cell culture medium was
extracted for ELISA assays. Restore western blot stripping buffer
was obtained from Pierce. All in vitro experiments were
performed at least three times in triplicate.
Measurement of cell viability
The BeWo cells were cultured in 24-well plate with
density of 3×104 cells/well in triplicate and 10% FBS
for 24 h under various conditions: Vehicle control group, hypoxic
condition group, L-NAME (100 µM) group, and COMT-I (10 µM) group.
Then methyl thiazolyl tetrazolium (MTT; 5 mg/ml; Sigma-Aldrich) was
added and incubated 4 h, after that we used 100% DMSO to dissolved
the generated formazan crystals. The absorbance of the crystalline
at 570 nm was analyzed with a microplate spectrophotometer (Epoch
Bioteck, Instruments). The cell viability of treatment groups were
represented as the percentage of viable cells relative to cell
viability of the control group.
Animals
Animal studies were approved (approval number;
PNU-2017-1452) by the Ethics Committee of Pusan National University
(Busan). Twenty-five pregnant female Sprague-Dawley [gestational
day (GD) 1] rats were purchased from Central Lab. Animal Inc.
(Seoul). The rats were randomly divided into five groups as
follows: Control group (CON group; n=5), L-NAME group (n=5),
COMT-inhibitor group (COMT-I; n=5), Sham group (n=5), and RUPP
group (n=5). The rats were housed under standard laboratory
conditions with controlled temperature/humidity, a 12:12-h
light/dark cycle, and free access to food and water at the Pusan
National University Laboratory Animal Resources Center. This
facility is accredited by the Korea Food and Drug Administration
(AFDA) in accordance with the Laboratory Animal Act (Accredited
Unit Number-000231) and AAALAC International according to the
National Institutes of Health guidelines (Accredited Unit
Number;001525).
Drugs and chemicals
The L-NAME and COMT-I groups were subcutaneously
(SC) injected daily with L-NAME (50 mg/kg/day/200 µl) and COMT-I
(2.5 mg/kg/day/200 µl) from GD 10 to 17 (17,18).
The CON group was administrated 0.9% saline (200 µl). The dosage
was adjusted according to changes in body weight (BW). BW, clinical
signs, and abnormal behavior were recorded daily throughout the
experimental period.
RUPP operation
On GD 14, Sham and RUPP groups were anesthetized
with intraperitoneal injection of Zoletil 50 (Virbac, Carros,
France) 20 mg/kg and Rompun (Bayer Korea) 5 mg/kg solution. A
mid-abdominal incision was made as suggested by Fushima et
al (3). After the midline
incision, a ligature was tied around the aorta above the iliac
bifurcation. Since compensatory blood flow to the placenta occurs
via an adaptive increase in ovarian blood flow, both right and left
uterine arcades are also tied. As complete ligation killed the dams
within a day or two of the operation, the aorta and arcades were
tied with a nylon thread 0.35 mm in diameter, followed by removal
of the thread to provide a small space for minimal circulation of
blood. These procedures reduce uterine blood flow in the gravid rat
by 40%. The sham group was operated on similarly, but without
ligation. The abdominal incision was then sutured.
Measurement of blood pressure and
urinary protein
Blood pressure, especially the systolic blood
pressure, is the key criterion when evaluating a pre-eclamptic
model (19). From GD 10, the
baseline systolic blood pressures were obtained with a blood
pressure monitor for mice and rats (CODA™ Monitor; Kent Scientific
Corporation) after the rats were pre-warmed on a warming platform
in rat holders. The systolic blood pressure measurements were
performed in triplicate, and the mean for each rat was recorded
till GD 18. To measure the urinary protein level, rats were placed
in metabolic cages on GD 17, and urine was collected over 24 h. The
urine was centrifuged at 10,000 × g for 10 min at 4°C and
immediately stored at −80°C. Protein levels in the urine were
measured using a BCA protein assay kit (Pierce Biotechnology).
Sample collection
On GD 18, the rats were euthanized in gradually
filled CO2 gas chamber with a flow rate of ≤30%
CO2 of the chamber volume/min. Rats were euthanized in
2018, which corresponds to the period approved by the Ethics
Committee of Pusan National University (approval number;
PNU-2017-1452). Approximately 1 ml of blood from the inferior vena
cava was collected in plastic tubes under aseptic conditions with
EDTA as an anticoagulant and centrifuged at 12,000 rpm for 10 min
at 4°C to separate the plasma. The plasma was collected and
immediately frozen at −80°C until analysis. The placentas were
weighed and stored at −80°C for western blot analysis. The kidneys
were harvested, fixed in 10% formalin and embedded in paraffin for
histological analysis. The fetuses were isolated to be weighed and
counted. The fetal resorption rate was obtained as percent fetal
resorption = (number of absorbed fetuses/total number of fetuses)
×100, as previously reported (20).
Western blot analysis
Protein samples were prepared with a Pro-prep
solution (iNtRON Biotechnology). A total of 20 µg protein was
analyzed by 10–12% sodium dodecyl sulfate polyacrylamide gel
electrophoresis (SDS-PAGE) and then transferred to nitrocellulose
membranes (Daeillab Service Co, Ltd.). Membranes were subsequently
blocked for 1 h with 5% skim milk (Difco) in Tris-buffered saline
(TBS) with 0.05% Tween-20 (TBS-T). Following blocking, membranes
were incubated with hypoxia inducible factor 1 subunit α (HIF1A,
1:500; Santa Cruz Biotechnology, Inc.; cat. no. sc53546), VEGF
(1:1,000; Abcam; cat. no. ab46154), sFlt-1 (1:1,000; Abcam; cat.
no. ab32152), CYP11A1 (1:1,000; Cell Signaling Technology Inc.;
cat. no. 14217), CYP17A1 (1:500; Santa Cruz Biotechnology, Inc.;
cat. no. sc46084), HSD3B1 (1:2,000; Santa Cruz Biotechnology, Inc.;
cat. no. sc30820), HSD17B3 (1:2,000; Abcam; cat. no. ab55268), and
CYP19A1 (1:500; Santa Cruz Biotechnology, Inc.; cat no. sc14244)
antibodies overnight at 4°C, followed by horseradish peroxidase
(HRP)-conjugated secondary antibodies (all 1:2,000; Santa Cruz
Biotechnology, Inc.; cat. no. sc2313, sc2005, sc2020). Luminol
reagent (Bio-Rad) was used to visualize antibody binding. Membranes
were subsequently probed with an antibody against β-actin (1:3,000;
Cell Signaling Technology Inc.; cat. no. 8457) as an internal
control. For direct comparisons between the concentration levels of
different signaling molecules, membranes were stripped and
re-probed using Restore western blot stripping buffer as detailed
by the manufacturer and re-probed after verifying the absence of
residual HRP activity of the membrane. Each blot was scanned using
a Bio-Rad ChemiDoc XRS (Bio-Rad), and band intensities were
normalized to β-actin levels.
ELISA assays
Concentrations of P5 (Alpco; cat. no. KA1912), P4
(Cayman Chemical Company; cat. no. 582601), DHEA (Enzo Life
Sciences, Inc.; cat. no. 20-DHEHU-E01), T (Cayman Chemical Company;
cat. no. 582701), E2 (Cayman Chemical Company; cat. no. 582251),
VEGF (R&D Systems, Inc.; cat. no. RRV00), and sFlt-1 (R&D
Systems, Inc.; cat. no. MVR100) were measured using competitive
enzyme immunoassay kits, according to the manufacturers' protocols.
The culture medium from BeWo cells, plasma and placenta tissues
were added to 96-well plates. The plate was incubated for 1 h at
room temperature on an orbital shaker. Following incubation at room
temperature for 90 min with Ellman's reagent, the optical density
was measured spectrophotometrically at 420 nm. The final
concentrations were calculated using standard curve analysis.
Histological analysis
Four micrometer paraffin sections of rat kidney
tissues were stained with hematoxylin and eosin (Sigma-Aldrich)
according to the standard protocol. Tissue images were captured at
×400 using a Leica DM 500 microscope (Leica).
Statistical analysis
Results are presented as the mean ± standard
deviation (SD). Data were analyzed for statistical significance
using one-way analysis of variance followed by Turkey's multiple
comparison test using SPSS version 10.10 for Windows (SPSS, Inc.).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Induction of PE conditions in
vitro
To generate an in vitro PE environment, human
placenta-derived BeWo cells were exposed to hypoxia by incubating
the cells in a hypoxic chamber for 24 h. For other models, the
cells were treated with L-NAME or COMT-I. The cytotoxicity of all
groups was assessed in MTT assay. According to the MTT viability
test, hypoxia condition, L-NAME and COMT-I did not exert any effect
on the viability of BeWo cells (Fig.
1A). Since HIF1A is a well-known biomarker of hypoxia (21), we examined HIF1A expression to test
our experimental conditions by western blot analysis (Fig. 1B and C). HIF1A was dramatically
over-expressed in the hypoxic group as expected, whereas it was
weakly expressed in other groups. To confirm PE conditions, the
expression levels of VEGF and sFlt-1 were examined by western blot
analysis (Fig. 1B and C). The
expression of VEGF was downregulated by hypoxia, whereas that of
sFlt-1 was upregulated by both L-NAME and COMT-I. These results
suggest that our in vitro PE models mimic some of the
clinical characteristics observed in PE patients.
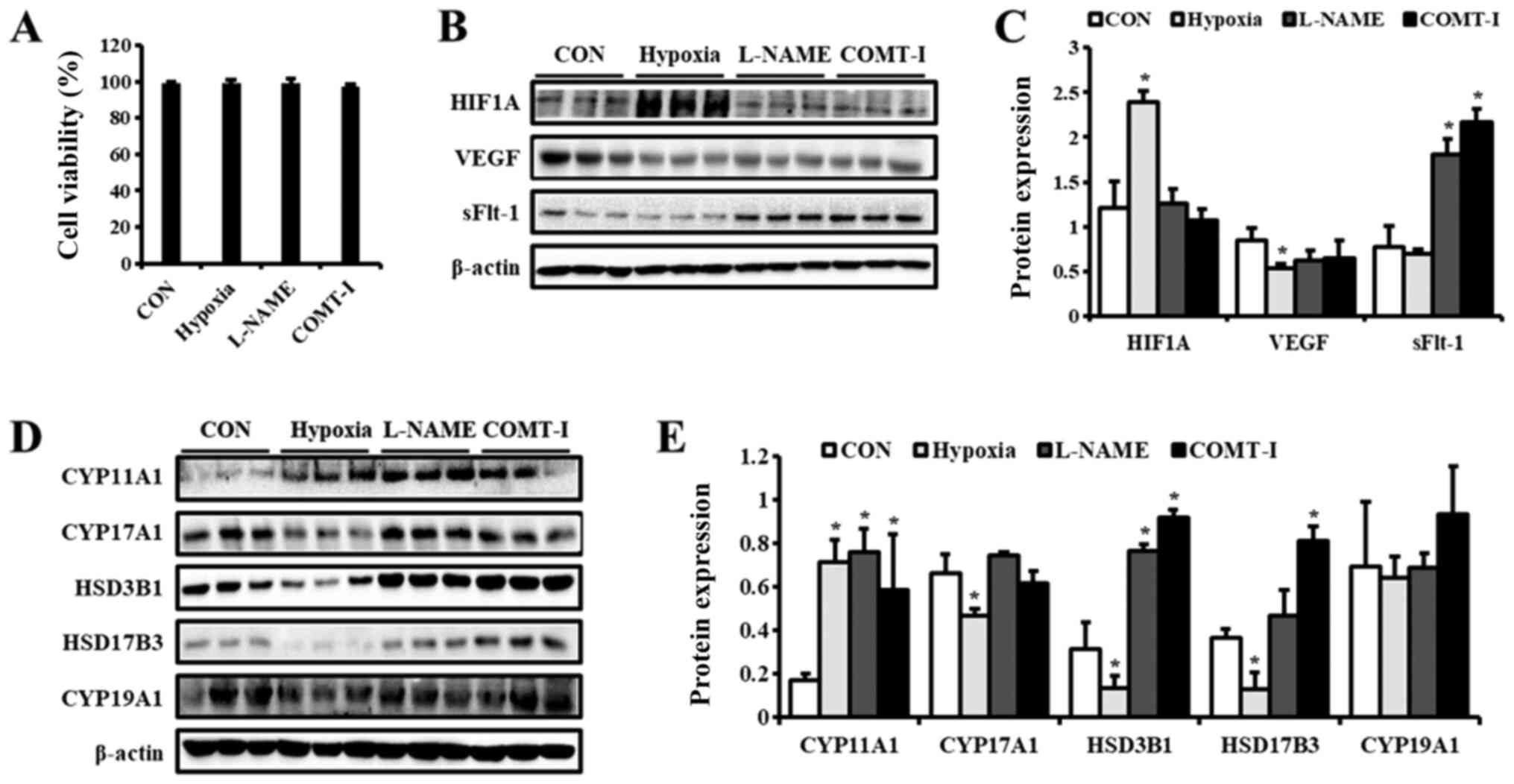 | Figure 1.Levels of PE biomarkers and
steroidogenic enzymes in in vitro PE models. Cell viability
was measured using the MTT assay and demonstrated as a (A)
percentage of the CON group. The expression of (B) PE biomarkers in
BeWo cells exposed to hypoxia or treated with either L-NAME or
COMT-I were analyzed by western blotting. (C) Expression levels of
HIF1A, VEGF, and sFlt-1. (D) Representative blot images of
steroidogenic enzymes in BeWo cells exposed to hypoxia or treated
with either L-NAME or COMT-I. (E) The levels of CYP11A1, CYP17A1,
HSD3B1, HSD17B3 and CYP19A1. The individual protein expression
level was normalized to that of β-actin. Data are expressed as the
mean ± SD. *P<0.05 vs. CON group. CON, control; PE,
preeclampsia; L-NAME, N-nitro-L-arginine methyl ester
hydrocholride; COMT-I, catechol-o-methyltransferase inhibitor;
HIF1A, Hypoxia inducible factor 1 subunit α; sFlt-1, soluble form
of fetal liver kinase 1; CYP11A1, cholesterol side-chain cleavage
enzyme; CYP17A1, 17α-hydroxylase/17,20-lyase; HSD3B1,
3β-hydroxysteroid dehydrogenase/δ5 4-isomerase type 1; HSD17B3,
17β-dehydrogenase 3; CYP19A1, aromatase cytochrome P450. |
Expression of steroidogenic enzymes in
in vitro PE models
To elucidate the effect of in vitro PE
conditions on the steroidogenesis in placental cells, the
expression of steroidogenic enzymes was examined by western blot
analysis (Fig. 1D and E). CYP11A1
was upregulated in all groups compared to the control, whereas the
levels of CYP19A1 were not altered. Interestingly, the expression
of other steroidogenic enzymes was regulated differentially by
hypoxia, L-NAME, and COMT-I. Under hypoxic conditions, levels of
CYP17A1, HSD3B1, and HSD17B3 were significantly downregulated.
L-NAME increased the protein levels of HSD3B1, whereas COMT-I
elevated both HSD3B1 and HSD17B3 expression.
Production of steroid hormones in in
vitro PE models
Next, we examined the synthesis of steroid hormones
in in vitro PE models. We determined the concentration of
steroid hormones, including P5, P4, DHEA, T and E2, by ELISA
(Table I). The concentration of
steroid hormones tended to decline in every group compared to the
CON group. L-NAME and hypoxia significantly decreased the
concentration of P5, P4, DHEA, and T. Further, the concentration of
DHEA and T were downregulated by COMT-I. These results suggest that
in vitro PE models, especially those established by hypoxia
and L-NAME treatment, affect the production of steroid
hormones.
 | Table I.Concentration of steroid hormones in
BeWo cells. |
Table I.
Concentration of steroid hormones in
BeWo cells.
| Hormone
(ng/ml) | CON | Hypoxia | L-NAME | COMT-I |
|---|
| P5 | 24.1±3.3 |
8.3±1.6a |
14.0±2.4a | 24.6±2.4 |
| P4 | 5.6±0.1 |
4.6±0.2a |
5.0±0.0a | 5.3±0.0 |
| DHEA | 0.8±0.1 |
0.1±0.0a |
0.1±0.0a |
0.5±0.1a |
| T | 0.3±0.0 |
0.1±0.0a |
0.2±0.0a |
0.2±0.0a |
| E2 | 11.9±2.1 | 10.7±1.0 | 10.4±1.9 | 11.0±0.8 |
Evaluation of PE characteristics in in
vivo PE models
Our next objective was to investigate the
association of steroid hormones and PE in animal models. Pregnant
rats were treated with L-NAME and COMT-I or underwent RUPP
operation. For the biological conditions, symptoms including BW,
placental and fetal weight, resorption rate, and urinary protein
concentration were evaluated (Table
II). All groups showed decreased BW compared to the CON group
and the Sham group during the period of treatment. Treatment with
L-NAME caused a significant reduction in placental and fetal weight
relative to saline solution control. Urinary protein excretion was
also higher in the L-NAME group compared to the CON group. Compared
to the Sham group, the RUPP group showed relatively lower placental
weight and a higher resorption rate and urinary protein level.
However, there were no significant changes in the COMT-I group. The
average weight gain was not significantly different for each
experimental group (Fig. 2A).
| Table II.Characteristics of rats on GD 18. |
Table II.
Characteristics of rats on GD 18.
| Characteristic | CON | L-NAME | COMT-I | Sham | RUPP |
|---|
| BW (g) | 312.8±17.2 |
279.3±15.4a |
276.7±26.4a | 275.0±20.2 |
214.8±20.3b |
| Placental weight
(g) | 0.5±0.1 |
0.4±0.00a | 0.5±0.1 | 0.36±0.05 |
0.20±0.06b |
| Fetal weight
(g) | 1.9±0.2 |
1.4±0.3a | 1.8±0.2 | 1.32±0.08 | 1.36±0.21 |
| Fetus (n) | 12.8±0.8 | 12.3±1.5 | 12.8±2.9 | 11.5±1.5 | 8.0±3.39 |
| Litter (n) | 0.0±0.0 | 0.1±0.4 | 2.0±4.9 | 1.29±1.91 | 6.8±2.38 |
| Fetal resorption
rate (%) | 0.0±0.0 | 1.0±2.5 | 16.7±40.8 | 13.88±15.23 |
89.4±18.04b |
| Urine protein
excretion (µg/µl) | 4.5±1.0 |
6.2±0.1a | 4.6±1.1 | 4.20±0.74 |
13.29±1.29b |
Systolic blood pressures for each experimental group
are shown in Fig. 2B. The normal
range for systolic blood pressure in rats is around 126 mmHg
(22). On GD 14, the systolic blood
pressures were significantly elevated in the L-NAME group compared
to the CON group (147.49±6.68 mmHg vs. 122.79±8.97 mmHg,
P<0.05), while it recovered from GD 14 to GD 16. On GD 18, both
L-NAME and COMT-I groups showed mild hypertension (135.39±6.88 mmHg
and 130.21±9.7 mmHg vs. 117.33±9.63 mmHg, P<0.05). The systolic
blood pressure of the RUPP group was marginally increased compared
to the Sham group on GD 18, but the increase was not significant
(Fig. 2B).
The effect on renal function observed in the PE rat
models was confirmed at the tissue level (Fig. 2C). Histological examination of the
kidney revealed that the COMT-I and RUPP groups had endotheliosis
involving swelling of the glomerulus and capillary occlusion,
whereas it was not observed in the CON and Sham groups. Moreover,
hemorrhage was detected in the RUPP group. The concentration of
VEGF and sFlt-1 in the plasma of PE models was examined by ELISA
(Fig. 3A). The levels of VEGF were
significantly lower in the L-NAME and COMT-I groups, whereas there
were no significant changes in sFlt-1 levels. To further
investigate the placental expression of VEGF and sFlt-1, their
levels were analyzed by western blot analysis (Fig. 3C). We found that sFlt-1 was
upregulated by COMT-I and VEGF was downregulated by RUPP, whereas
L-NAME did not affect the expression of either.
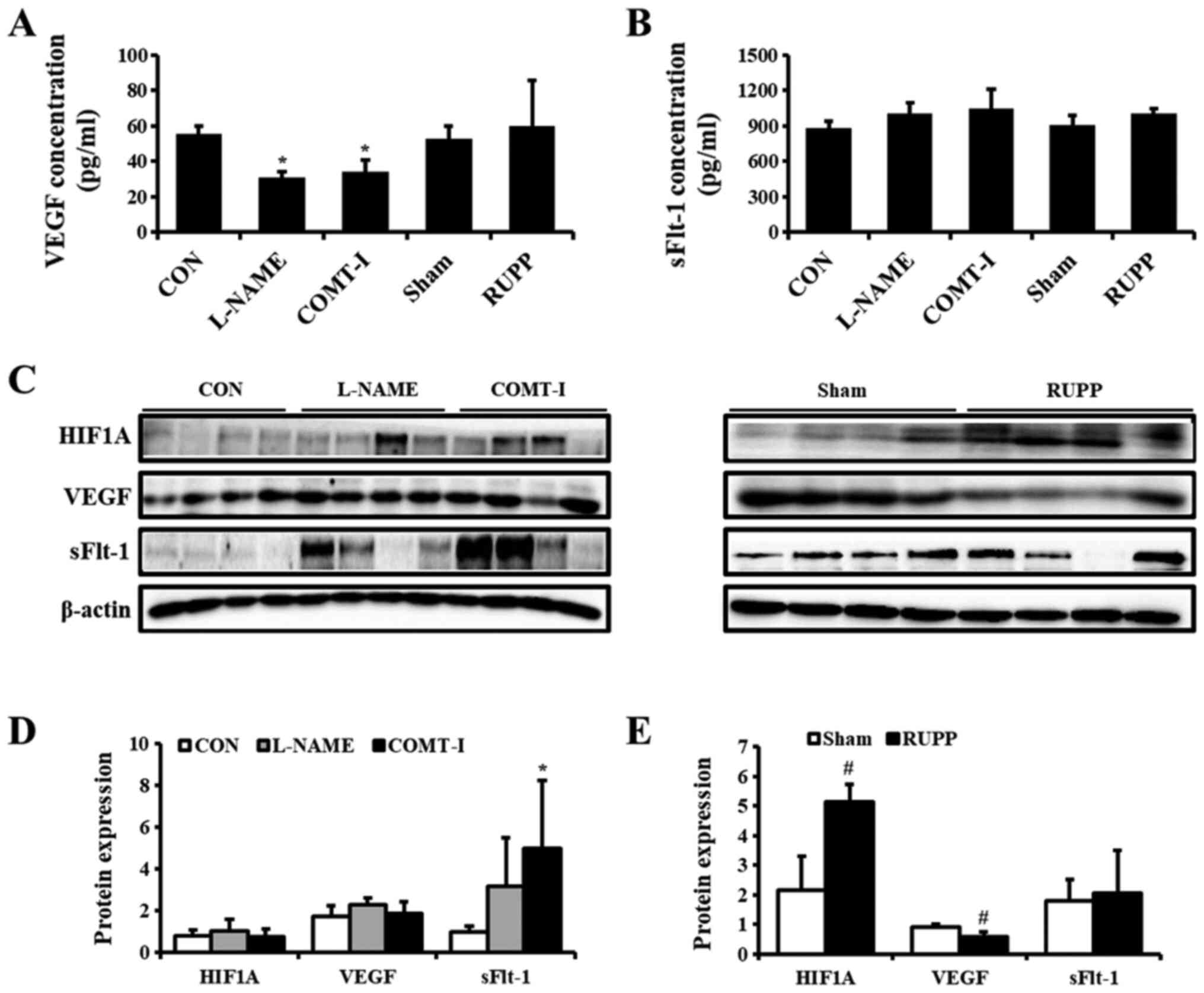 | Figure 3.Effects of angiogenic factors on
in vivo PE models. (A and B) The plasma concentrations of
VEGF and sFlt-1 in L-NAME, COMT-I and RUPP groups were analyzed by
ELISA and compared with the CON or Sham group. (C) The expression
of PE biomarkers in the placenta of in vivo rat models was
analyzed by western blot analysis. (D and E) Expression levels of
HIF1A, VEGF and sFlt-1 in the L-NAME, COMT-I, and RUPP groups. Data
are expressed as the mean ± SD. *P<0.05 vs. CON group;
#P<0.05 vs. Sham group. sFlt-1, soluble form of fetal
liver kinase 1; L-NAME, N-nitro-L-arginine methyl ester
hydrocholride; COMT-I, catechol-o-methyltransferase inhibitor;
RUPP, reduced uterine perfusion pressure; CON, control; PE,
Preeclampsia; HIF1α, hypoxia inducible factor 1 subunit α. |
Expression of steroidogenic enzymes in
in vivo PE models
As the PE rat models showed features and symptoms
similar to human PE, the steroidogenesis process was investigated
by western blot analysis (Fig. 4).
The CYP11A1 protein levels were decreased in all groups compared
with CON and Sham groups. The expression of HSD3B1 was
significantly upregulated (up to 1.8-fold) by L-NAME. The levels of
CYP19A1 were elevated by COMT-I and RUPP. CYP17A1 and HSD17B3
levels were unaltered in all in vivo PE models.
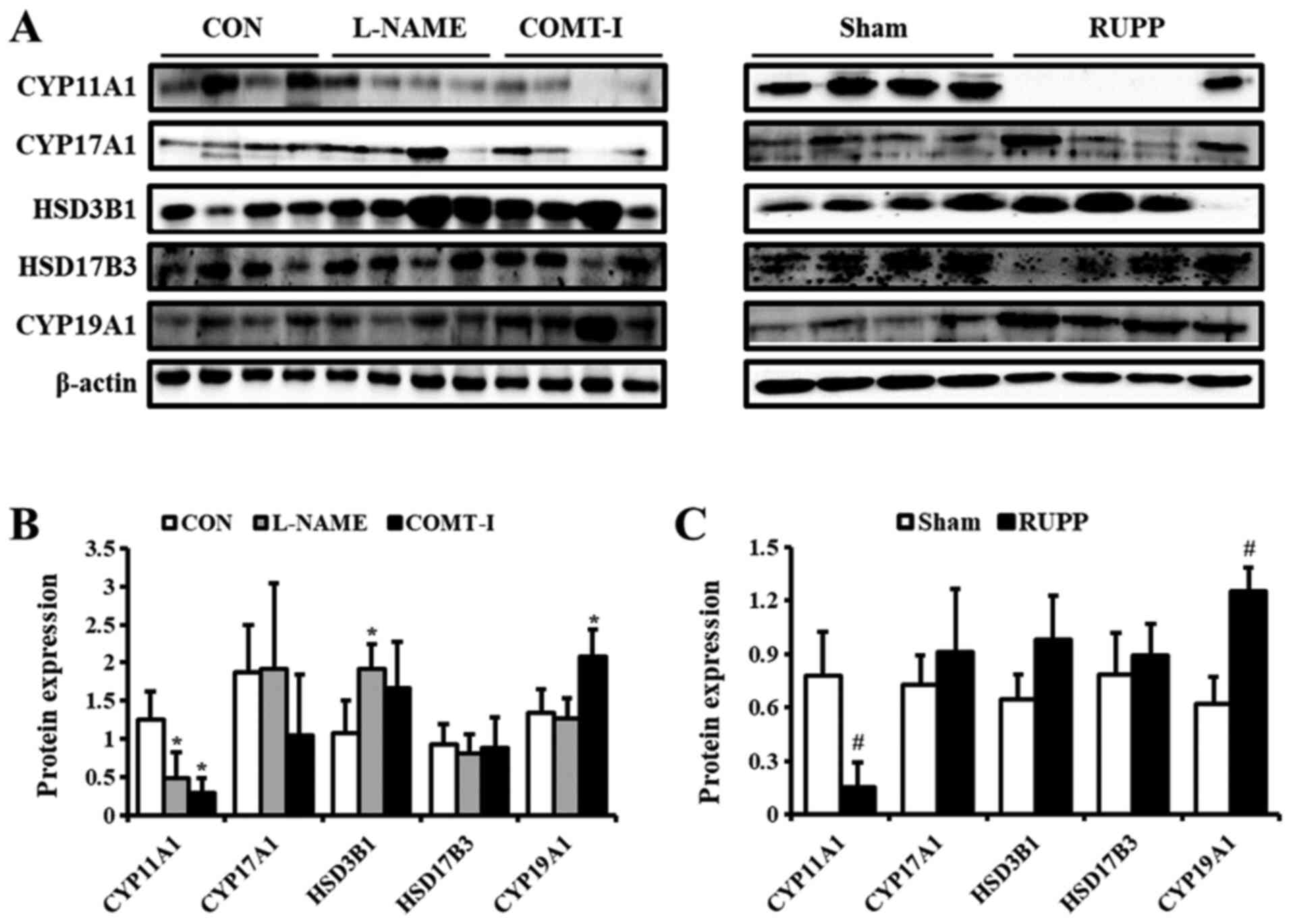 | Figure 4.Levels of steroidogenic enzymes in
in vivo PE models. (A) The expression of steroidogenic
enzymes in the L-NAME, COMT-I, and RUPP groups was analyzed by
western blot analysis. (B and C) The levels of CYP11A1, CYP17A1,
HSD3B1, HSD17B3 and CYP19A1. The individual protein expression was
normalized to that of β-actin. Data are expressed as the mean ± SD.
*P<0.05 vs. CON group; #P<0.05 vs. Sham group.
L-NAME, N-nitro-L-arginine methyl ester hydrocholride; COMT-I,
catechol-o-methyltransferase inhibitor; RUPP, reduced uterine
perfusion pressure; CYP11A1, cholesterol side-chain cleavage
enzyme; CYP17A1, 17α-hydroxylase/17,20-lyase; HSD3B1,
3β-hydroxysteroid dehydrogenase/δ5 4-isomerase type 1; HSD17B3,
17β-dehydrogenase 3; CYP19A1, aromatase cytochrome P450; CON,
control. |
Concentration of steroid hormones in
in vivo PE models
Based on the regulation of steroidogenesis in the
placenta of rats, the concentration of steroid hormones was
determined by ELISA (Tables III
and IV). L-NAME reduced the levels
of E2 2-fold, while COMT-I significantly increased concentration of
P5 and DHEA in the plasma (Table
III). The concentration of other steroid hormones were not
significantly altered. The placental levels of steroid hormones
were different compared to their levels in the plasma (Table IV). The levels of P4 in the
placenta of the L-NAME group were 2-fold lower than those in the
CON group. Other placental steroid hormones were not significantly
altered. The RUPP operation did not affect the levels of any
steroid hormones in both plasma and placenta.
| Table III.Plasma concentration of steroid
hormones in vivo PE models. |
Table III.
Plasma concentration of steroid
hormones in vivo PE models.
| Hormone
(ng/ml) | CON | L-NAME | COMT-I | Sham | RUPP |
|---|
| P5 | 1.6±0.1 | 1.6±0.4 |
3.4±0.5a | 1.4±0.8 | 1.3±0.4 |
| P4 | 42.2±7.9 | 37.8±4.4 | 45.8±6.4 | 38.8±13.0 | 34.9±7.8 |
| DHEA | 0.5±0.1 | 0.5±0.10 |
0.9±0.1a | 0.6±0.2 | 0.4±0.1 |
| T | 8.7±2.3 | 8.9±2.1 | 9.6±1.5 | 8.6±4.3 | 7.6±4.7 |
| E2 | 0.8±0.2 |
0.4±0.0a | 0.7±0.1 | 0.7±0.4 | 0.8±0.3 |
| Table IV.Placental concentration of steroid
hormones in vivo PE models. |
Table IV.
Placental concentration of steroid
hormones in vivo PE models.
| Hormone | CON | L-NAME | COMT-I | Sham | RUPP |
|---|
| P5 (ng/ml) | 30.0±0.5 | 30.3±2.0 | 29.7±2.7 | 28.6±0.8 | 25.9±2.1 |
| P4 (ng/ml) | 0.2±0.0 |
0.1±0.0a | 0.3±0.0 | 0.2±0.1 | 0.3±0.1 |
| DHEA (ng/ml) | 0.5±0.0 | 0.5±0.0 | 0.6±0.1 | 0.4±0.1 | 0.3±0.1 |
| T (ng/ml) | 4.1±0.5 | 4.5±0.6 | 4.7±0.8 | 4.0±0.5 | 4.6±1.9 |
| E2 (ng/ml) | 5.0±0.6 | 4.5±0.6 | 4.5±0.6 | 4.5±0.8 | 4.5±0.2 |
Discussion
It is well-known that hormonal changes are
associated with the pathology of PE. Therefore, the assessment of
steroid hormone levels in the blood has been the focus of intense
study to find potential biomarkers to predict PE (23). Multiple PE models have been
developed to mimic the physiological conditions of PE. However,
these models are limited in their ability to represent a disease
with complicated interactions and the effects on PE-related
tissues, and thus single model can't reproduce all the clinical
aspects of the PE syndrome (9).
Considering this, we established three in vitro and in
vivo PE models via inducing hypoxia and via L-NAME, and COMT-1
treatments, and evaluated PE-related biomarkers and pathological
characteristics. In addition, we examined how the three PE models
affect steroidogenesis in the placenta and hormone levels in the
blood.
For our in vitro models, placental cells were
exposed to hypoxic conditions or treated with L-NAME and COMT-I.
The expression of HIF1A, VEGF, and sFlt-1 in the placenta are
predictive biomarkers of PE (24).
HIF1 acts as a central regulator of oxygen homeostasis in most
mammalian cells. In placenta, HIF1 is essential for placental
development regulating several genes that control cell growth,
differentiation, and metabolism including proangiogenic factors
(25). HIF1A expression can be
increased by the inhibition of NOS and COMT activity in the
placenta, and consequently result in ubiquitous placental tissue
damage (26,27). One study demonstrated that the
levels of mRNA and protein expression of placental HIF1A were
significantly increased in rats administered 0.5 mg/ml of L-NAME
via drinking water from GD 6 to GD 10. Also, in another study,
dramatically increased placental HIF1 protein level is observed in
KO mice with a deficiency in COMT (28,29).
However, in our study, hypoxia increased the levels of HIF1A, while
L-NAME and COMT-I did not. These results were also seen in our
in vivo models, where L-NAME and COMT-I did not alter levels
of HIF1A while RUPP enhanced HIF1A in the placenta. These distinct
results may be due to differences in treatment period and dosage
compared to previous studies.
VEGF, known as a target gene of HIF-1A, initiate
angiogenesis and induce placental hypoxia (30). The VEGF/sFlt-1 pathway may be
induced by hypoxia through HIF-dependent and HIF-independent
pathways and the relative overproduction of sFlt-1 than VEGF by
placental tissue is one of the symptoms of PE (31,32).
In our in vitro study, the VEGF was decreased by hypoxic
condition, but not by L-NAME and COMT-I, which suggest that the
process of angiogenesis was regulated by hypoxia. However, both
L-NAME and COMT-I upregulated the expression of sFlt-1, meaning
that the treatments also inhibit angiogenesis in a different way
from hypoxia in placental cell. Collectively, our COMT-I- and
VEGF-induced PE models showed higher levels of sFlt-1 than VEGF and
we suggest that these results may be mediated by HIF1A-independent
pathway in vitro and in vivo models.
The pathological characteristics, including
placental and fetal weight, proteinuria, hypertension, and
glomerular endotheliosis, were analyzed in the in vivo
models. Lower venous capacitance and compliance have been suggested
to increase blood pressure and protein excretion in PE (33). Fetal growth restriction is a result
of reduced placental blood flow due to altered placental
vasculature caused by angiogenic factors and impaired development
of the placenta (34). Consistent
with previous findings, the RUPP operation caused a significant
reduction in fetal and placental weights and fetal death. Although
blood pressure elevation was not significant in the RUPP group,
proteinuria was observed due to endotheliosis and massive bleeding,
which had spread beyond the glomerulus. L-NAME administration in
pregnant rats led to hypertension, proteinuria, and fetal growth
restriction. The rats treated with COMT-I showed hypertension,
glomerular endotheliosis, and glomerular injury. The L-NAME and
COMT-1 groups showed a lower concentration of VEGF in the blood.
These results indicate that the three different PE models showed
more than one pathological characteristic of PE, although the
underlying mechanisms are different. Among the models, the L-NAME
group was closest to representing PE symptoms, because hypertension
and proteinuria are considered as the most critical factors for
PE.
To investigate the effect of PE conditions on the
production of steroid hormones, we examined their levels in the
culture-supernatant of placental cells. The levels of steroid
hormones, including P5, P4, DHEA, and T, tended to decrease in the
hypoxia and L-NAME groups, while the COMT-I group showed lower DHEA
and T levels. We also evaluated the expression of
steroidogenesis-related enzymes in the placental cells. Although
the enzymes were regulated differentially by individual PE models,
CYP11A1 was upregulated in all in vitro PE models. CYP11A1
is a rate-limiting enzyme for the production of P5 that is the
precursor hormone of all other steroid hormones. This could be a
compensatory regulation because P5 and its downstream steroid
hormones, such as P4, DHEA, and T, were downregulated. These
results indicate that the symptoms of PE are closely linked with
the levels of steroid hormones and different pathological symptoms
of PE may modulate the process of steroidogenesis in distinct ways
in the placental cells. The regulation of steroidogenesis in the
cellular models of PE in this study were different from the human
placenta. It has been demonstrated that P4 and E2 were decreased,
whereas other steroid hormones were not altered in human PE
(10,35). The steroidogenic process in the
human placenta is often influenced by the fetal endocrine system.
The fetal adrenal cortex mostly synthesizes byproducts, DHEA and
DHEA-S, which are delivered to the placenta for the production of
androgens and E2 (36). Since
feto-placental communication is absent in in vitro models,
there are bound to be differences between the in vitro data
and the results from animal and human studies.
We next investigated the steroidogenic process under
in vivo PE conditions. Because of the different species and
PE conditions, the regulation of steroid hormones in our in
vivo PE models is different from that seen in in vitro
models and human with PE. Both rat and human placentas are
anatomically classified as discoid and hemochorial type. However,
there are differences between rats and humans in terms of the
histological structure of the placenta, the feto-maternal
interface, and the function of the yolk sac (37).
We found that lower placental blood flow induced by
RUPP affects the levels of CYP11A1 and CYP19A1 in placental
tissues, which may, in turn, affect the production of P5 and E2.
However, the quantity of steroid hormones produced was not
significantly altered in our study. L-NAME treatment in pregnant
rats led to a downregulation of VEGF, lower placental weight, and
fetal growth restriction. Additionally, L-NAME decreased plasma E2
levels, although the levels were not regulated in the placenta.
These results indicated that the plasma E2 was not directly
regulated by placenta. It was possibly modulated by fetus or
extra-placental tissues in mother. Indeed, the levels of E2 is
regulated by complicated maternal-placental-fetal unit (38). L-NAME inhibits the production of NO
which is associated with E2 production (39). Previous studies have demonstrated
that fetal- and placental-VEGF were stimulated by E2, indicating
that the decrease in E2 production obstructs angiogenesis and
causes oxidative stress in the placenta (40,41).
The damaged placenta by oxidative stress can affect the growth and
function of the placenta, including the production of steroid
hormones. Further, oxidative stress plays a role as an
anti-angiogenic factor in blood vessels.
As we mentioned, the rats deficient in COMT showed
lower levels of VEGF and elevated sFlt-1. It has been reported that
COMT deficiency induces defects in both angiogenesis and the
placenta by dysfunction of angiogenic factors (42). Our results from the COMT-I model
showed that disrupting angiogenesis in the placenta may lead to the
overproduction of P5 and DHEA through the regulation of CYP11A1 and
CYP19A1. It is widely accepted that DHEA is associated with
vascular angiogenesis (43,44).
In conclusion, we compared steroid hormones and
steroidogenic enzymes in three different in vitro and in
vivo PE models. The steroid hormones were dynamically
dysregulated depending on the symptoms of PE. The overall in
vitro and in vivo results are illustrated in Fig. 5. Our findings provide new insights
into the correlation of steroid hormones with PE and may contribute
to the development of new diagnostic and therapeutic methodologies
for PE.
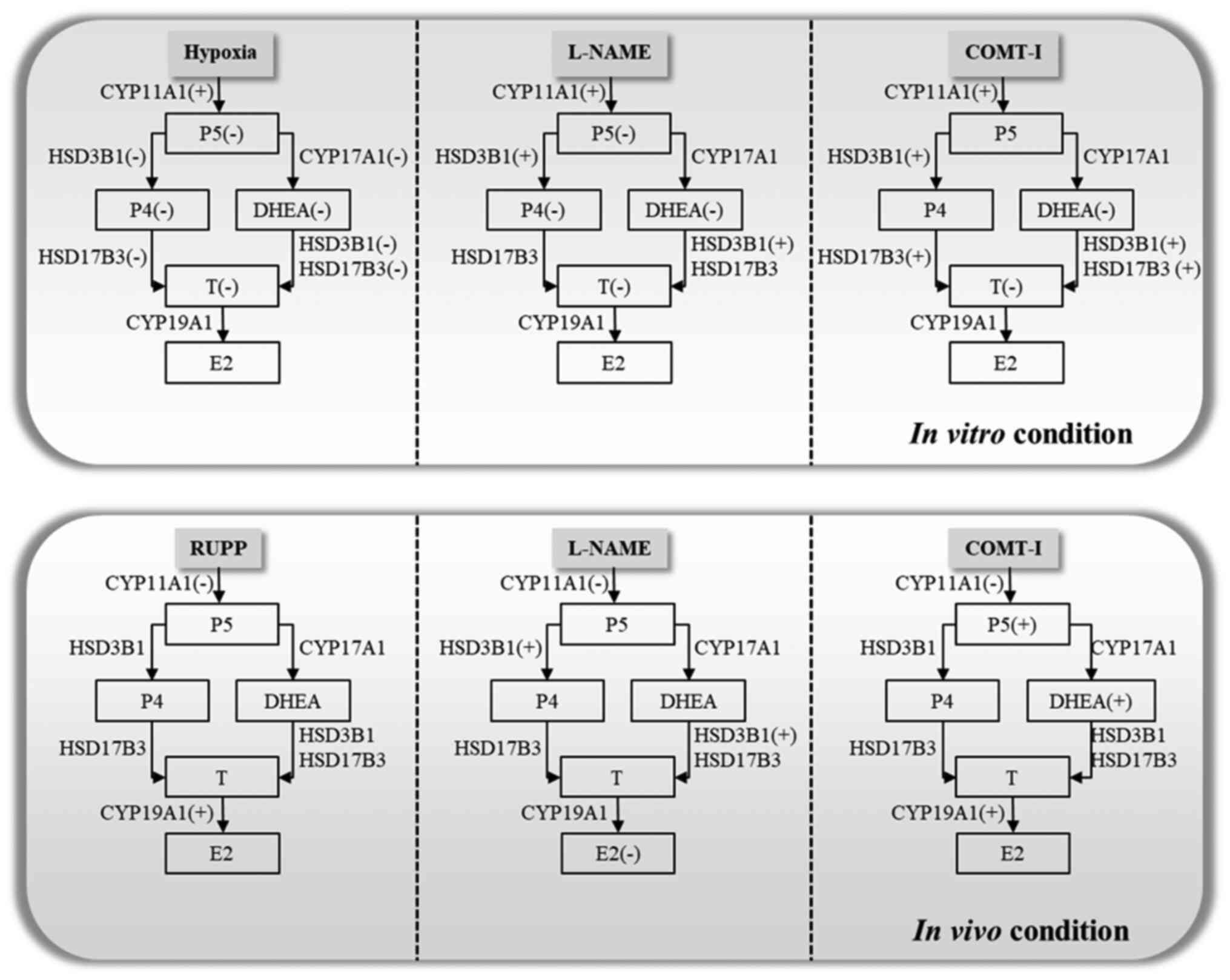 | Figure 5.Scheme illustrating how various
symptoms of PE regulate steroidogenesis in vitro and in
vivo based on the changes observed in plasma. L-NAME,
N-nitro-L-arginine methyl ester hydrocholride; COMT-I,
catechol-o-methyltransferase inhibitor; RUPP, reduced uterine
perfusion pressure; CYP11A1, cholesterol side-chain cleavage
enzyme; CYP17A1, 17α-hydroxylase/17,20-lyase; HSD3B1,
3β-hydroxysteroid dehydrogenase/δ5 4-isomerase type 1; HSD17B3,
17β-dehydrogenase 3; CYP19A1, aromatase cytochrome P450; E2,
estradiol; P4, progesterone; P5, pregnenolone; T, testosterone;
DHEA, dehydroepiandrosterone. |
Acknowledgements
Not applicable.
Funding
The present study was supported by a 2-year research
grant from Pusan National University (grant no. 201912470002) and
also partially supported by the BK21 FOUR Program through the
National Research Foundation (NRF) of Korea funded by the Ministry
of Education, Korea.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
YYS, SMA and BSA designed the experiments and wrote
the manuscript. YYS, SMA, JSJ, SYY and SCK performed the
experiments and analyzed the data. GSL, EJH, EBJ and BSA analyzed
the data and revised the manuscript. All authors read and approved
the final manuscript.
Ethics approval and informed consent
The animal protocol used in this study was reviewed
and approved based on ethical procedures and scientific care by the
Pusan National University-Institutional Animal Care and Use
Committee (PNU-IACUC) (Approval no. PNU-2017-1452).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Li J, LaMarca B and Reckelhoff JF: A model
of preeclampsia in rats: The reduced uterine perfusion pressure
(RUPP) model. Am J Physiol Heart Circ Physiol. 303:H1–H8. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dragun D and Haase-Fielitz A: Low
catechol-O-methyltransferase and 2-methoxyestradiol in
preeclampsia: More than a unifying hypothesis. Nephrol Dial
Transplant. 24:31–32. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fushima T, Sekimoto A, Minato T, Ito T, Oe
Y, Kisu K, Sato E, Funamoto K, Hayase T, Kimura Y, et al: Reduced
uterine perfusion pressure (RUPP) model of preeclampsia in mice.
PLoS One. 11:e01554262016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Talebianpoor MS and Mirkhani H: The effect
of tempol administration on the aortic contractile responses in rat
preeclampsia model. ISRN Pharmacol. 2012:1872082012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
AbdAlla S, Lother H, el Massiery A and
Quitterer U: Increased AT(1) receptor heterodimers in preeclampsia
mediate enhanced angiotensin II responsiveness. Nat Med.
7:1003–1009. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
McKinney D, Boyd H, Langager A, Oswald M,
Pfister A and Warshak CR: The impact of fetal growth restriction on
latency in the setting of expectant management of preeclampsia. Am
J Obstet Gynecol. 214:395.e1–e7. 2016. View Article : Google Scholar
|
|
7
|
Sato K, Iemitsu M, Matsutani K, Kurihara
T, Hamaoka T and Fujita S: Resistance training restores muscle sex
steroid hormone steroidogenesis in older men. FASEB J.
28:1891–1897. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shin YY, Jeong JS, Park MN, Lee JE, An SM,
Cho WS, Kim SC, An BS and Lee KS: Regulation of steroid hormones in
the placenta and serum of women with preeclampsia. Mol Med Rep.
17:2681–2688. 2018.PubMed/NCBI
|
|
9
|
Pennington KA, Schlitt JM, Jackson DL,
Schulz LC and Schust DJ: Preeclampsia: Multiple approaches for a
multifactorial disease. Dis Model Mech. 5:9–18. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hertig A, Liere P, Chabbert-Buffet N, Fort
J, Pianos A, Eychenne B, Cambourg A, Schumacher M, Berkane N,
Lefevre G, et al: Steroid profiling in preeclamptic women: Evidence
for aromatase deficiency. Am J Obstet Gynecol. 203:477.e1–e9. 2010.
View Article : Google Scholar
|
|
11
|
Lowe DT: Nitric oxide dysfunction in the
pathophysiology of preeclampsia. Nitric Oxide. 4:441–458. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kanasaki K, Palmsten K, Sugimoto H, Ahmad
S, Hamano Y, Xie L, Parry S, Augustin HG, Gattone VH, Folkman J, et
al: Deficiency in catechol-O-methyltransferase and
2-methoxyoestradiol is associated with pre-eclampsia. Nature.
453:1117–1121. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Suzuki H, Ohkuchi A, Shirasuna K,
Takahashi H, Usui R and Matsubara S: Animal models of preeclampsia:
Insight into possible biomarker candidates for predicting
preeclampsia. Med J Obstet Gynecol. 2:10312014.
|
|
14
|
Gilbert JS, Babcock SA and Granger JP:
Hypertension produced by reduced uterine perfusion in pregnant rats
is associated with increased soluble fms-like tyrosine kinase-1
expression. Hypertension. 50:1142–1147. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kumasawa K: Animal models in preeclampsia.
Preeclampsia. Springer Nature Singapore; pp. 141–155. 2018,
View Article : Google Scholar
|
|
16
|
Zeisler H, Jirecek S, Hohlagschwandtner M,
Knofler M, Tempfer C and Livingston JC: Concentrations of estrogens
in patients with preeclampsia. Wien Klin Wochenschr. 114:458–461.
2002.PubMed/NCBI
|
|
17
|
Sun MN and Zi Y: Effects of
preeclampsia-like symptoms at early gestational stage on
feto-placental outcomes in a mouse model. Chin Med J (Engl).
123:707–712. 2010.PubMed/NCBI
|
|
18
|
Yang H, Ahn C and Jeung EB: Differential
expression of calcium transport genes caused by COMT inhibition in
the duodenum, kidney and placenta of pregnant mice. Mol Cell
Endocrinol. 401:45–55. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nathan HL, Duhig K, Hezelgrave NL,
Chappell LC and Shennan AH: Blood pressure measurement in
pregnancy. Obstetr Gynaecol. 17:91–98. 2015.
|
|
20
|
Spradley FT, Tan AY, Joo WS, Daniels G,
Kussie P, Karumanchi SA and Granger JP: Placental growth factor
administration abolishes placental ischemia-induced hypertension.
Hypertension. 67:740–747. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Berg A, Fasmer KE, Mauland KK, Ytre-Hauge
S, Hoivik EA, Husby JA, Tangen IL, Trovik J, Halle MK, Woie K, et
al: Tissue and imaging biomarkers for hypoxia predict poor outcome
in endometrial cancer. Oncotarget. 7:69844–69856. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yasujima M, Abe K, Kohzuki M, Tanno M,
Kasai Y, Sato M, Omata K, Kudo K, Takeuchi K, Hiwatari M, et al:
Effect of atrial natriuretic factor on angiotensin II-induced
hypertension in rats. Hypertension. 8:748–753. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Costa RA, Hoshida MS, Alves EA, Zugaib M
and Francisco RP: Preeclampsia and superimposed preeclampsia: The
same disease? The role of angiogenic biomarkers. Hypertens
Pregnancy. 35:139–149. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tal R: The role of hypoxia and
hypoxia-inducible factor-1alpha in preeclampsia pathogenesis. Biol
Reprod. 87:1342012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gaspar JM and Velloso LA: Hypoxia
inducible factor as a central regulator of metabolism-implications
for the development of obesity. Front Neurosci. 12:8132018.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Olson N and Van Der Vliet A: Interactions
between nitric oxide and hypoxia-inducible factor signaling
pathways in inflammatory disease. Nitric Oxide. 25:125–137. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Arendt KW and Garovic VD: Association of
deficiencies of catechol-O-methyltransferase and 2-methoxyestradiol
with preeclampsia. Expert Rev Obstetr Gynecol. 4:379–381. 2009.
View Article : Google Scholar
|
|
28
|
Zheng L, Huang J, Su Y, Wang F, Kong H and
Xin H: Vitexin ameliorates preeclampsia phenotypes by inhibiting
TFPI-2 and HIF-1α/VEGF in al-NAME induced rat model. Drug Dev Res.
80:1120–1127. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kulkarni K: HIF-1 alpha: A master
regulator of trophoblast differentiation and placental development.
Browse Theses Dissertations. 943:2009, https://corescholar.libraries.wright.edu/etd_all/943September
3–2020
|
|
30
|
Saravani M, Rokni M, Mehrbani M,
Amirkhosravi A, Faramarz S, Fatemi I, Esmaeili Tarzi M and
Nematollahi MH: The evaluation of VEGF and HIF-1α gene
polymorphisms and multiple sclerosis susceptibility. J Gene Med.
21:e31322019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mizukami Y, Li J, Zhang X, Zimmer MA,
Iliopoulos O and Chung DC: Hypoxia-inducible factor-1-independent
regulation of vascular endothelial growth factor by hypoxia in
colon cancer. Cancer Res. 64:1765–1772. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pant V, Yadav BK and Sharma J: A cross
sectional study to assess the sFlt-1:PlGF ratio in pregnant women
with and without preeclampsia. BMC Pregnancy Childbirth.
19:2662019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schrier RW: Renal and Electrolyte
Disorders. Lippincott Williams & Wilkins; Philadelphia, PA:
2010
|
|
34
|
Banek CT, Bauer AJ, Gingery A and Gilbert
JS: Timing of ischemic insult alters fetal growth trajectory,
maternal angiogenic balance, and markers of renal oxidative stress
in the pregnant rat. Am J Physiol Regul Integr Comp Physiol.
303:R658–R664. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Salari S, Eskandari M, Nanbakhsh F and
Dabiri A: Evaluation of androgen and progesterone levels in women
with preeclampsia. Iranian J Med Sci. 30:186–189. 2015.
|
|
36
|
Kaludjerovic J and Ward WE: The interplay
between estrogen and fetal adrenal cortex. J Nutr Metab.
2012:8379012012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Furukawa S, Tsuji N and Sugiyama A:
Morphology and physiology of rat placenta for toxicological
evaluation. J Toxicol Pathol. 32:1–17. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Buster JE: Gestational changes in steroid
hormone biosynthesis, secretion, metabolism, and action. Clin
Perinatol. 10:527–552. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hu XQ, Song R and Zhang L: Effect of
oxidative stress on the estrogen-NOS-NO-KCa channel pathway in
uteroplacental dysfunction: Its implication in pregnancy
complications. Oxid Med Cell Longev. 2019:91942692019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Vonnahme KA, Wilson ME and Ford SP:
Relationship between placental vascular endothelial growth factor
expression and placental/endometrial vascularity in the pig. Biol
Reprod. 64:1821–1825. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Maliqueo M, Echiburú B and Crisosto N: Sex
steroids modulate uterine-placental vasculature: Implications for
obstetrics and neonatal outcomes. Front Physiol. 7:1522016.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kanasaki K and Kanasaki M: Angiogenic
defects in preeclampsia: What is known, and how are such defects
relevant to preeclampsia pathogenesis? Hypertension Res Pregnancy.
1:57–65. 2013. View Article : Google Scholar
|
|
43
|
Liu D, Iruthayanathan M, Hosman LL, Wang
Y, Yang L, Wang Y and Dillon JS: Dehydroepiandrosterone stimulates
endothelial proliferation and angiogenesis through extracellular
signal-regulated kinase 1/2-mediated mechanisms. Endocrinology.
149:889–898. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Barnabas O, Wang H and Gao XM: Role of
estrogen in angiogenesis in cardiovascular diseases. J Geriatr
Cardiol. 10:377–382. 2013.PubMed/NCBI
|















