Introduction
Diffuse large B-cell lymphoma (DLBCL) is a
genetically and clinically heterogeneous lymphoid malignancy type,
which represents the most common category and disease entity of
non-Hodgkin lymphoma in adults and accounts for ~30-40% of all
cases in different geographic regions, such as Europe and the USA
(1,2). In total, two major molecular subtypes
of DLBCL have been identified via gene expression profiling:
Germinal center B cell-like (GCB) and activated B cell-like (ABC),
which represent lymphomas derived from different stages of B-cell
lymphoid differentiation (3). GCB-
and ABC-subtype DLBCLs have different oncogenic pathways but they
share similar morphologies such as a white, ‘fish-flesh’,
homogeneous cut surface (4). Thus
far, the use of the R-CHOP regimen (including rituximab,
cyclophosphamide, doxorubicin, vincristine and prednisone) as a
standard component of immunochemotherapy has greatly improved the
prognosis of patients with DLBCL, with a response rate of ~80%
(5). However, some patients with
DLBCL do not respond to the R-CHOP treatment due to disease
heterogeneity (6). Thus,
alternative or supplemental strategies are required for these
subsets of patients with DLBCL.
STAT3 is a transcription activation factor, and
participates in the malignant progression of multiple tumors,
including DLBCL (7). RNA in
situ hybridization (RNA scope) has revealed that there are more
STAT3 positive cells in the ABC-subtype of DLBCL tissue samples
compared with those of the GCB-subtype of DLBCL tissue samples
(8). Activation or phosphorylation
of STAT3 is strongly associated with more advanced clinical stage
and overall poor survival of patients with DLBCL (9). Therefore, inhibition of STAT3
activation provides an additional therapeutic target for DLBCL,
especially for ABC-subtype DLBCL (10).
Chidamide is a benzamide histone deacetylase (HDAC)
inhibitor, and it has been marketed in China for the treatment of
peripheral T-cell lymphoma (PTCL) since 2015 (11). Chidamide has also been used to treat
myeloma, non-small lung cancer types and breast cancer due to its
antitumor potency (12–14). Although the antitumor mechanism of
chidamide has been widely investigated, studies examining the
target profiling for chidamide are sparse, which limits its further
application in the treatment of multiple tumors.
To provide evidence for the clinical application of
chidamide in DLBCL, the present study examined the cytotoxic effect
of chidamide on DLBCL cells and its possible underlying molecular
mechanism.
Materials and methods
Cells and cell culture
Human DLBCL cell lines SU-DHL2 and MZ were purchased
from Jennio-Bio, Co., Ltd., and the OCI-LY3 cell line was from
Beina Chuanglian Biological Research Institute (Beijing, China).
SU-DHL2 and OCI-LY3 cells of the ABC-subtype, and the MZ cells of
the GCB-subtype of DLBCL cells were cultured in RPMI-1640 medium
(Gibco; Thermo Fisher Scientific, Inc.) containing 10% FBS (Gibco;
Thermo Fisher Scientific, Inc.) and 1% penicillin-streptomycin
(Gibco; Thermo Fisher Scientific, Inc.). All cells were cultured in
a 37°C incubator with 5% CO2. All three cell lines were
tested for mycoplasma contamination and authenticated by STR
profiling.
Reagents and antibodies
Chidamide was purchased from Absin Bioscience, Inc.,
and dissolved in DMSO (Sigma-Aldrich; Merck KGaA) to prepare the
stock solution (30 mM), which was stored at −20°C in the dark. A
Cell Counting Kit-8 (CCK-8) was obtained from Beijing Solarbio
Biotechnology Co., Ltd. The Annexin V-FITC/PI kit, which was used
for apoptotic cell death determination via flow cytometry, was
purchased from Beyotime Institute of Biotechnology. Antibodies
against cleaved caspase-3 (cat. no. 9664S; 1:1,000), Bcl-2 (cat.
no. 15071S; 1:1,000), HDAC1 (cat. no. 34589S; 1:1,000), HDAC2 (cat.
no. 57156S; 1:1,000), HDAC3 (cat. no. 85057S; 1:1,000), STAT3 (cat.
no. 9139S; 1:1,000), phosphorylated (P)-STAT3-705 (cat. no. 9145S;
1:1,000), P-STAT3-727 (cat. no. 49081S; 1:1,000) and β-actin (cat.
no. 3700S; 1:5,000) were purchased from Cell Signaling Technology,
Inc. An antibody against HDAC8 (cat. no. JJ0845; 1:1,000) was
purchased from Novus Biologicals, LLC.
Cell viability assay
SU-DHL2, OCI-LY3 and MZ DLBCL cells
(2×105 cells/ml) were seeded into 96-well plates at a
volume of 100 µl/well and then treated with 5 µl/well chidamide at
different final concentrations (0, 0.1, 0.3, 1, 3, 10 and 30 µM)
for 24, 48 and 72 h in a 37°C incubator with 5% CO2,
respectively, to examine the concentration- and time-dependence of
cellular chidamide effects. Equal molar concentrations of DMSO were
used in the control group. At the indicated time of the reaction,
10 µl CCK-8 reagent was added to each well and incubated at 37°C
for 2 h, according to the manufacturer's instructions. The final
absorbance [optical density (OD) value] of the cultured cells was
measured at a wavelength of 450 nm with a SynergyHTX microplate
reader (BioTek Instruments, Inc.). Cell viability was calculated
and expressed as the ratio of the OD450 values for
chidamide-treated cells over those of the DMSO-treated control
cells. The drug concentration and cell survival rate were entered
into GraphPad Prism software 5.0 (GraphPad Software, Inc.) to
obtain a concentration-survival rate curve. The half maximal
inhibitory concentration (IC50), which represents the
concentration of an inhibitor (drug) that is required for 50%
inhibition of cells (15), was
calculated. The concentration corresponding to the point where this
line and the curve intersect is the IC50. The specific
value is directly obtained using the software and displayed in the
results.
Flow cytometry analysis
Cells were seeded in 6-well flat-bottomed plates at
a density of 3×105 cells/well and treated with chidamide
at different concentrations (0, 1, 2.5, 5 and 10 µM) in a 37°C
incubator with 5% CO2 for 48 h. The cells were then
collected, and their density was adjusted to 5×105
cells/ml. Then, 100 µl cell suspension was incubated with 5 µl
Annexin V-FITC at room temperature for 10 min in the dark, followed
by centrifugation at 1,000 × g for 5 min at room temperature.
Subsequently, 10 µl PI staining solution was added to the Annexin
V-FITC stained cells for 15 min at room temperature in dark, which
were then analyzed via flow cytometry (FACSCanto II; BD
Biosciences) and FACSCanto Clinical Software (v3.0; BD Biosciences)
to determine the percentage of Annexin V+/PI−
(early apoptotic) and Annexin V+/PI+ (late
apoptotic) cells. The Annexin V-FITC apoptotic cell detection kit
(Beijing TransGen Biotech Co., Ltd.) used included PI to label the
cellular DNA of dead cells with a compromised total membrane. Thus,
the kit allows the differentiation among early apoptotic cells
(Annexin V+, PI−), late apoptotic cells
(Annexin V+, PI+) and viable cells (Annexin
V−, PI−) in chidamide-treated DLBCL cells.
Early and late apoptosis were analyzed, and the apoptotic rate was
calculated as the percentage of early and late apoptotic cells.
RNA extraction and reverse
transcription-quantitative (RT-q)PCR analysis
SU-DHL2, OCI-LY3 and MZ DLBCL cells were treated for
48 h with chidamide (0, 2.5, 5 and 10 µM) in a 37°C incubator with
5% CO2, and total RNAs were extracted from the cells
using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocols.
RT-qPCR was performed with a UltraSYBR One Step RT-qPCR kit (cat.
no. CW0659S; CWBio) using the ABI 7500 Real-time PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.) in duplicate
for each reaction. The reverse transcription of total RNA into cDNA
was performed at 45°C for 10 min. The following thermocycling
conditions were used for qPCR: Initial denaturation at 95°C for 5
min; followed by 30 cycles at 95°C for 10 sec and 60°C for 45 sec.
Averages of the obtained Cq values were used for further
calculations. Gene expression levels were normalized to the
expression of the endogenous control gene, β-actin. The gene
expression levels were calculated using the 2−ΔΔCq
method (16). The primer sequences
used for the qPCR were as follows: STAT3 forward,
5′-GGAGGAGTTGCAGCAAAAAG-3′ and reverse, 5′-TGTGTTTGTGCCCAGAATGT-3′;
and β-actin forward, 5′-CTGGCACCACACCTTCTACA-3′ and reverse,
5′-AGCACAGCCTGGATAGCAAC-3′.
Western blot analysis
After SU-DHL2, OCI-LY3 and MZ DLBCL cells were
treated for 48 h with chidamide (at 0, 2.5, 5 and 10 µM) in a 37°C
incubator with 5% CO2, proteins were extracted from
these cells using RIPA lysis buffer (Beyotime Institute of
Biotechnology) and quantified using the BCA protein assay reagent
(CWBio). SDS-PAGE with 10% gels was used to isolate proteins (30 µg
per lane). The proteins were transferred to PVDF membranes, which
were then blocked with 5% BSA (Sigma-Aldrich; Merck KGaA) for 1 h
at room temperature. After incubation with the primary antibodies
overnight at 4°C, the membranes were washed three times with
Tween-20 (1:1,000 dilution)-PBS for 10 min each. Finally, the
membranes were treated with the appropriate HRP-conjugated
secondary antibodies (cat. nos. CW0103S and CW0102S; 1:5,000;
CWBio) for 1 h at room temperature, and protein bands were
visualized with ECL reagents (Corning, Inc.). Images were captured
and analyzed with a Universal Hood II Chemiluminescence Imaging
system (Bio-Rad Laboratories, Inc.). Densitometric analysis was
performed on the instrument.
Statistical analysis
Data are presented as the mean ± SD for the number
of indicated replicates. Statistical analyses were conducted using
GraphPad Prism version 5.0 (GraphPad Software, Inc.). A Student's
t-test was used to compare the difference between two groups.
Homogeneity of variance was assessed using Brown-Forsythe test.
Comparisons among multiple groups were performed using one-way
ANOVA followed by Tukey's test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Chidamide inhibits the viability of
DLBCL cells
To investigate the cellular effect of chidamide on
DLBCL cells, three cell lines of SU-DHL2, OCI-LY3 and MZ were
selected for this study. These cells were treated with chidamide at
different final concentrations (0, 0.1, 0.3, 1, 3, 10 and 30 µM)
for 24, 48 and 72 h. As presented in Fig. 1, the exposure of these DLBCL cells
to chidamide reduced their viability in both concentration- and
time-dependent manners, as determined via the cellular CCK-8 assay.
For instance, after 72 h of treatment, the calculated
IC50 values for the concentration-dependent effect of
chidamide inhibition were 2.722 µM for SU-DHL20 cells, 1.353 µM for
OCI-LY3 cells and 4.183 µM for MZ cells (Fig. 1). Studies of the time dependence of
cell viability by chidamide indicated that, for SU-DHL20 cells, the
IC50 values were 94.02 µM at 24 h, 4.984 µM at 48 h and
2.722 µM at 72 h (Fig. 1A). The
IC50 decreased with increasing chidamide concentration
and treatment time in these DLBCL cell lines. These data suggested
that chidamide can inhibit or decrease the cell viabilities of
these three DLBCL cell lines with different potency.
Chidamide induces apoptosis in DLBCL
cells
As the malignancy of DLBCL cells has been associated
with the inhibition of apoptosis (17), it was next determined whether
apoptosis was associated with the chidamide-induced reduction of
the DLBCL cell viability. SU-DHL2, OCI-LY3 and MZ cells were
treated with chidamide at different concentrations (0, 1, 2.5, 5
and 10 µM) for 48 h, and the apoptotic nature of cell death was
determined via flow cytometry assays and western blot analysis of
apoptosis-related proteins. As presented in Fig. 2, quantitative flow cytometry
indicated that Annexin V-FITC staining of apoptotic cells was
evident in all the three types of chidamide-treated DLBCL cells in
a dose-dependent manner (Fig. 2).
Early apoptotic cells were consistently present in all three types
of chidamide-treated DLBCL cells, and the percentages of late
apoptotic cells were increased as the chidamide concentration was
increased after 48 h exposure (Fig.
2). Moreover, flow cytometry assays demonstrated that SU-DHL2
cells were more sensitive to chidamide.
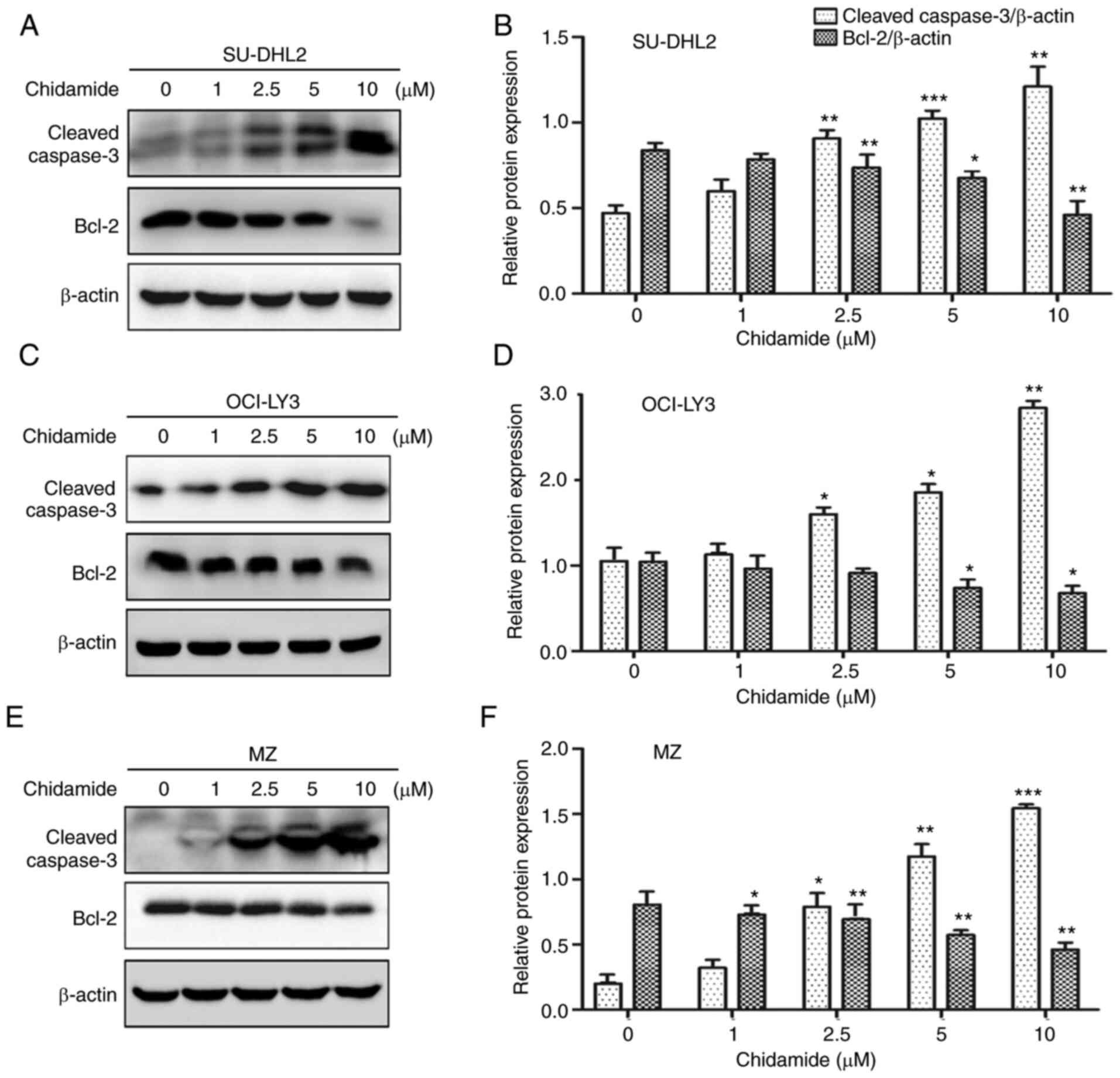
Western blot analysis of total proteins extracted
from the cells exposed to chidamide revealed an enhanced expression
level of cleaved caspase-3 and a decreased expression level of
Bcl-2 (Fig. 3), a further hallmark
of apoptosis (18); these effects
were exerted in a concentration-dependent manner. These results
suggest that chidamide inhibits cell viability in all three types
of DLBCL cells by stimulating a mechanism of apoptotic cell
death.
Chidamide suppresses the expression
levels of class I HDACs
Chidamide is a selective and potent inhibitor of the
activities of class I HDAC enzymes, including HDAC1, HDAC2, HDAC3
and HDAC8 (19). To investigate the
possible mechanism of chidamide-evoked apoptosis in DLBCL cells,
the expression levels of these class I HDACs were evaluated via
semi-quantitative western blot analysis in chidamide-treated
SU-DHL2, OCI-LY3 and MZ cells. Chidamide administration
significantly decreased the expression levels of HDAC1, HDAC2,
HDAC3 and HDAC8 in a concentration-dependent manner after 48 h
(Fig. 4). These results demonstrate
that chidamide can suppress the expression levels of class I HDAC
enzymes in all the three DLBCL cell lines tested under the current
experimental conditions.
Chidamide inhibits the activation of
STAT3 in DLBCL cells
Activation or phosphorylation of STAT3 has been
reported to serve a critical role in the malignant progression of
multiple tumors, including DLBCL (7). To further examine the possible
mechanism of chidamide-induced death of DLBCL cells, it was next
investigated whether chidamide influences the expression levels of
STAT3 and its phosphorylation in the DLBCL cells. The results
demonstrated that chidamide exposure significantly decrease both
the mRNA and protein expression levels of STAT3 in SU-DHL2, OCI-LY3
and MZ cells in a concentration-dependent manner during a 48 h
treatment, as determined via RT-qPCR (Fig. 5) and western blotting (Fig. 6). The levels of STATs
phosphorylation in these chidamide-treated DLBCL cells were
determined via western blotting, and a concentration-dependent
reduction of STAT3 phosphorylation was also identified in SU-DHL2,
OCI-LY3 and MZ cells. Collectively, these results indicate that
chidamide treatment suppresses STAT3 protein expression and also
inhibits its activation by phosphorylation in the tested DLBCL
cells.
Discussion
It has been well documented that histone
acetylation, which is regulated by the opposing activities of
histone acetyltransferases and HDACs, serves a critical role in the
development and progression of cancer by modulating gene
transcription, chromatin remodeling and nuclear architecture
(20). Along with histones, HDACs
can also deacetylate numerous non-histone cellular substrates, such
as the transcription factors p53 and NF-κB (21), to regulate a wide variety of
biological processes, including cancer initiation and progression
(20). Therefore, HDAC inhibitors,
acting as a potential therapeutic agent, are becoming increasing
promising in cancer treatment.
A high level of HDAC expression has been well
established in DLBCL cell lines and tissue sections, especially for
class I HDACs that are closely associated with the survival of
patients with DLBCL (22,23). Accordingly, HDAC inhibitors have
been widely used in clinical trials for DLBCL management (24). As a novel and orally active
benzamide class of HDAC inhibitor, chidamide has thus been approved
by the Chinese Food and Drug Administration authority in 2015 for
the treatment of lymphoma, in particular for PTCL, since it can
selectively inhibit the activities of class I HDACs, including
HDAC2, 3 and 10 (25–27). Importantly, the present study has
further demonstrated that chidamide can suppress the expression of
HDAC1, 2, 3 and 8 of the class I HDACs in three different DLBCL
cell lines in a concentration-dependent manner. These data suggest
that chidamide regulates the functions of HDACs by influencing
their abundance and activities, and thus enhances the current
understanding of the role of chidamide in HDACs.
To evaluate the clinical potential of chidamide in
the treatment of DLBCL, the current study determined whether
chidamide affects the viability of three different DLBCL cell
lines; SU-DHL2 and OCI-LY3 cells of the ABC-subtype and MZ cells of
the GCB-subtype. The present study found that exposure of these
DLBCL cells to chidamide for 72 h evoked both concentration- and
time-dependent inhibition of cell viability, displaying
IC50 values of 2.722 µM for SU-DHL2 cells, 1.353 µM for
OCI-LY3 cells and 4.183 µM for MZ cells. While these data do
suggest that chidamide is a potent inhibitor of cell viability in
DLBCL cells, they cannot provide conclusive information on which
subtype of DLBCL cells is more sensitive to chidamide as only three
cell lines were analyzed here.
To further examine the mechanism of chidamide-evoked
inhibition of cell viability in DLBCL cells, flow cytometry and
western blot analyses were performed to determine whether chidamide
induces apoptosis in these DLBCL cells. There is evidence that
apoptosis is involved in the initiation, progress and chemotherapy
resistance of DLBCL (28), and that
apoptosis can serve a vital role in governing the turnover of
normal cells to transformed cells. The present flow cytometry
analysis results of Annexin V-FITC staining of apoptotic cells
identified that early apoptotic cells (Annexin V+,
PI−) were consistently evident in all three types of
chidamide-treated DLBCL cells, and that the percentages of their
late apoptotic cells (Annexin V+, PI+) were
increased in a chidamide concentration-dependent manner following
48 h exposure. Herein, flow cytometry assays demonstrated that
SU-DHL2 cells were more sensitive to chidamide, possibly due to the
heterogeneity of these DLBCL cells. It is well established that
OCI-Ly3 cells contain the MYD88-L256P mutation, while SU-DHL2 cells
possess the mutations of MYD88-S222R and TNFAIP3/A20 (29,30).
The diverse mutation statuses of these cell lines may be the
primary causes of their various sensitivities to chidamide
treatment. Western blot analysis of total proteins extracted from
these chidamide-exposed cells further revealed the apoptotic nature
of cells, as indicated by enhanced cleavage of caspase-3 and
reduction of Bcl-2 protein expression. These data support the
previously reported pro-apoptotic effect of chidamide on DLBCL
cells (27,31). Taken together, these results suggest
that inhibition of cell viability by chidamide is executed by
stimulating apoptotic cell death in DLBCL cells.
As a transcription factor, STAT3 is constitutively
activated in various cancer cell lines and tumor tissues, where it
promotes tumor cell proliferation, invasion and migration (32). The activity of STAT3 is regulated by
diverse mechanisms, including an array of post-translational
modifications, such as phosphorylation (e.g., Tyr705 and Ser727)
(33). Since STAT3 serves vital
roles in the progression of most human cancer types, it has been
suggested that STAT3 could be an important candidate therapeutic
target for tumor treatment (34).
It has also been reported that different mechanisms are involved in
STAT3 inhibition, including suppression of tyrosine enzymes that
contribute to the phosphorylation or activation of STAT3,
inhibition of STAT3-mediated transcription, repression of nuclear
localization of STAT3 and inhibition of the interaction between
STAT3 and its target gene (35).
Nevertheless, remains unknown if and how chidamide influences STAT3
in DLBCL cells.
The present study demonstrated that the total mRNA
and protein expression levels of STAT3 were progressively decreased
by the administration of increasing concentrations of chidamide in
these DLBCL cells. A concentration dependent suppression of STAT3
phosphorylation (Tyr705 and Ser727) was also induced by chidamide
treatment in DLBCL cells. Interestingly, recent evidence has
indicated that valproic acid, another HDAC inhibitor, can inhibit
STAT3 Tyr705 phosphorylation, but had no effect on the total
protein expression level of STAT3 in natural killer cells (36). This difference with the current
findings may be due to the fact that different HDAC inhibitors and
cell types were used. The present results indicated that chidamide
could regulate STAT3 activities via its protein expression,
post-translational modification or both. It has been reported that
p65 and p50 NF-κB interact physically with STAT3, thereby
facilitating NF-κB recruitment to STAT3 promoters to modulate STAT3
expression (37). Thus, it was
suggested that administration of chidamide may attenuate the
transcriptional activity of NF-κB along with its inhibition of
HDACs activities to suppress STAT3 transcription. Although the
underlying mechanism via which chidamide restrains the expression
of STAT3 requires further study, the present results also
demonstrated that expression level of the anti-apoptotic Bcl-2
protein, a downstream target of STAT3 (38), was attenuated by chidamide
treatment, which decreased the expression level STAT3 and its
phosphorylation activities in DLBCL cells. This was consistent with
the finding that phosphorylation is a pattern of activity
regulation for STAT3 (39). In
future studies, deletion of Bcl-2 and STAT3 using small interfering
RNA will be performed to further reveal the mechanism of the
anticancer effects of chidamide.
In conclusion, the present study demonstrated that
chidamide can have a tumor-suppressive effect on DLBCL cells by
inducing apoptotic cell death, which is regulated by the
HDACs/STAT3/Bcl-2 pathway. As well as inhibiting the activity of
HDAC, chidamide may also inhibit the activity of STAT3 to function
as an apoptosis inducer in these DLBCL cells. Therefore, the
present results suggested that chidamide has great potential as a
therapeutic agent for the management of DLBCL. While these results
are encouraging, the current research is limited to determine which
of the ABC- and GCB-subtype cells or both are more sensitive to
chidamide as only three cell lines (two ABC subtypes and one GCB
subtype) were used. Additionally, the current data were only
obtained from these cell lines, which possess their inherent
limitations as any other in vitro models (40). Consequently, further research,
including sufficient numbers of two subtypes of DLBCL cells and
in vivo studies, is necessary to validate the antitumor
effect of chidamide and its precise underlying cellular mechanism
for its true clinical value to be revealed.
Acknowledgements
Not applicable.
Funding
This work was supported by Nature Science Project of
Shanxi, China (grant nos. 201701D121165 and 201901D111190), the
Research Project Supported by Shanxi Scholarship Council of China
(grant no. 2020-194), Key R & D Projects in Shanxi, China
(grant no. 201703D421023), the Open Fund from Key Laboratory of
Cellular Physiology (Shanxi Medical University), Ministry of
Education, China (grant no. KLMEC/SXMU-202011) and the Shanxi ‘1331
Project’ Key Subjects Construction, China (grant no. 1331KSC).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
BYu and LS designed the project. HZ, FC, KQ, XM and
LW performed the experiments. HZ, BYa, YW, MB and ZL analyzed the
data. HZ and BYa wrote the manuscript. All authors read and
approved the final manuscript. BYu and LS confirm the authenticity
of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
DLBCL
|
diffuse large B-cell lymphoma
|
|
CCK-8
|
Cell Counting Kit-8
|
|
HDAC
|
histone deacetylase
|
References
|
1
|
Kubuschok B, Held G and Pfreundschuh M:
Management of diffuse large B-cell lymphoma (DLBCL). Cancer Treat
Res. 165:271–288. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li S, Young KH and Medeiros LJ: Diffuse
large B-cell lymphoma. Pathology. 50:74–87. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kong Y, Chen G, Xu Z, Yang G, Li B, Wu X,
Xiao W, Xie B, Hu L, Sun X, et al: Pterostilbene induces apoptosis
and cell cycle arrest in diffuse large B-cell lymphoma cells. Sci
Rep. 6:374172016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hunt KE and Reichard KK: Diffuse large
B-cell lymphoma. Arch Pathol Lab Med. 132:118–124. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sehn LH and Gascoyne RD: Diffuse large
B-cell lymphoma: Optimizing outcome in the context of clinical and
biologic heterogeneity. Blood. 125:22–32. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mondello P and Mian M: Frontline treatment
of diffuse large B-cell lymphoma: Beyond R-CHOP. Hematol Oncol.
37:333–344. 2019. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pan YR, Chen CC, Chan YT, Wang HJ, Chien
FT, Chen YL, Liu JL and Yang MH: STAT3-coordinated migration
facilitates the dissemination of diffuse large B-cell lymphomas.
Nat Commun. 9:36962018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tamma R, Ingravallo G, Albano F, Gaudio F,
Annese T, Ruggieri S, Lorusso L, Errede M, Maiorano E, Specchia G
and Ribatti D: STAT-3 RNA scope determination in human diffuse
large B-cell lymphoma. Transl Oncol. 12:545–549. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lu K, Li B, Zhang H, Xu Z, Song D, Gao L,
Sun H, Li L, Wang Y, Feng Q, et al: A novel silicone derivative of
natural osalmid (DCZ0858) induces apoptosis and cell cycle arrest
in diffuse large B-cell lymphoma via the JAK2/STAT3 pathway. Signal
Transduct Target Ther. 5:312020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ding BB, Yu JJ, Yu RY, Mendez LM,
Shaknovich R, Zhang Y, Cattoretti G and Ye BH: Constitutively
activated STAT3 promotes cell proliferation and survival in the
activated B-cell subtype of diffuse large B-cell lymphomas. Blood.
111:1515–1523. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Moskowitz AJ and Horwitz SM: Targeting
histone deacetylases in T-cell lymphoma. Leuk Lymphoma.
58:1306–1319. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bai X, Jiang H, Han G and He Q: Chidamide
suppresses the glycolysis of triple negative breast cancer cells
partially by targeting the miR33a5pLDHA axis. Mol Med Rep.
20:1857–1865. 2019.PubMed/NCBI
|
|
13
|
Sun Y, Li J, Xu Z, Xu J, Shi M and Liu P:
Chidamide, a novel histone deacetylase inhibitor, inhibits multiple
myeloma cells proliferation through succinate dehydrogenase subunit
A. Am J Cancer Res. 9:574–584. 2019.PubMed/NCBI
|
|
14
|
Wu YF, Ou CC, Chien PJ, Chang HY, Ko JL
and Wang BY: Chidamide-induced ROS accumulation and
miR-129-3p-dependent cell cycle arrest in non-small lung cancer
cells. Phytomedicine. 56:94–102. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ubink I, Bolhaqueiro ACF, Elias SG, Raats
DAE, Constantinides A, Peters NA, Wassenaar ECE, de Hingh IHJT,
Rovers KP, van Grevenstein WMU, et al: Organoids from colorectal
peritoneal metastases as a platform for improving hyperthermic
intraperitoneal chemotherapy. Br J Surg. 106:1404–1414. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Best S, Hashiguchi T, Kittai A, Bruss N,
Paiva C, Okada C, Liu T, Berger A and Danilov AV: Targeting
ubiquitin-activating enzyme induces ER stress-mediated apoptosis in
B-cell lymphoma cells. Blood Adv. 3:51–62. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jin CY, Moon DO, Choi YH, Lee JD and Kim
GY: Bcl-2 and caspase-3 are major regulators in Agaricus
blazei-induced human leukemic U937 cell apoptosis through
dephoshorylation of Akt. Biol Pharm Bull. 30:1432–1437. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ning ZQ, Li ZB, Newman MJ, Shan S, Wang
XH, Pan DS, Zhang J, Dong M, Du X and Lu XP: Chidamide
(CS055/HBI-8000): A new histone deacetylase inhibitor of the
benzamide class with antitumor activity and the ability to enhance
immune cell-mediated tumor cell cytotoxicity. Cancer Chemother
Pharmacol. 69:901–909. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li Y and Seto E: HDACs and HDAC inhibitors
in cancer development and therapy. Cold Spring Harb Perspect Med.
6:a0268312016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Williams SA, Chen LF, Kwon H, Ruiz-Jarabo
CM, Verdin E and Greene WC: NF-kappaB p50 promotes HIV latency
through HDAC recruitment and repression of transcriptional
initiation. EMBO J. 25:139–149. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee SH, Yoo C, Im S, Jung JH, Choi HJ and
Yoo J: Expression of histone deacetylases in diffuse large B-cell
lymphoma and its clinical significance. Int J Med Sci. 11:994–1000.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gloghini A, Buglio D, Khaskhely NM,
Georgakis G, Orlowski RZ, Neelapu SS, Carbone A and Younes A:
Expression of histone deacetylases in lymphoma: Implication for the
development of selective inhibitors. Br J Haematol. 147:515–525.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang M, Fang X and Wang X: Emerging role
of histone deacetylase inhibitors in the treatment of diffuse large
B-cell lymphoma. Leuk Lymphoma. 61:763–775. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gao S, Li X, Zang J, Xu W and Zhang Y:
Preclinical and clinical studies of chidamide (CS055/HBI-8000), an
orally available subtype-selective HDAC inhibitor for cancer
therapy. Anticancer Agents Med Chem. 17:802–812. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chan TS, Tse E and Kwong YL: Chidamide in
the treatment of peripheral T-cell lymphoma. Onco Targets Therapy.
10:347–352. 2017. View Article : Google Scholar
|
|
27
|
Li Q, Huang J, Ou Y, Li Y and Wu Y:
Progressive diffuse large B-cell lymphoma with TP53 gene mutation
treated with chidamide-based chemotherapy. Immunotherapy.
11:265–272. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Miao Y, Medeiros LJ, Li Y, Li J and Young
KH: Genetic alterations and their clinical implications in DLBCL.
Nat Rev Clin Oncol. 16:634–652. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ngo VN, Young RM, Schmitz R, Jhavar S,
Xiao W, Lim KH, Kohlhammer H, Xu W, Yang Y, Zhao H, et al:
Oncogenically active MYD88 mutations in human lymphoma. Nature.
470:115–119. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Compagno M, Lim WK, Grunn A, Nandula SV,
Brahmachary M, Shen Q, Bertoni F, Ponzoni M, Scandurra M, Califano
A, et al: Mutations of multiple genes cause deregulation of
NF-kappaB in diffuse large B-cell lymphoma. Nature. 459:717–721.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guan XW, Wang HQ, Ban WW, Chang Z, Chen
HZ, Jia L and Liu FT: Novel HDAC inhibitor Chidamide synergizes
with Rituximab to inhibit diffuse large B-cell lymphoma tumour
growth by upregulating CD20. Cell Death Dis. 11:202020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Teng Y, Ross JL and Cowell JK: The
involvement of JAK-STAT3 in cell motility, invasion, and
metastasis. JAKSTAT. 3:e280862014.PubMed/NCBI
|
|
33
|
Sgrignani J, Garofalo M, Matkovic M,
Merulla J, Catapano CV and Cavalli A: Structural biology of STAT3
and its implications for anticancer therapies development. Int J
Mol Sci. 19:15912018. View Article : Google Scholar
|
|
34
|
Gharibi T, Babaloo Z, Hosseini A,
Abdollahpour-Alitappeh M, Hashemi V, Marofi F, Nejati K and
Baradaran B: Targeting STAT3 in cancer and autoimmune diseases. Eur
J Pharmacol. 878:1731072020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mohassab AM, Hassan HA, Abdelhamid D and
Abdel-Aziz M: STAT3 transcription factor as target for anti-cancer
therapy. Pharmacol Rep. 72:1101–1124. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ni L, Wang L, Yao C, Ni Z, Liu F, Gong C,
Zhu X, Yan X, Watowich SS, Lee DA and Zhu S: The histone
deacetylase inhibitor valproic acid inhibits NKG2D expression in
natural killer cells through suppression of STAT3 and HDAC3. Sci
Rep. 7:452662017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hoesel B and Schmid JA: The complexity of
NF-κB signaling in inflammation and cancer. Mol Cancer. 12:862013.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lu L, Zhu F, Li Y, Kimpara S, Hoang NM,
Pourdashti S and Rui L: Inhibition of the STAT3 target SGK1
sensitizes diffuse large B cell lymphoma cells to AKT inhibitors.
Blood Cancer J. 9:432019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Belo Y, Mielko Z, Nudelman H, Afek A,
Ben-David O, Shahar A, Zarivach R, Gordan R and Arbely E:
Unexpected implications of STAT3 acetylation revealed by genetic
encoding of acetyl-lysine. Biochim Biophys Acta Gen Subj.
1863:1343–1350. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang R, Lyu C, Lu W, Pu Y, Jiang Y and
Deng Q: Synergistic effect of programmed death-1 inhibitor and
programmed death-1 ligand-1 inhibitor combined with
chemotherapeutic drugs on DLBCL cell lines in vitro and in vivo. Am
J Cancer Res. 10:2800–2812. 2020.PubMed/NCBI
|