Introduction
Inflammatory bowel diseases (IBDs) are immune
disorders that cause chronic inflammation in the gastrointestinal
tract, and include ulcerative colitis (UC) and Crohn's disease (CD)
(1–3). UC is limited to the colon and
consists of diffuse mucosal inflammation, whereas CD can involve
inflammation of any part of the gastrointestinal tract (4,5).
It has been reported that China had the highest incidence of IBD in
Asia in recent years (6).
Numerous studies have reported that inflammation,
apoptosis, genetics, immune dysregulation and stress participate in
the development of UC (3,7–10).
Among these causes, apoptosis is considered to serve a critical
role in the pathogenesis of UC and multiple strategies have been
developed to treat IBD through the targeting of apoptosis. For
example, it has been reported that maggot extracts (11), conjugated linoleic acid (12) and oxymatrine (13) can inhibit the initiation and
progression of IBD. Mechanistic analysis has demonstrated that
these compounds exert their protective effects through the
inhibition of oxidative stress and inflammation in UC (11–13).
Acteoside (ACT) is a lipase inhibitor and a major
component of Ligustrum purpurascens (kudingcha tea)
(14). ACT has been reported to
regulate inflammation and immune responses (15,16). For example, it has been reported
that ACT inhibits the IL-1β-induced inflammation and apoptosis of
chondrocytes through inhibition of Janus kinase/STAT, thereby
protecting against surgery-induced osteoarthritis (17). Another study reported that ACT
inhibits lipopolysaccharide (LPS)-induced inflammation in acute
lung injury via regulation of the NF-κB signaling pathway (18). Hausmann et al (19) reported that ACT may ameliorate
intestinal inflammation in a dextran sulphate sodium (DSS)-induced
colitis model. However, the underlying mechanism via which ACT
exerts a protective role remains largely unknown. The present study
evaluated the mechanism underlying the protective effect of ACT in
UC.
Materials and methods
Mice
All experiments were performed according to the
Institutional Guidelines for the Care and Use of Laboratory Animals
in Research (20) and were
approved by the Biomedical Ethics Committee of Peking University
(approval no. LA2020356; Beijing, China). The 8-week-old C57BL/6
male mice (weight, 20–24 g; n=24) were obtained from the Department
of Laboratory Animal Science, Peking University Health Science
Center. The mice were housed at a temperature of 18–22°C and a
relative humidity of 50–60%. All mice had free access to food and
water, and were maintained under a fixed 12-h light/dark cycle.
Induction and evaluation of UC
Mice were randomly divided into four groups as
follows (n=6 per group): i) Control; ii) DSS; iii) DSS + 30 mg/kg
ACT; and iv) DSS + 60 mg/kg ACT. Mice were treated with drinking
water containing 4% DSS (FUJIFILM Wako Pure Chemical Corporation)
to induce UC (21,22). Mice had no restriction on the dose
of DSS solution for 7 consecutive days and control mice received
standard drinking water throughout the 7 days of the experiment.
ACT (Fig. 1A; CAS 61276-17-3;
Shanghai Aladdin Biochemical Technology Co., Ltd.) was administered
intraperitoneally (i.p.) every day for 7 consecutive days (the same
days the mice were given DSS). The body weight of the mice was
evaluated daily. The disease activity index (DAI) was used to
assess the mice daily as previously described (23); DAI=[(score of weight loss rate) +
(score of stool consistency) + (score of hematochezia)]/3.
Humane endpoint criteria for all experiments that
involved mice were assessed daily. Euthanasia was performed when
mice exhibited signs of distress, such as weight loss after
treatment ≥25%, hunched posture or loss of active movements.
Animals with diarrhea, but weight loss <25%, received daily i.p.
injections of saline (1 ml three times at 8-h intervals) to avoid
dehydration as previously described (24). Animals were euthanized using an
overdose of pentobarbital (i.p.; 150 mg/kg supplemented as
required) (25). The loss of
pedal withdrawal reflexes, and the cessation of respiration and
heartbeat for 6 min in mice were used to confirm that they were
dead. No mice died during the experiment.
Histological analysis and
immunohistochemistry
Hematoxylin and eosin (H&E) staining was
performed to assess the induction of IBD. After being euthanized,
the colon tissues from normal and IBD mice were collected and fixed
in 4% paraformaldehyde for 24 h at 37°C. The tissues were then
embedded using paraffin and were sectioned (thickness, 6 µm) using
an SM2500 microtome (Leica Microsystems GmbH). The sections were
stained using H&E for 10 min at 37°C according to a standard
protocol. For immunohistochemistry, unstained 4-mm sections were
cut from paraffin blocks and incubated at 65°C for 30 min. The
slides were deparaffinized in xylene followed by absolute ethanol
and subsequent rehydration in graded ethanol. Antigen retrieval was
performed by immersing slides in boiling citric acid buffer (pH
6.0) for 15 min. Following blocking of endogenous peroxidase
activity for 20 min in 3% hydrogen peroxide at room temperature,
the sections were incubated with the following primary antibodies
for 1 h at room temperature: Occludin (1:200; cat. no. 27260-1-AP;
ProteinTech Group, Inc.), zonula occludens-1 (ZO-1; 1:200; cat. no.
ab216880; Abcam) and claudin-2 (1:200; cat. no. ab53032; Abcam).
Subsequently, after three washes with PBS, the sections were
incubated with HRP-conjugated goat anti-rabbit IgG H&L
(1:1,000; cat. no. ab6721; Abcam) secondary antibodies at 37°C for
10 min. Sections were incubated with the strept-ABC complex reagent
for 15 min, and subsequently exposed to 3,3′-diaminobenzidine,
counterstained with hematoxylin and assessed using a confocal
microscope (Olympus Corporation).
Measurement of malondialdehyde (MDA),
superoxide dismutase (SOD), catalase (CAT) and glutathione (GSH)
levels
MDA, SOD, CAT and GSH in colon tissues were assessed
using the Lipid Oxidation MDA Assay kit (cat. no. S0131S; Beyotime
Institute of Biotechnology), Total SOD Assay kit (cat. no. S0101S;
Beyotime Institute of Biotechnology), CAT Detection kit (cat. no.
S0051; Beyotime Institute of Biotechnology) and Total GSH Assay kit
(cat. no. S0052; Beyotime Institute of Biotechnology),
respectively. All procedures were performed according to the
manufacturer's protocols. The GSH content, and the activities of
MDA, SOD and CAT were normalized to the corresponding total protein
content.
TUNEL assay
Paraffin-embedded colon sections were stained using
a TUNEL assay kit (Roche Diagnostics) at 37°C for 1 h and
counterstained with 2 µg/ml DAPI at room temperature for 10 min.
Mouse colon tissues were fixed in 4% paraformaldehyde at room
temperature for 24 h. Then, the colon tissues were cut into 5-µm
thick tissue sections which were treated with 0.1% Triton X-100 for
15 min (cat. no. X10010; Shanghai Angyi Biotechnology Co., Ltd.)
before staining. The apoptotic cells were quantified by counting
the number of positive cells in four fields with at least 400 cells
per field in each group. The apoptotic cells were assessed using a
fluorescence microscope (Olympus Corporation).
Western blotting
Caco-2 cell samples and mouse colon tissues were
collected and lysed using RIPA lysis buffer containing protease
inhibitor (cat. no. P0013C; Beyotime Institute of Biotechnology).
The protein concentration was assessed using a BCA protein assay
kit (cat. no. P0012; Beyotime Institute of Biotechnology). The
protein samples (30 µg/lane) were then separated by SDS-PAGE on
8–15% gels. The proteins were transferred onto nitrocellulose
membranes, which were blocked in 5% skimmed milk for 2 h at room
temperature and incubated with the following specific primary
antibodies overnight at 4°C: Occludin (1:3,000; cat. no.
27260-1-AP; ProteinTech Group, Inc.), ZO-1 (1:5,000; cat. no.
21773-1-AP; ProteinTech Group, Inc.), claudin-2 (1:500; cat. no.
ab53032; Abcam), Bcl-2 (1:5,000; cat. no. 12789-1-AP; ProteinTech
Group, Inc.), Bax (1:5,000; cat. no. 50599-2-Ig; ProteinTech Group,
Inc.), cleaved caspase-3 (1:500; cat. no. ab32042; Abcam),
caspase-3 (1:5,000; cat. no. ab32351; Abcam), heme oxygenase-1
(HO-1; 1:1,500; cat. no. 10701-1-AP; ProteinTech Group, Inc.), high
mobility group box 1 protein (HMGB1; 1:1,500; cat. no. 10829-1-AP;
ProteinTech Group, Inc.) and GAPDH (1:2,500; cat. no. ab9485;
Abcam). Then, the membranes were incubated with HRP-conjugated goat
anti-rabbit IgG H&L (1:3,000; cat. no. ab6721; Abcam) secondary
antibodies at room temperature for 1 h. The membranes were washed
using TBS with 0.05% Tween-20 three times and the protein levels
were assessed using the BeyoECL Star kit (cat. no. P0018AS;
Beyotime Institute of Biotechnology). Signals were quantified using
ImageJ software (version 1.47; National Institutes of Health).
Cell culture
The human colorectal adenocarcinoma Caco-2 cell line
with epithelial morphology was purchased from the American Type
Culture Collection. Cells were maintained in Dulbecco's Modified
Eagle Medium (Gibco; Thermo Fisher Scientific) supplemented with
10% FBS (Gibco; Thermo Fisher Scientific), 100 U/ml penicillin and
100 µg/ml streptomycin at 37°C under 5% CO2. When the
confluence reached 80% and the cells were highly differentiated,
they were treated with 2% DSS or sterilized water (control) for 24
h at 37°C. In addition, cells were pretreated with ACT (1, 10 and
100 µM) for 6 h at 37°C. Based on previous reports, the commonly
used HO-1 inhibitor tin protoporphyrin (SnPP; 2 µM; Shanghai
Aladdin Biochemical Technology Co., Ltd.) was used to pretreat
cells for 30 min to inhibit HO-1 expression before ACT treatment
(26,27). The experimental grouping of cells
was as follows: Control, DSS, DSS + ACT and SnPP + DSS + ACT
groups.
MTT assay
The viability of Caco-2 cells was evaluated using an
MTT assay kit (Beyotime Institute of Biotechnology) according to
the manufacturer's protocol. Briefly, cells were seeded into
96-well plates (2×103 cells/well) and incubated at 37°C
under 5% CO2 for 24 h. Subsequently, 20 µl MTT solution
was added to the cells for 4 h at 37°C. The culture medium was
carefully removed and 150 µl DMSO was used to dissolve the purple
formazan. The absorbance was measured at 570 nm.
ELISA
A total of 50 mg colonic tissues and Caco-2 cells
seeded into 96-well plates (5×103 cells/well) were
collected and added to 2–3 ml pre-cooled saline. After
homogenization with a glass homogenizer on ice, the tissue was
centrifuged at 5,000 × g for 8 min at 4°C and the supernatant was
collected to determine the total protein amount using the BCA
method. The concentrations of TNF-α, IL-1β and IL-6 were assessed
using the Mouse TNF-α ELISA kit (cat. no. PT512), Human TNF-α ELISA
kit (cat. no. PT518), Mouse IL-1β ELISA kit (cat. no. PI301), Human
IL-1β ELISA kit (cat. no. PI305), Mouse IL-6 ELISA kit (cat. no.
PI326) and Human IL-6 ELISA kit (cat. no. PI330) from Beyotime
Institute of Biotechnology, respectively. All procedures were
performed according to the manufacturer's protocols.
Bioinformatics and statistical
analyses
The potential targets of ACT were assessed using the
STITCH website (http://stitch.embl.de; version 5.0), a database of
protein-chemical interactions. The data are presented as the mean ±
SD and were analyzed using GraphPad Prism 8.0 software (GraphPad
Software, Inc.) using one-way ANOVA with Tukey's multiple
comparison test. All experiments were repeated at least three
times, unless otherwise stated. P<0.05 was considered to
indicate a statistically significant difference.
Results
ACT inhibits DSS-induced UC in
mice
The present study evaluated the potential protective
effect of ACT on DSS-induced UC. It was demonstrated that mice
displayed slow body weight gain at day 1–4 of DSS treatment and
exhibited gradual body weight loss during day 5, 6 and 7 of DSS
treatment. Further, 30 or 60 mg/kg ACT treatment significantly
elevated the body weight of DSS mice from day 4 onwards (Fig. 1B). DSS-treated mice also
demonstrated typical UC characteristics, including significant
shortening of the colon (Fig. 1C and
D), an acute inflammatory response with mucosal erosion,
congestion, edema, reduction of crypts and infiltration of
inflammatory cells such as neutrophils (Fig. 1E) and significantly increased DAI
(Fig. 1F) compared with in the
control group. Compared with the DSS group, DSS-induced colonic
shortening was also markedly improved by ACT treatment at 60 mg/kg
(Fig. 1C and D). Additionally,
mucosal inflammatory cell infiltration, erosion and edema were
significantly improved (Fig. 1E)
and DAI score was notably decreased (Fig. 1F) in the DSS + 30 mg/kg ACT group
and DSS + 60 mg/kg ACT group. These results suggested that UC
models were successfully established. Furthermore, these UC-related
changes were markedly attenuated by ACT, which suggested that ACT
prevented the progression of UC.
ACT inhibits DSS-induced inflammation
and oxidative stress in the colon tissues
Consistent with the protective effect of ACT on the
progression of UC, DSS significantly increased the levels of
inflammatory cytokines, including TNF-α, IL-1β and IL-6, compared
with those in the control group, and this was significantly
inhibited by 60 mg/kg ACT (Fig.
2A). Furthermore, the levels of oxidative stress markers were
assessed. As shown in Fig. 2B,
the levels of MDA were significantly increased in the DSS group
compared with those in the control group, which suggested that ROS
levels were increased in DSS-treated mice. Furthermore, the levels
of CAT, GSH and SOD were significantly decreased in the colon of
DSS-treated mice compared with those in the control group. As
expected, the alterations in MDA, CAT, GSH and SOD were markedly
reversed following treatment with 60 mg/kg ACT compared with in the
DSS group. These results suggested that ACT inhibited inflammation
and oxidative stress in the colon of mice with UC.
 | Figure 2.Inhibitory effect of ACT on
DSS-induced inflammation of the colon tissues. (A) Inhibitory
effect of ACT on DSS-induced levels of inflammatory cytokines,
including TNF-α, IL-1β and IL-6, assessed using ELISA. (B)
Inhibitory effect of ACT on DSS-induced alterations in the levels
of MDA, CAT, GSH and SOD. n=6. ***P<0.001 vs. control;
#P<0.05, ##P<0.01 and
###P<0.001 vs. DSS. ACT, acteoside; DSS, dextran
sulphate sodium; MDA, malondialdehyde; CAT, catalase; GSH,
glutathione; SOD, superoxide dismutase. |
ACT prevents DSS-induced colonic
barrier dysfunction
The effect of ACT on DSS-induced colonic barrier
dysfunction was evaluated. As presented in Fig. 3A and B, immunohistochemistry and
western blotting demonstrated that the protein expression levels of
occludin and ZO-1, two tight junction proteins, were significantly
decreased, whereas claudin-2 protein expression levels were
significantly increased in the colon of DSS-treated mice compared
with those in the control group. Occludin and ZO-1 expression were
noticeably stimulated and claudin-2 expression was markedly
inhibited by 60 mg/kg ACT. These results further demonstrated that
ACT prevented colonic barrier injury in mice with DSS-induced
UC.
ACT inhibits DSS-induced apoptosis in
mice colon tissues
To improve the understanding of the mechanism of
action of ACT, apoptosis was investigated. As presented in Fig. 4A, the TUNEL assay demonstrated
that DSS enhanced the number of TUNEL-positive cells which was then
reduced by 40 or 60 mg/kg ACT. Consistently, DSS significantly
increased the expression levels of apoptosis-related proteins,
including Bax and cleaved caspase-3/caspase-3, compared with those
in the control group, whereas DSS significantly decreased the
protein expression levels of Bcl-2 (Fig. 4B). Notably, these changes were
markedly altered by pretreatment with 60 mg/kg ACT. These results
suggested that ACT inhibited DSS-induced apoptosis.
ACT reverses the effect of DSS on the
protein expression levels of HO-1 and HMGB1 in mouse colon
tissues
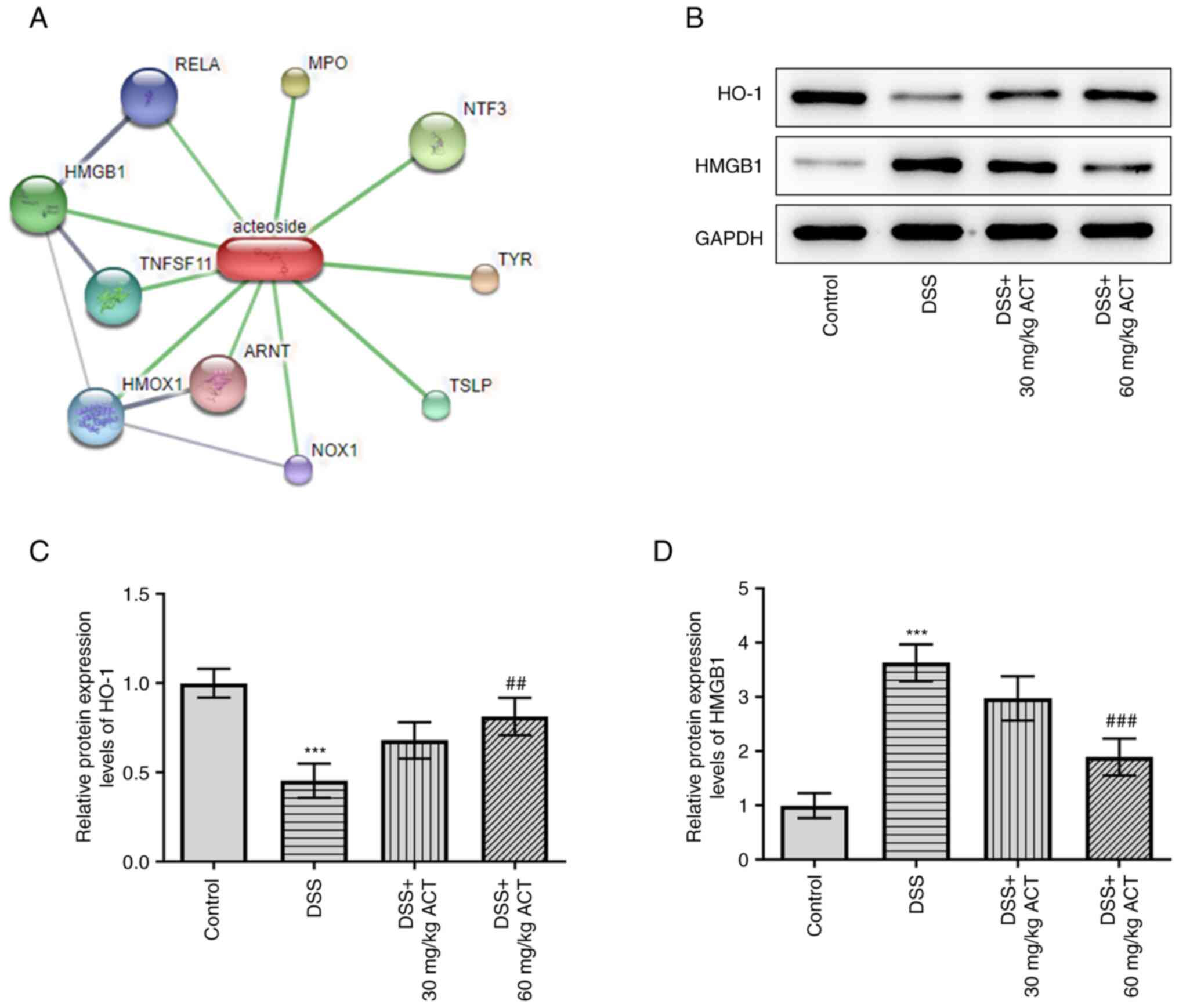
STITCH analysis was subsequently performed to
identify the potential binding protein of ACT (http://stitch.embl.de/cgi/network.pl?taskId=TjD9OOCPlkMM)
(Fig. 5A). HMGB1 was identified
as a potential binding target of ACT. Moreover, it has previously
been reported that HMGB1 can be regulated by HO-1 (28). The present study evaluated the
changes in the protein expression levels of HO-1/HMGB1 and the
potential effect of ACT on these changes. DSS significantly
decreased the protein expression levels of HO-1 compared with those
in the control group, whereas it markedly increased HMGB1 protein
expression levels (Fig. 5B-D).
Notably, the effect of DSS on the altered expression levels of HO-1
and HMGB1 was significantly reversed by 60 mg/kg ACT. These results
suggested that ACT may inhibit HMGB1 by promoting HO-1 expression
in colon tissues. Therefore, in subsequent experiments, the
relationship between ACT and HO-1 was further assessed using the
HO-1 inhibitor SnPP.
ACT prevents the DSS-induced decrease
of cell viability and inflammation via HMGB1 inhibition in a
HO-1-dependent manner in Caco-2 cells
The present study constructed an in vitro
colon injury model using DSS-induced Caco-2 cells, and a HO-1
inhibitor was used to further study whether the effects of ACT were
associated with regulation of the HO-1/HMGB1 signaling pathway. As
presented in Fig. 6A, ACT at 1,
10 and 100 µM had no significant effect on the viability of Caco-2
cells compared with that in the control group. However, DSS
significantly decreased the viability of Caco-2 cells compared with
that in the control group and the inhibitory effect was markedly
reversed by ACT in a dose-dependent manner with the most
significant effect at 100 µM ACT (Fig. 6B). Therefore, subsequent studies
were performed using ACT at a concentration of 100 µM. In addition,
DSS significantly increased the levels of inflammatory cytokines,
including TNF-α, IL-1β and IL-6, which were markedly inhibited by
ACT (Fig. 6C). However, in the
presence of SnPP, the inhibitory effect of ACT was markedly
reduced. Furthermore, DSS significantly increased MDA levels
compared with those in the control group, and significantly
decreased the levels of anti-oxidative stress proteins, including
CAT, GSH and SOD (Fig. 6D). ACT
markedly reversed the effect of DSS on the levels of these four
proteins; however, in the presence of SnPP, the inhibitory effects
of ACT were markedly reduced (Fig.
6D). These results were consistent with those of the in
vivo study (Fig. 2B) and
demonstrated that the addition of the HO-1 inhibitor effectively
reversed the effects of ACT.
 | Figure 6.ACT prevents DSS-induced inflammatory
response and oxidative stress through high mobility group box 1
protein inhibition via a heme oxygenase-1-dependent manner in
Caco-2 cells. (A) ACT demonstrated no significant effect on cell
viability in the MTT assay. (B) ACT had a dose-dependent inhibitory
effect on the DSS-induced reduction of cell viability. (C) SnPP
partially reversed the inhibitory effect of ACT on the DSS-induced
production of inflammatory cytokines, including TNF-α, IL-1β and
IL-6, in Caco-2 cells. (D) SnPP partially reversed the inhibitory
effect of ACT on DSS-induced oxidative stress. n=3. **P<0.01 and
***P<0.001 vs. control; #P<0.05,
##P<0.01 and ##P<0.001 vs. DSS;
@P<0.05 vs. DSS + ACT. ACT, acteoside; DSS, dextran
sulphate sodium; MDA, malondialdehyde; CAT, catalase; GSH,
glutathione; SOD, superoxide dismutase; SnPP, tin
protoporphyrin. |
ACT prevents DSS-induced cell injury
and alterations of apoptosis-related proteins through HMGB1
inhibition in a HO-1-dependent manner in Caco-2 cells
Besides inflammation, the present study evaluated
the effect of ACT on DSS-induced cell injury and apoptosis in
Caco-2 cells. As presented in Fig.
7A, DSS significantly decreased the protein expression levels
of occludin and ZO-1, and significantly increased the protein
expression levels of claudin-2 compared with those in the control
group. Pretreatment with ACT significantly inhibited the effects of
DSS and the inhibitory effect of ACT was markedly reduced in the
presence of SnPP. In addition, DSS significantly increased the
expression levels of apoptosis-related proteins, including Bax and
cleaved caspase-3, whereas DSS significantly decreased the protein
expression levels of Bcl-2 compared with those in the control group
(Fig. 7B). These changes were
significantly reversed by pretreatment with ACT compared with in
the DSS group. However, in the presence of SnPP, the effect of ACT
was significantly reduced compared with that in the DSS + ACT
group. Finally, the effect of ACT on the protein expression levels
of HMGB1 was evaluated. DSS significantly increased HMGB1
expression compared with that in the control group, which was
significantly inhibited by ACT. The inhibitory effect of ACT was
significantly attenuated by SnPP compared with that in the DSS +
ACT group. These results suggested that ACT prevented DSS-induced
cell injury in a HO-1-dependent manner in Caco-2 cells.
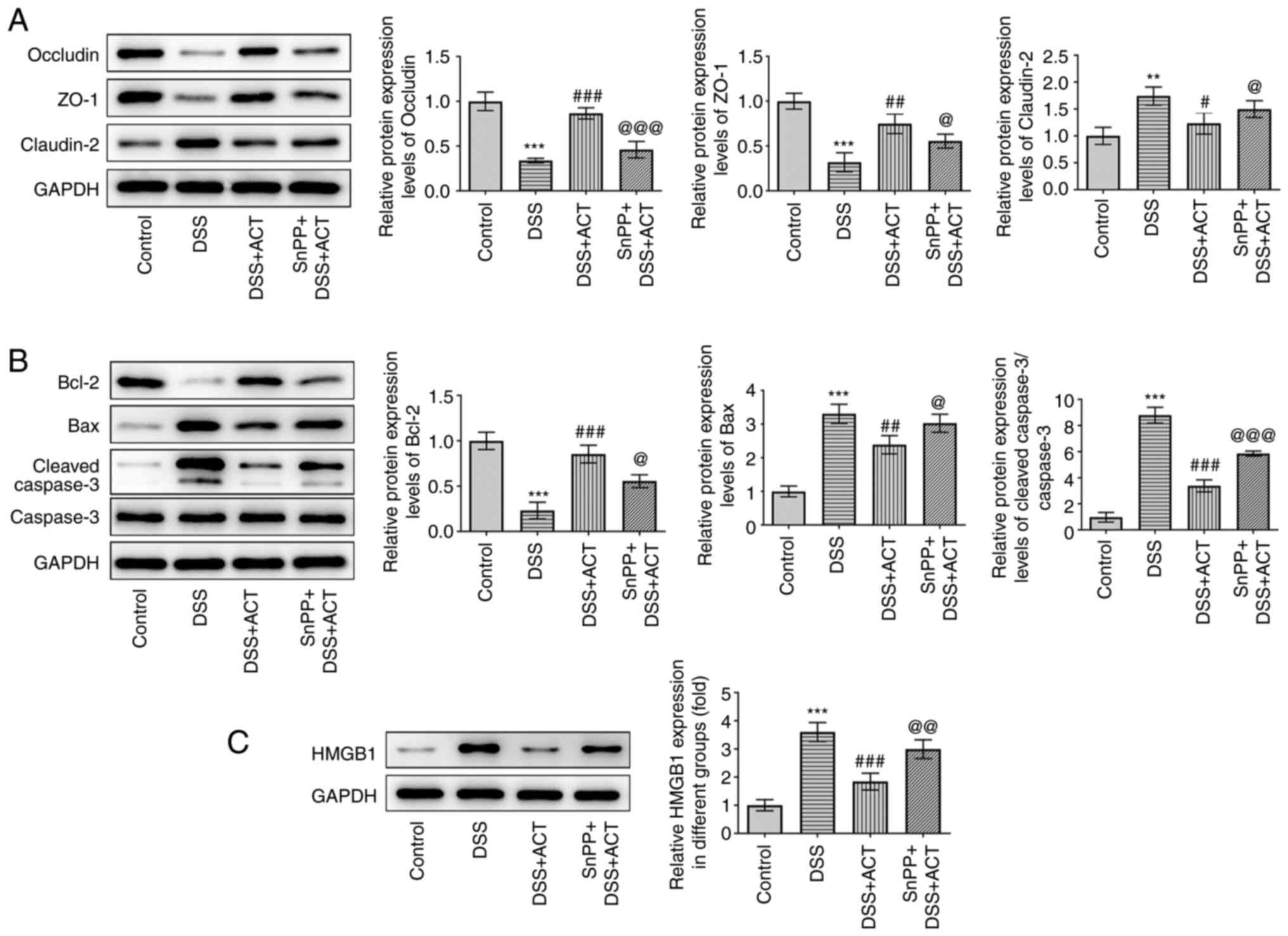 | Figure 7.ACT prevents DSS-induced barrier
dysfunction and apoptosis through HMGB1 inhibition in a heme
oxygenase-1-dependent manner in Caco-2 cells. (A) SnPP partially
reversed the inhibitory effect of ACT on DSS-induced changes of
tight junction components, including occludin, ZO-1 and claudin-2.
(B) SnPP partially reversed the inhibitory effect of ACT on the
expression of apoptosis-related proteins, including Bcl-2, Bax,
cleaved caspase-3 and caspase-3. (C) Representative western blot
images demonstrating the inhibitory effect of ACT on DSS-induced
HMGB1 expression with or without SnPP treatment. n=3. **P<0.01
and ***P<0.001 vs. control; #P<0.05,
##P<0.01 and ###P<0.001 vs. DSS;
@P<0.05, @@P<0.001 and
@@@P<0.001 vs. DSS + ACT. ACT, acteoside; DSS,
dextran sulphate sodium; HMGB1, high mobility group box 1 protein;
SnPP, tin protoporphyrin; ZO-1, zonula occludens-1. |
Discussion
An increasing number of studies have reported that
ACT inhibits inflammation in osteoarthritis (17), the gastrointestinal tract
(19,29) and LPS-induced acute lung injury
(18). Therefore, ACT may be
considered a promising drug for the treatment of
inflammation-related diseases. However, the underlying mechanisms
by which ACT exerts its protective effects remain largely unknown.
In the present study, research models were constructed using DSS
induction, and the effects and mechanisms were assessed using in
vivo and in vitro experiments. The results demonstrated
that ACT prevented the progression of UC and colonic barrier
dysfunction, and decreased inflammation and oxidative stress.
It has been reported that the degree of oxidative
stress, inflammation and apoptosis are aberrantly increased in UC
(30). Furthermore, apoptosis
serves an important role in the initiation and progression of UC,
which is at least partially activated by oxidative stress and
inflammation (30). In the
present study, ACT ameliorated DSS-induced UC injury, while also
demonstrating significant inhibitory effects on the inflammatory
factors TNF-α, IL-1β and IL-6. Consistent with previous reports
(31,32), the present study demonstrated that
ACT markedly inhibited DSS-induced oxidative stress and apoptosis.
More importantly, high concentrations of ACT (60 mg/kg)
significantly increased HO-1 expression and inhibited HMGB1
expression compared with those in the DSS group.
An increasing number of studies have reported that
HMGB1 serves a critical role in the initiation and progression of
UC, and that HMGB1 may be a potential marker for the diagnosis of
UC and gut inflammation (33,34). For example, it has been reported
that HMGB1 expression is increased in patients with severe UC
(35), and that the application
of HMGB1 antagonists may provide novel insights into the diagnosis
and treatment of UC (36).
Consistent with these previous studies, the present study
demonstrated that ACT markedly inhibited the increased HMGB1
expression in mice with DSS-induced UC. Considering the central
role of HMGB1 in UC, the findings of the present study, that ACT
inhibited HMGB1 expression, suggested that ACT may have potential
usage in the prevention and treatment of UC.
It has been reported that HO-1 is an upstream
regulator of HMGB1 and that gastrodin increases HO-1 expression,
leading to inhibition of HMGB1 and NF-κB in Tourette syndrome
(37). Wang et al
(38) reported that HO-1 inducers
(hemin and cobalt protoporphyrin IX) or transfection with HO-1
markedly inhibited LPS-induced HMGB1 release and translocation of
HMGB1 from the nucleus to the cytosol in RAW264.7 cells, a
monocyte/macrophage like cell lineage. Further in vivo study
demonstrated that HO-1 inducers improved the survival of mice in a
LPS- and cecal ligation and puncture-induced sepsis model (38). In the present study, the effects
of ACT were interfered with using the HO-1 inhibitor SnPP.
Consistent with the previous reports, the results of the present
study demonstrated that ACT significantly decreased the protein
expression levels of HMGB1 via the activation of HO-1.
In conclusion, the present study demonstrated that
ACT protected against the progression of UC. Notably, it was
suggested that the molecular mechanism by which ACT may exert its
protective effect was via the inhibition of HMGB1 in a
HO-1-dependent manner. This finding may provide the fundamental
basis for the potential clinical usage of this component in the
treatment of UC.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Beijing Natural Science
Foundation (grant no. 7204303).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WG, XW, FL, SC, SW and SD conceived and designed the
study, and acquired and interpreted the data. WG, XW, QZ, LY and FL
performed the experiments. SC, SW, QZ and LY wrote the manuscript.
WG, XW, FL, SC and SD confirm the authenticity of all the raw data.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments were performed according to
the Institutional Guidelines for the Care and Use of Laboratory
Animals in Research and were approved by the Biomedical Ethics
Committee of Peking University (approval no. LA2020356).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hodson R: Inflammatory bowel disease.
Nature. 540:S972016. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chu H, Khosravi A, Kusumawardhani IP, Kwon
AH, Vasconcelos AC, Cunha LD, Mayer AE, Shen Y, Wu WL, Kambal A, et
al: Gene-microbiota interactions contribute to the pathogenesis of
inflammatory bowel disease. Science. 352:1116–1120. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Neurath MF: Cytokines in inflammatory
bowel disease. Nat Rev Immunol. 14:329–342. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang YZ and Li YY: Inflammatory bowel
disease: Pathogenesis. World J Gastroenterol. 20:91–99. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ungar B and Kopylov U: Advances in the
development of new biologics in inflammatory bowel disease. Ann
Gastroenterol. 29:243–248. 2016.PubMed/NCBI
|
|
6
|
Ng SC, Tang W, Ching JY, Wong M, Chow CM,
Hui AJ, Wong TC, Leung VK, Tsang SW, Yu HH, et al: Incidence and
phenotype of inflammatory bowel disease based on results from the
Asia-pacific Crohn's and colitis epidemiology study.
Gastroenterology. 145:158–165.e152. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kaser A, Zeissig S and Blumberg RS:
Inflammatory bowel disease. Annu Rev Immunol. 28:573–621. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Maloy KJ and Powrie F: Intestinal
homeostasis and its breakdown in inflammatory bowel disease.
Nature. 474:298–306. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cho JH: The genetics and
immunopathogenesis of inflammatory bowel disease. Nat Rev Immunol.
8:458–466. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Khor B, Gardet A and Xavier RJ: Genetics
and pathogenesis of inflammatory bowel disease. Nature.
474:307–317. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang R, Luo Y, Lu Y, Wang D, Wang T, Pu W
and Wang Y: Maggot extracts alleviate inflammation and oxidative
stress in acute experimental colitis via the activation of Nrf2.
Oxid Med Cell Longev. 2019:47032532019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Saito M, Chen-Yoshikawa TF, Suetsugu K,
Okabe R, Takahagi A, Masuda S and Date H: Pirfenidone alleviates
lung ischemia-reperfusion injury in a rat model. J Thorac
Cardiovasc Surg. 158:289–296. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Y, Shou Z, Fan H, Xu M, Chen Q, Tang
Q, Liu X, Wu H, Zhang M, Yu T, et al: Protective effects of
oxymatrine against DSS-induced acute intestinal inflammation in
mice via blocking the RhoA/ROCK signaling pathway. Biosci Rep. Jul
18–2019.(Epub ahead of print). View Article : Google Scholar
|
|
14
|
Wu X, He W, Zhang H, Li Y, Liu Z and He Z:
Acteoside: A lipase inhibitor from the Chinese tea Ligustrum
purpurascens kudingcha. Food Chem. 142:306–310. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Esposito E, Mazzon E, Paterniti I, Dal
Toso R, Pressi G, Caminiti R and Cuzzocrea S: PPAR-alpha
contributes to the anti-inflammatory activity of verbascoside in a
model of inflammatory bowel disease in mice. PPAR Res.
2010:9173122010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gao H, Cui Y, Kang N, Liu X, Liu Y, Zou Y,
Zhang Z, Li X, Yang S, Li J, et al: Isoacteoside, a
dihydroxyphenylethyl glycoside, exhibits anti-inflammatory effects
through blocking toll-like receptor 4 dimerization. Br J Pharmacol.
174:2880–2896. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Qiao Z, Tang J, Wu W, Tang J and Liu M:
Acteoside inhibits inflammatory response via JAK/STAT signaling
pathway in osteoarthritic rats. BMC Complement Altern Med.
19:2642019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jing W, Chunhua M and Shumin W: Effects of
acteoside on lipopolysaccharide-induced inflammation in acute lung
injury via regulation of NF-kappaB pathway in vivo and in vitro.
Toxicol Appl Pharmacol. 285:128–135. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hausmann M, Obermeier F, Paper DH, Balan
K, Dunger N, Menzel K, Falk W, Schoelmerich J, Herfarth H and
Rogler G: In vivo treatment with the herbal phenylethanoid
acteoside ameliorates intestinal inflammation in dextran sulphate
sodium-induced colitis. Clin Exp Immunol. 148:373–381. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Committee for the Update of the Guide for
the Care and Use of Laboratory Animals, . Guide for the Care and
Use of Laboratory Animals. 8th edition. The National Academies
Press; Washington, DC: 2011
|
|
21
|
Liu YJ, Tang B, Wang FC, Tang L, Lei YY,
Luo Y, Huang SJ, Yang M, Wu LY, Wang W, et al: Parthenolide
ameliorates colon inflammation through regulating Treg/Th17 balance
in a gut microbiota-dependent manner. Theranostics. 10:5225–5241.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Eichele DD and Kharbanda KK: Dextran
sodium sulfate colitis murine model: An indispensable tool for
advancing our understanding of inflammatory bowel diseases
pathogenesis. World J Gastroenterol. 23:6016–6029. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Park YH, Kim N, Shim YK, Choi YJ, Nam RH,
Choi YJ, Ham MH, Suh JH, Lee SM, Lee CM, et al: Adequate dextran
sodium sulfate-induced colitis model in mice and effective outcome
measurement method. J Cancer Prev. 20:260–267. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen Z, Yu K, Zhu F and Gorczynski R:
Over-expression of CD200 protects mice from dextran sodium sulfate
induced colitis. PLoS One. 11:e01466812016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dutton JW III, Artwohl JE, Huang X and
Fortman JD: Assessment of pain associated with the injection of
sodium pentobarbital in laboratory mice (mus musculus). J Am Assoc
Lab Anim Sci. 58:373–379. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ren J, Su D, Li L, Cai H, Zhang M, Zhai J,
Li M, Wu X and Hu K: Anti-inflammatory effects of aureusidin in
LPS-stimulated RAW264.7 macrophages via suppressing NF-κB and
activating ROS- and MAPKs-dependent Nrf2/HO-1 signaling pathways.
Toxicol Appl Pharmacol. 387:1148462020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lien GS, Wu MS, Bien MY, Chen CH, Lin CH
and Chen BC: Epidermal growth factor stimulates nuclear factor-κB
activation and heme oxygenase-1 expression via c-Src, NADPH
oxidase, PI3K, and Akt in human colon cancer cells. PLoS One.
9:e1048912014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tsoyi K, Lee TY, Lee YS, Kim HJ, Seo HG,
Lee JH and Chang KC: Heme-oxygenase-1 induction and carbon
monoxide-releasing molecule inhibit lipopolysaccharide
(LPS)-induced high-mobility group box 1 release in vitro and
improve survival of mice in LPS- and cecal ligation and
puncture-induced sepsis model in vivo. Mol Pharmacol. 76:173–182.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Reinke D, Kritas S, Polychronopoulos P,
Skaltsounis AL, Aligiannis N and Tran CD: Herbal substance,
acteoside, alleviates intestinal mucositis in mice. Gastroenterol
Res Pract. 2015:3278722015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tian T, Wang Z and Zhang J:
Pathomechanisms of oxidative stress in inflammatory bowel disease
and potential antioxidant therapies. Oxid Med Cell Longev.
2017:45351942017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xia D, Zhang Z and Zhao Y: Acteoside
attenuates oxidative stress and neuronal apoptosis in rats with
focal cerebral ischemia-reperfusion injury. Biol Pharm Bull.
41:1645–1651. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Peerzada KJ, Faridi AH, Sharma L, Bhardwaj
SC, Satti NK, Shashi B and Tasduq SA: Acteoside-mediates
chemoprevention of experimental liver carcinogenesis through STAT-3
regulated oxidative stress and apoptosis. Environ Toxicol.
31:782–798. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nakov R: New markers in ulcerative
colitis. Clin Chim Acta. 497:141–146. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Palone F, Vitali R, Cucchiara S,
Pierdomenico M, Negroni A, Aloi M, Nuti F, Felice C, Armuzzi A and
Stronati L: Role of HMGB1 as a suitable biomarker of subclinical
intestinal inflammation and mucosal healing in patients with
inflammatory bowel disease. Inflamm Bowel Dis. 20:1448–1457. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen Y, Wu D and Sun L: Clinical
significance of high-mobility group box 1 protein (HMGB1) and
Nod-like receptor protein 3 (NLRP3) in patients with ulcerative
colitis. Med Sci Monit. 26:e9195302020.PubMed/NCBI
|
|
36
|
Hu Z, Wang X, Gong L, Wu G, Peng X and
Tang X: Role of high-mobility group box 1 protein in inflammatory
bowel disease. Inflamm Res. 64:557–563. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Long H, Ruan J, Zhang M, Wang C and Huang
Y: Gastrodin alleviates Tourette syndrome via
Nrf-2/HO-1/HMGB1/NF-small ka, CyrillicB pathway. J Biochem Mol
Toxicol. 33:e223892019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang J, Hu X, Xie J, Xu W and Jiang H:
Beta-1-adrenergic receptors mediate Nrf2-HO-1-HMGB1 axis regulation
to attenuate hypoxia/reoxygenation-induced cardiomyocytes injury in
vitro. Cell Physiol Biochem. 35:767–777. 2015. View Article : Google Scholar : PubMed/NCBI
|