Introduction
All-trans retinoic acid (ATRA) plays a vital role in
the differentiation of hepatic stellate cells (HSCs) (1). In advanced liver fibrosis, HSCs are
activated to shift from a quiescent phenotype into myofibroblasts
expressing α-smooth muscle actin (α-SMA), a marker of activated
hepatic stellate cells), in the absence of ATRA. The activation of
HSCs is inhibited when extracellular ATRA levels increase (2–4). In
the liver, ATRA binds to retinoic acid receptors β and α of HSCs to
restrain the production of collagen fibers (5–7). A
recent study revealed that ATRA can constrain HSC proliferation and
extracellular matrix (ECM) deposition via the JNK-dependent NF-κB
signaling pathway (8). Moreover,
ATRA can enhance the effect of cisplatin by inducing the
differentiation of tumor initiating cells and decreasing the
chemoresistant subpopulations of hepatocellular carcinoma (HCC)
cells (9).
HCC cells are located in a complex microenvironment
in which stromal cells, including HSCs, endothelial cells and
immune cells, participate in the development of tumors (10,11).
Among them, HSCs can be activated by tumor cells. In the tumor
microenvironment (TME), HSCs not only induce ECM deposition by
producing type I collagen, but also secrete various factors, such
as interleukin 6 (IL-6), IL-8 and monocyte chemoattractant
protein-1, to induce cancer development (12–17).
Hence, inhibition of HSC-induced tumor growth is an effective
strategy for anti-HCC therapy. Considering that activated HSCs are
located in the TME of HCC, it is possible that a combination
therapy based on ATRA and chemotherapeutic drugs might display a
synergistic anticancer therapeutic approach through HSCs
redifferentiation and tumor apoptosis.
However, free drug formulations are limited in their
clinical use due to systemic toxicity, short half-life and low
solubility (18). Nano-sized drug
delivery systems (NDDSs) have been demonstrated to be potential
carriers for the delivery of antitumor drugs (19,20).
Compared with traditional formulations, NDDSs possess a lower
systemic toxicity, longer circulation time in the blood and higher
bioavailability. Moreover, nano-sized formulations can improve the
selective solubility of insoluble drugs to tumor cells. Hyaluronic
acid (HA), a natural polysaccharide, is abundantly present in the
ECM and has non-toxic, non-immunogenic properties (21,22).
High-molecular weight HA has been shown to inhibit tumor
development, while a simple degradation process to low-molecular
weight HA by hyaluronidase in the TME allows for the low-molecular
weight HA to facilitate tumor migration (23). sHA (sulfated HA) has been
previously synthesized by introducing sulphation to the-OH groups
of HA polymers and used to block degradation by hyaluronidase, thus
inhibiting the proliferation, motility and invasion of tumor cells
(24,25). Amphiphilic conjugates based on HA
polymers are self-assembled into nanoparticles composed of
hydrophobic cores and hydrophilic shells (26,27).
Moreover, drug-loaded HA nanoparticles specifically bind to CD44
receptors, which are highly expressed on activated HSCs, thereby
enhancing the antitumor effect via CD44 receptor-mediated
endocytosis (28,29). The use of drug-loaded HA
nanoparticles has been demonstrated in cancer treatment, such as
that of breast cancer, HCC and colorectal cancer (30–32).
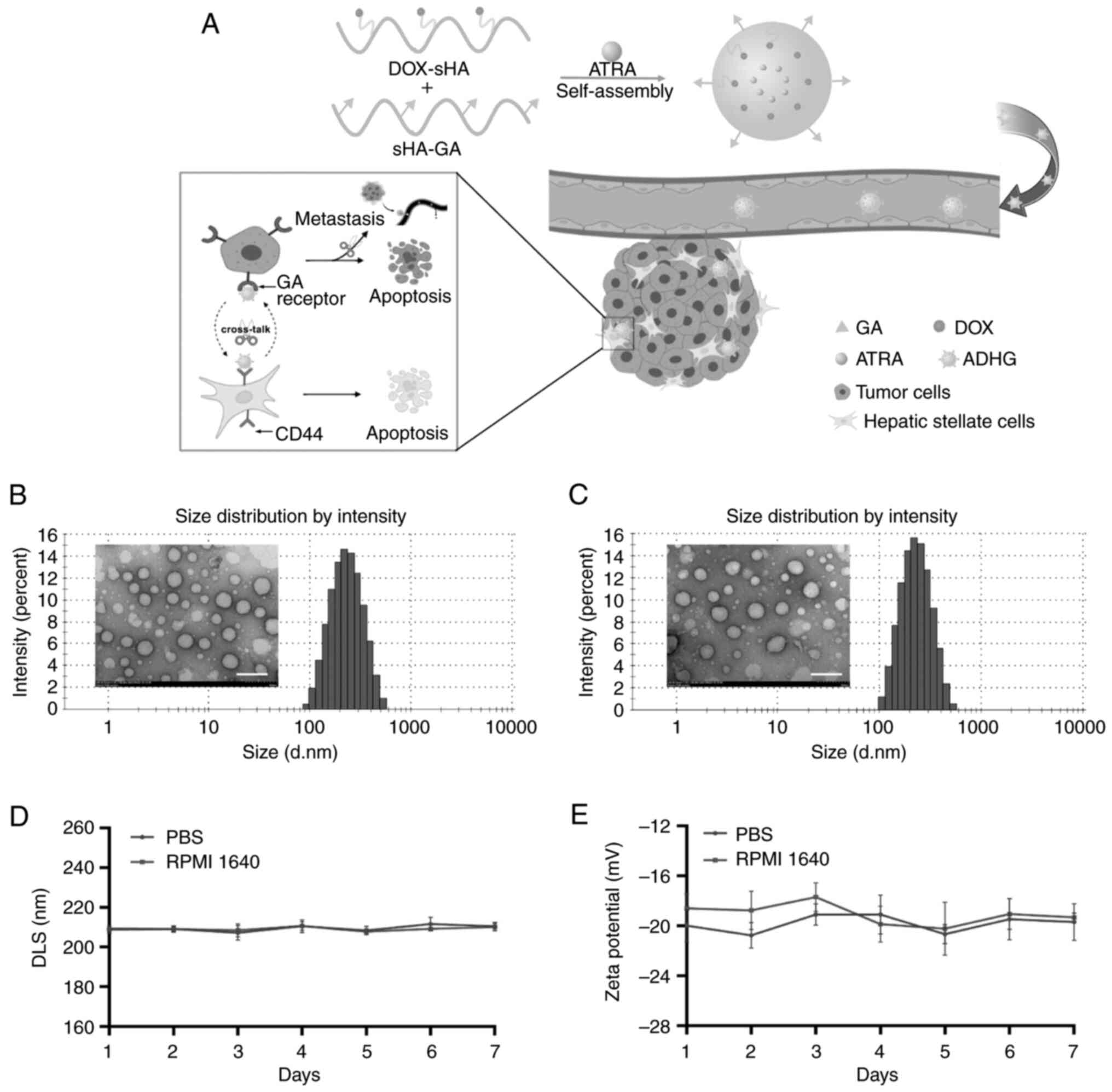
In the present study, liver-targeting co-loaded HA
nanoparticles (ADHG) were prepared to inhibit HSC-induced tumor
proliferation and metastasis (Fig.
1A). To mimic the TME, an in vitro dual-cell research
model and an in vivo co-implantation mouse model composed of
stromal cells and cancer cells were established for antitumor
studies. The anti-proliferation and the anti-migration effects of
ADHG were evaluated against cancer cells alone and the dual-cell
model. The drug distribution and the antitumor efficacy of ADHG was
also investigated in co-implantation mice.
Materials and methods
Materials
ATRA was purchased from Shanghai Aladdin Biochemical
Technology Co., Ltd. Doxorubicin (DOX), fluorescence probes
1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine iodide
(DiR), RPMI-1640 medium and the Calcein-AM/PI kit were obtained
from Dalian Meilun Biology Technology Co., Ltd. Cell culture medium
and H&E and Masson staining kit were acquired from Beijing
Solarbio Science & Technology Co., Ltd. The
5(6)-carboxyfluorescein diacetate succinimidyl ester (CFSE)
fluorescence dye was obtained from Abcam. Anhydrous ethanol and
xylene were of analytical grade (Analytical Reagent; Red
Label).
Cell lines and animals
Huh-7 cells (human HCC line; cat. no.
1101HUM-PUMC000679) were obtained from China Infrastructure of Cell
Line Resources, Institute of Basic Medical Sciences, Chinese
Academy of Medical Sciences. H22 (mouse HCC line; cat. no.
GDC0091), LX-2 (human HSC line; cat. no. GPC0076) and m-HSC (mouse
HSC line; cat. no. GPC0414) cells were purchased from the China
Center for Type Culture Collection. Human-derived cells and
mouse-derived cells were used for in vitro and in
vivo experiments, respectively. A total of 80 female BALB/c
mice (weight, 18 g; age, 6–8 weeks) were obtained from Jinan
Pengyue Experimental Animal Breeding Co., Ltd.
Preparation and characterization of
ADHG nanoparticles
The sHA is a polymer synthesized by grafting
chlorosulfonic acid onto HA. Considering that sHA could inhibit
hyaluronidase-induced HA degradation, sHA was used as the drug
carrier in the present study. The DOX-sHA (DH) polymers were
laboratory-made. DH and ATRA were dissolved in distilled water and
anhydrous ethanol solution, respectively. The two solutions were
mixed (final volumes, 10 ml) at different mass ratios of DH and
ATRA (10:1, 5:1 and 2.5:1), followed by ultrasonication for 10 min
(20 KHz) under ice water bath to prepare ATRA/DH (ADH)
nanoparticles using a probe-type sonicator (VCX-750; Sonics
Vibra-Cell™). To remove the unloaded ATRA, the mixed
solution was dialyzed in distilled water using a dialysis bag
(molecular weight cut-off, 3,500) at room temperature for 24 h.
Subsequently, the obtained solutions were freeze-dried at −45°C for
further study. Similarly, ADHG nanoparticles were prepared by
adding glycyrrhetinic acid (GA)-modified HA polymers to ADH
solution and sonicated (20 KHz) in an ice water bath for 15 min.
DOX-sHA-GA (DHG) nanoparticles were prepared by adding GA-modified
HA polymer to DH solution and sonicated using the same method.
The average particle size of ADHG was detected by a
Malvern Nano-ZS90 analyzer (33).
The morphology of nanoparticles was observed using a transmission
electron microscope (TEM; model no. HT7700; Hitachi
High-Technologies Corporation). The stability of ADHG nanoparticles
was examined in PBS solution (pH 7.4) or RPMI 1640 medium
containing 10% fetal bovine serum (FBS; ExCell Bio) for 7 days. The
ATRA content was calculated according to a standard curve, and the
drug-loading rate (DL) and encapsulation efficiency rate (EE) of
ATRA were measured using a UV spectrophotometer at 340 nm.
In vitro cytotoxicity
The in vitro dual-cell research model was
established by mixing cancer cells and HSCs at a ratio of 5:1
(34). The MTT assay was then used
to detect the cytotoxicity of ADHG. In brief, free ATRA, free DOX,
DOX + ATRA, DH, ADH and ADHG solutions were added into the 96-well
plates (5×103 cells/well) to achieve DOX concentrations
ranging from 0.01 to 10 µg/ml and incubated for 48 h at 37°C.
Dimethyl sulfoxide was used to dissolve the purple formazan and
cell viability was detected using a microplate reader at 490 nm.
The experiment was repeated in triplicate. Similarly, a live/dead
assay was performed using a Calcein-AM/PI kit (Meilun Biotechnology
Co., Ltd.), and the images were obtained using a fluorescent
microscope.
Wound-healing assay
To distinguish Huh-7 and LX-2 cells under a
microscope, LX-2 cells were labeled with CFSE. The wound-healing
assay was conducted using cancer cells and the dual-cell system
(35). Prior to the assay, the
cells were subjected to serum starvation (2% FBS) for 24 h in 37°C.
When the cells reached ~80% confluency in each well, the cells were
scratched using a 200 µl pipette tip to form a wound area and
subsequently treated with different drug solutions of DOX, ATRA,
DOX + ATRA, DH, ADH, ADHG (equivalent DOX, 2 µg/ml; ATRA, 1.47
µg/ml) for 24 h at 37°C. The wound-healing images were observed
with a Nikon fluorescence microscope, (Nikon eclipse Ti-S; Nikon
Corporation), and the cell migration rate was analyzed by the
edge-finding method using Image J 1.8.0 (National Institutes of
Health).
Cellular uptake
The liver-targeting ability of ADHG was measured
against Huh-7 cells and the dual-cell model in vitro. The
cells were treated with DOX, DH and DHG (equivalent DOX, 10 µg/ml)
for 3 h at 37°C, followed by DAPI (1 µg/ml) staining for 10 min and
observation using a confocal laser microscope (CLMS). The
intracellular DOX fluorescence intensity was also examined using
analyte reporter PE by flow cytometry (BD accuri c6 plus; BD
Biosciences) and analyzed by FlowJo v10 (BD Biosciences).
In vivo drug distribution
The liver-targeting capacity of ADHG was also
studied using an in vivo imaging system. An in vivo
co-implantation model was established by subcutaneous injection of
0.2 ml of 2×106 H22/m-HSC cells into the right flank,
and DiR was loaded in DHG nanoparticles to evaluate drug
distribution in vivo. The subcutaneous tumor-bearing mice
were injected intravenously with 0.2 ml free DiR, DH + DiR and DHG
+ DiR (DiR; 4 mg/kg). In vivo fluorescent images were
captured at different time points by the PerkinElmer IVIS Spectrum
system.
In vivo antitumor efficacy
The in vivo antitumor studies were performed
using tumor-bearing BALB/c mice. To mimic the TME, the
H22/m-HSC-bearing mice model was established. In the TME, the
activated HSCs could promote tumor growth, while the traditional
HCC-bearing mouse model was established by subcutaneously injecting
H22 cells. Each mouse was subcutaneously injected with 0.2 ml
2×106 H22/m-HSC cells (the ratio of H22 to m-HSC cells
was 5:1) or 2×106 H22 cells into the right flank. The
anticancer effect of ADHG was studied using these co-implantation
mice. When the cancerous volume grew to 150 mm3, the
mice were randomly divided into seven groups (n=5 per group), and
free DOX, free ATRA, DOX + ATRA, DH, ADH and ADHG were
administrated through the tail vein (equivalent DOX, 3 mg/kg; ATRA,
2.2 mg/kg) while the control group was injected with 0.2 ml of
saline. The body weight and tumor volume were detected every other
day. The mice were euthanized when they lost 20% of their original
body weight or when the tumor volume grew beyond 2,000
mm3. Mice health and behavior, including activity level,
appetite, body temperature, daily behavior, body weight and
respiratory rate, were monitored every 2 days. During the
experiment, mice were provided with comfortable and clean housing
conditions and were checked daily for any abnormal behavior. Mice
were anesthetized by intraperitoneal injection of 2% sodium
pentobarbital (45 mg/kg) and sacrificed by cervical dislocation
when the corneal response had disappeared, the skin pinch response
had disappeared and the muscles had relaxed. Following euthanasia,
the mice were verified as deceased by checking that the pain
response had disappeared and the heartbeat and respiration had
stopped. The mice were sacrificed 14 days after drug treatment, and
the tumors and major organs were removed for H&E staining,
Masson staining and immunohistochemistry assays. Following
euthanasia, tumors and major organs were removed and immersed in 4%
paraformaldehyde and fixed for 48 h at 4°C. After the tissue was
paraffin-embedded, sections of 4 µm thickness were prepared for
H&E staining and immunohistochemistry. The slices were dewaxed
in xylene twice for 10 min, followed by rehydration with 100%
ethanol twice for 10 min and 95, 80 and 70% ethanol for 5 min each.
For H&E staining, hematoxylin staining was performed for 8 min
and eosin staining for 2 min at room temperature using an H&E
staining kit (cat. no. G1120; Solarbio). Masson staining was
performed using a Masson staining kit (cat. no. G1346; Solarbio).
For immunohistochemistry, antigen repair by microwave heating at
98°C for 5 min in citrate buffer solution, permeabilization by 0.1%
Triton-100 (cat. no. T8200; Solarbio) for 15 min, endogenous
peroxidase blocker (cat. PV-6001; OriGene) at room temperature for
10 min and blocking in 5% albumin bovine V (cat. no. A8020;
Solarbio) for 60 min at 37°C were performed. Samples were incubated
with primary antibody at 37°C for 1 h [α-SMA antibody (cat. no.
AF1032; dilution, 1:200; Affinity Biosciences); anti-CD31 antibody
(cat. no. ab182981; dilution, 1:2,000; Abcam)]. As secondary
antibodies, enzyme-labeled goat anti-rabbit IgG polymer (cat. no.
PV-6001; OriGene) was applied for 20 min at room temperature. A DAB
color development kit (cat. no. ZLI-9017; OriGene) was used for
color development, and counterstaining was performed with
hematoxylin for 2 min at room temperature. The microscopy samples
were all observed under a light microscope (magnification,
×200).
Inhibition of lung metastases
In total, 0.2 ml of 2×106 H22 cells were
intravenously injected into the tail vein of BALB/c mice to
establish a lung metastasis model. The mice (n=4 per group) were
treated via the tail vein with physiological saline (control), free
DOX, free ATRA, DOX + ATRA, DH, ADH and ADHG (equivalent DOX, 3
mg/kg; ATRA, 2.2 mg/kg) every 2 days for 14 days. The health of the
mice was monitored every 2 days, including activity level,
appetite, body temperature, daily behavior, body weight and
respiratory rate. Mice were euthanized as described above.
Subsequently, the lungs were acquired for H&E staining assay
according to the protocol stated above.
Statistical analysis
All statistical analyses were performed using
GraphPad Prism 9.0.0 (Dotmatics) and are presented as mean ± SD.
All experiments were repeated at least three times. Results were
analyzed using one-way or two-way ANOVA followed by Bonferroni's
multiple-comparisons test. P<0.05 was considered to indicate a
statistically significant difference.
Results and Discussion
Morphology and characteristics of ADHG
nanoparticles
The amphiphilic HA conjugates self-assembled into
nano-sized particles. ADHG nanoparticles based on DHG were prepared
for co-encapsulating ATRA and DOX, and GA-free co-loaded
nanoparticles were named ADH. As shown in Table I, when the ratio of DHG to ATRA was
5:1, the average size of the nanoparticles was 211.95 nm, which was
smaller than that of other groups. The DL and EE were 8.79 and
52.72%, respectively. In addition, the morphology of DHG and ADHG
were investigated by TEM. In Fig. 1B
and C, the dual drug-loaded nanoparticles are shown to be
similarly spherical in shape.
 | Table I.Characterization of the different
nanoparticle formulations. |
Table I.
Characterization of the different
nanoparticle formulations.
| DHG:ATRA | DLS, nm | PDI | ζ-Potential,
mV | EE, % | DL, % |
|---|
| 10:1 | 258.40±3.41 | 0.149±0.02 | −18.01±0.55 | 84.85±7.18 | 7.71±0.65 |
| 5:1 | 211.95±2.61 | 0.110±0.01 | −18.97±0.46 | 52.72±5.18 | 8.79±0.86 |
| 2.5:1 | 240.70±1.13 | 0.158±0.01 | −22.87±0.42 | 33.89±4.29 | 9.68±1.23 |
To evaluate the stability of the nano-delivery
system, the characteristics of ADHG were measured for 7 days.
Fig. 1D and E shows that no
notable changes were observed in particle size and zeta potential
within 7 days, suggesting that ADHG nanoparticles were stable under
physiological conditions.
Cellular uptake and cytotoxicity
assay
In the TME, stromal cells, such as HSCs, have been
proven to facilitate cancer development (36). To simulate the TME, a dual-cell
research model was established by mixing HSCs and HCC cells
together, while HCC cells alone were used as the control (Fig. 2A).
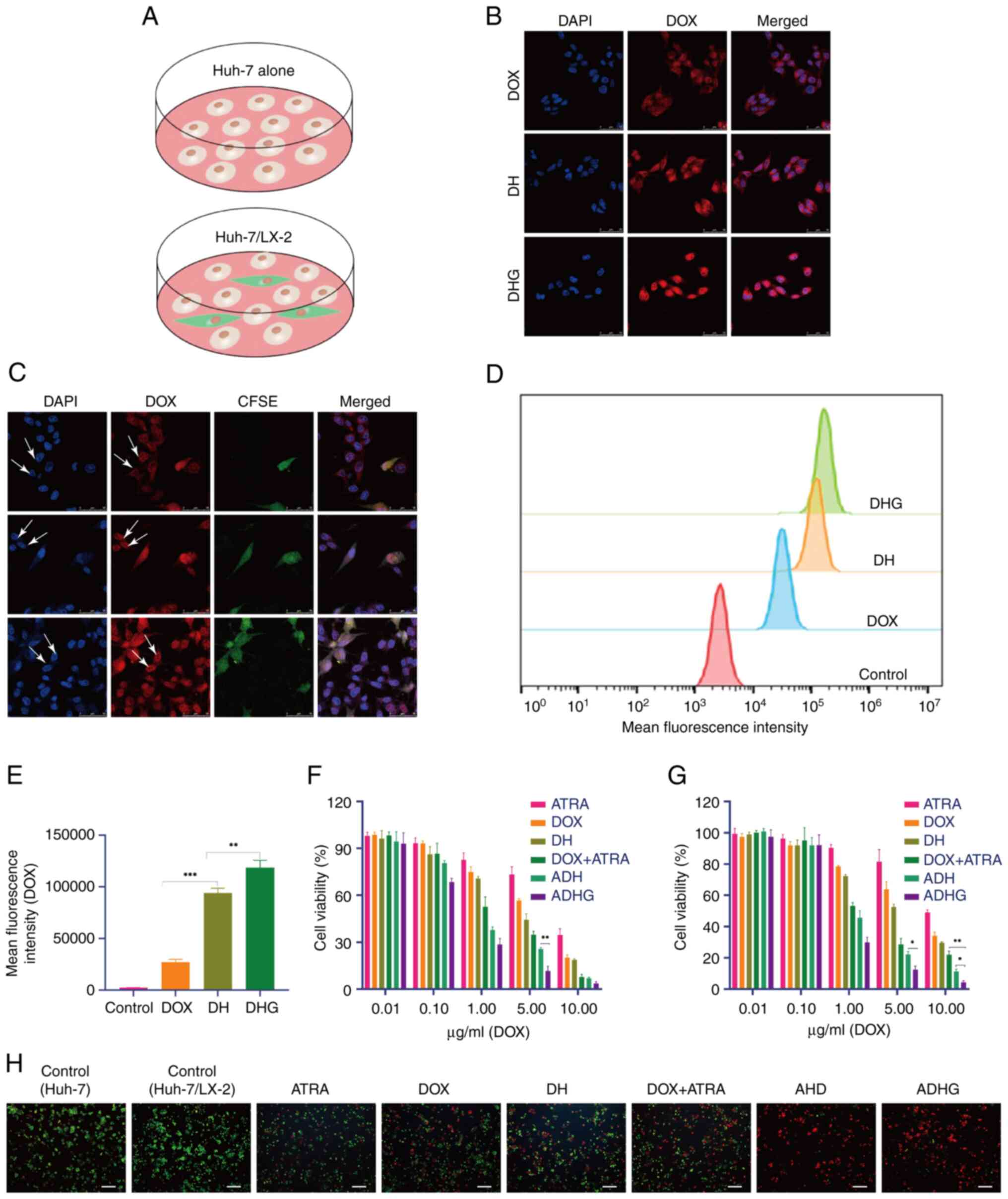 | Figure 2.Cellular uptake and cytotoxicity. (A)
Two research models used in the present study. Images of (B) Huh-7
cells and (C) the dual-cell model. White arrows indicate the
fluorescence intensity of Huh-7 cells. (D) Flow cytometry assay and
(E) its quantitative analysis. Cytotoxicity of (F) Huh-7 cells
alone and (G) the dual-cell model. (H) Live/dead staining test
(scale bar, 100 µm). *P<0.05, **P<0.01 and ***P<0.001.
DHG, DOX-sHA-GA; DH, DOX-sHA; DOX, doxorubicin; CFSE,
5(6)-carboxyfluorescein diacetate succinimidyl ester; ATRA,
all-trans retinoic acid; ADH, ATRA/DOX-sHA; ADHG, ATRA/DOX-sHA-GA;
sHA, sulfated hyaluronic acid. |
The cellular uptake of ADHG was detected in HCC
cells, in which red fluorescence (DOX) tracked the intracellular
drug distribution, and blue fluorescence (DAPI) indicated nuclear
location. Considering that GA receptors are highly expressed in
liver cancer cells, DHG nanoparticles were prepared for co-delivery
of DOX and ATRA. As shown in Fig.
2B, compared with ADH, there was more red fluorescence signal
in the cells treated with ADHG. One possible explanation for this
is that the GA ligand promoted ADHG internalization via GA/GA
receptor-mediated endocytosis.
In fact, the HCC-HSCs interaction has been shown to
facilitate cancer development; hence, targeting both HSCs and tumor
cells might be a potential anticancer strategy. In the present
study, a dual-cell model was established in which HSCs were marked
with CFSE dye. In Fig. 2C, strong
red fluorescence was observed in both HSCs and Huh-7 cells,
indicating that the drug was taken up by the cells. Considering
that the CD44 protein is highly expressed on HSCs (37), ADHG might have been taken up by
HSCs and Huh-7 cells through HA/CD44- and GA/GA receptor-mediated
internalization, respectively. This result was also confirmed by
FCM (Fig. 2D and E), in which more
drugs were detected in the cells treated with ADHG.
In the present study, MTT assays were performed on
HCC cells and the dual-cell model. In Fig. 2F and G, all drug-treated groups
exhibited dose-dependent cytotoxicity. Of note, the dual-drug
groups exhibited a greater inhibitory effect than single-drug
groups, suggesting that the combined treatment of DOX and ATRA
enhanced the antitumor effect. As expected, ADHG had lower cell
viability than the other groups. Since ATRA were modified by the GA
ligand, it is indicated that GA/GA receptor-mediated drug
internalization promoted apoptosis.
Similarly, the cytotoxicity of ADHG in the dual-cell
model was detected by MTT assay. On note, the IC50 of
free DOX was 6.627 µg/ml, which was 1.8-fold of that in Huh-7 cells
alone. This result suggested that HSCs might induce drug resistance
of the tumor cells in the dual-cell model. In fact, it has been
shown that cancer cells exist in a complex TME, in which HSCs have
a vital role in cancer development through the HSC-cancer cell
interaction (38). Hence, the
dual-cell research model was an appropriate model for anticancer
studies in vitro. Furthermore, in the two nano-sized drug
groups, particularly ADHG, higher cytotoxicity was observed than in
the DOX + ATRA group. Live/dead staining (Fig. 2H) was further performed on the
dual-cell model. Of note, more red-stained cells (dead cells) were
observed in the ADHG group, indicating that ADHG was able to
efficiently constrain cancer cell proliferation in the presence of
HSCs.
Wound-healing assay
To examine the anti-migration efficacy of ADHG
nanoparticles, a wound-healing assay was performed on the HCC
(Huh-7) cells (Fig. 3A). However,
it has been shown that HSCs can promote tumor migration by
secreting numerous pro-migration factors (39). To simulate the TME, a dual-cell
model composed of Huh-7 and CFSE-labeled HSCs was established
(Fig. 3B). Compared with the
control, different drug formulations had inhibitory effects on cell
migration in Huh-7 cells (Fig.
3C). As expected, ADHG treatment inferred a lower migration
rate in comparison with ADH or DOX + ATRA, suggesting that the
nano-sized therapeutic combination approach could promote
anti-migration efficacy.
Interestingly, in Fig.
3D, the migration rate in the dual-cell model (36.75%) was
higher than in Huh-7 cells alone (27.55%) in the control group,
suggesting that HSCs facilitated the migration of Huh-7 cells. ADHG
resulted in a greater inhibitory effect than other drug
formulations, indicating that the proliferation and migration of
HCC could be effectively restrained by the nano-sized combination
therapy based on DHG conjugates.
In vivo imaging experiments
To investigate the cancer-targeting property of DHG
nanoparticles, DiR-loaded nanoparticles were prepared and injected
into tumor-bearing mice. Fig. 4
shows that the fluorescent signals of free DiR groups in the tumor
area reached their peak at 12 h, then notably decreased. Compared
with the control, the dual drug-loaded nanoparticles showed
stronger fluorescence in the cancer region. A possible explanation
is that the nano-sized vehicles could accumulate in the tumor due
to enhanced permeability and retention effects (40,41).
As expected, the fluorescence intensity of the DHG + DiR group was
higher than that of DH + DiR at 48 h, suggesting that the GA ligand
promoted drug internalization in cancer cells.
In vivo antitumor effect against
HCC/HSC co-implanted mice
In the TME, the activated HSCs could promote tumor
growth, while the traditional HCC-bearing mouse model was
established by subcutaneously injecting H22 cells alone. To
simulate the TME, an in vivo HCC/HSC-bearing mice model was
established to examine the anticancer effect of ADHG (Fig. 5A). In a previous study, by our
group the DOX-loaded formulation at 3 mg/kg could inhibit HCC
development (42). Hence, in the
present study, 3 mg/kg of DOX was chosen for the in vivo
antitumor studies. In the present study, 5 mice in the
co-implantation control 2 group reached the humane endpoint on day
11, with tumor volumes exceeding 2,000 mm3, and these 5
mice were euthanized. Of note, compared with the traditional H22
tumor-bearing mice model (control 1, saline), the cancerous volume
in the co-implantation model (control 2, saline) was greater
(Fig. 5B and C). Since HSCs are
located in the TME, HCC/HSC-bearing mice are a more appropriate
model than HCC-bearing model for anti-hepatoma studies. After 14
days, cancer progression was constrained in all drug-treated
groups. Similar to the in vitro results, the dual-drug
groups exhibited smaller tumor sizes than the single-drug groups,
suggesting that ATRA promoted a DOX-induced anti-proliferative
effect. Moreover, the inhibition rate of the ADHG group was 85.8%,
which was higher than that in the DOX + ATRA (67.9%) or ADH (76.0%)
groups (Fig. 5D). The
anti-proliferative assay result was also confirmed by the results
of the H&E assay, in which severe apoptosis was observed in the
ADHG group (Fig. 5E). One
explanation may be that ADHG was internalized by the cancer cells
and HSCs in an active-targeting manner (HA/CD44 and/or GA/GA
receptor-mediated endocytosis), resulting in a greater inhibitory
effect.
 | Figure 5.Evaluation of anti-hepatoma activity
in H22/m-HSC-bearing mice. (A) Treatment schedule. (B) Tumor
images. (C) Tumor volumes. (D) Tumor growth inhibition rates. (E)
H&E and (F) Masson staining assays. Immunohistochemical assays
of (G) α-SMA and (H) CD31 proteins, with red arrows indicating the
tumor vessels (scale bars, 50 µm). *P<0.05 and ***P<0.001.
m-HSC, mouse hepatic stellate cells; SMA, smooth muscle actin;
ATRA, all-trans retinoic acid; ADH, ATRA/DOX-sHA; ADHG,
ATRA/DOX-sHA-GA; DOX, doxorubicin; DH, DOX-sHA; sHA, sulfated
hyaluronic acid. |
In addition, Masson staining and
immunohistochemistry assays were conducted to further investigate
the anticancer effect of ADHG. Studies have revealed that activated
HSCs have an important role in ECM deposition by secreting collagen
fibers (43,44). Fig.
5F indicated that the blue-colored region (collagen fiber) was
larger in Control 2 (H22/m-HSC) than that in Control 1 (H22), which
may be due to HSC activation. By contrast, there was almost no
blue-colored area in the ADHG group indicating negative ECM
deposition. In Fig. 5G, a notable
α-SMA+ region was present in Control 2 (H22/m-HSCS),
suggesting that activated HSCs were distributed in the cancerous
tissues. Furthermore, the α-SMA+ region was decreased in
the ADHG group, indicating that ADHG could block HSC activation,
resulting in lower ECM deposition. In addition, it has been proven
that tumor angiogenesis may promote tumor proliferation and
metastasis (45,46). The platelet endothelial cell
adhesion molecule CD31, a marker of blood vessels, was used to
examine angiogenesis. In Fig. 5H,
compared with the other drug-treated groups, there were fewer
vessels in the ADHG group, indicating that ADHG was able to inhibit
tumor angiogenesis. Hence, the combined therapeutic design based on
HA polymers could suppress HSCs-HCC interaction and constrain HCC
progression.
In vivo suppression of lung
metastasis
Mice with lung metastases were established to
examine the anti-metastatic effect of ADHG (Fig. 6A). Considering that the metastatic
nodules in the lungs were very small, it was difficult to monitor
the progression of internal tumors in real-time. According to
previous lung metastasis studies (47,48),
the lung tissues were harvested to evaluated the anti-metastasis
effect of different drug groups. Similar to the anti-proliferation
evaluation in vivo, no significant differences in mice
weight were observed between the healthy control and the
nanoparticle-treated mice (Fig.
6B). Notably, numerous metastatic nodules occurred in the
control model (Fig. 6C and D), and
the dual-drug treatment groups contained fewer metastatic nodules
than the free DOX group, indicating that ATRA improved the
anti-metastatic effect of DOX. Notably, the ADHG group presented
the smallest number of nodules among all drug-treated groups. This
result was consistent with the H&E assay. Fig. 6E showed that no notable tumor
regions were observed in the lungs of the mice treated with ADHG.
These results may be due to the fact that ADHG could be taken up by
metastatic HCC cells, resulting in an enhanced anti-proliferative
and anti-metastatic effect.
Conclusion
In summary, DHG conjugates were synthesized for
co-delivery of ATRA and DOX for anti-HCC therapy, and HCC/HSC
research models were established for the in vitro and the
in vivo anti-HCC studies. The results showed that ADHG
nanoparticles were spherical in shape with high stability and were
readily taken up by HSCs and HCC cells simultaneously. Compared
with free DOX, ATRA increased the inhibitory effect on tumor
proliferation and migration. The in vivo biodistribution
images indicated that DHG nanoparticles accumulated in the tumor
region. The in vivo anticancer results showed that, compared
with other groups, ADHG nanoparticles exhibited lower ECM
deposition, less tumor angiogenesis and stronger pro-apoptotic and
anti-metastatic effects. However, further studies are needed to
explore the anti-tumor mechanisms of ADHG nanoparticles, and there
are still many difficulties to overcome in terms of clinical
applications.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Science Foundation of
China (grant no. 81803464), the Sci-Technology Development Program
of Weifang (grant no. 2021GX053) and Scientific Research Project of
Weifang University of Science and Technology (grant nos. 2021XKJS12
and 2021XKJS19).
Availability of data and materials
The datasets used and/or analyzed in the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JW designed the experiments. QL, XG and GJ performed
the antitumor studies and drafted the manuscript. SL performed the
in vitro and in vivo mouse model experiments. KS and
GT analyzed the data. GT and SL confirm the authenticity of all the
raw data. JW edited the manuscript. All authors have read and
approved the final version manuscript.
Ethics approval and consent to
participate
The present study was approved by the Animal Ethics
Committee of Weifang Medical University (Weifang, China; approval
no. 2019SDL045).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ADH
|
ATRA/DOX-sHA
|
|
ADHG
|
ATRA/DOX-sHA-GA
|
|
ATRA
|
all-trans retinoic acid
|
|
CFSE
|
5(6)-carboxyfluorescein diacetate
succinimidyl ester
|
|
CLMS
|
confocal laser microscope
|
|
DH
|
DOX-sHA
|
|
DHG
|
DOX-sHA-GA
|
|
DiR
|
1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine iodide
|
|
DL
|
drug-loading rate
|
|
DOX
|
doxorubicin
|
|
ECM
|
extracellular matrix
|
|
EE
|
encapsulation efficiency rate
|
|
FCM
|
flow cytometry
|
|
GA
|
glycyrrhetinic acid
|
|
HA
|
hyaluronic acid
|
|
HCC
|
hepatocellular carcinoma
|
|
HSC
|
hepatic stellate cell
|
|
m-HSC
|
mouse HSC
|
|
NDDS
|
nano-sized drug delivery systems
|
|
sHA
|
sulfated HA
|
|
TME
|
tumor microenvironment
|
|
TEM
|
transmission electron microscope
|
References
|
1
|
Lee YS and Jeong WI: Retinoic acids and
hepatic stellate cells in liver disease. J Gastroenterol Hepatol.
27 (Suppl 2):S75–S79. 2012. View Article : Google Scholar
|
|
2
|
Hisamori S, Tabata C, Kadokawa Y, Okoshi
K, Tabata R, Mori A, Nagayama S, Watanabe G, Kubo H and Sakai Y:
All-trans-retinoic acid ameliorates carbon tetrachloride-induced
liver fibrosis in mice through modulating cytokine production.
Liver Int. 28:1217–1225. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mizobuchi Y, Shimizu I, Yasuda M, Hori H,
Shono M and Ito S: Retinyl palmitate reduces hepatic fibrosis in
rats induced by dimethylnitrosamine or pig serum. J Hepatol.
29:933–943. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Davis BH, Kramer RT and Davidson NO:
Retinoic acid modulates rat Ito cell proliferation, collagen, and
transforming growth factor beta production. J Clin Invest.
86:2062–2070. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tsuchida T and Friedman SL: Mechanisms of
hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol.
14:397–411. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Panebianco C, Oben JA, Vinciguerra M and
Pazienza V: Senescence in hepatic stellate cells as a mechanism of
liver fibrosis reversal: A putative synergy between retinoic acid
and PPAR-gamma signalings. Clin Exp Med. 17:269–280. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang L, Tankersley LR, Tang M, Potter JJ
and Mezey E: Regulation of the murine alpha(2)(I) collagen promoter
by retinoic acid and retinoid X receptors. Arch Biochem Biophys.
401:262–270. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim SJ, Park JH, Lee SA, Lee JG, Shin JM
and Lee HM: All-trans retinoic acid regulates TGF-β1-induced
extracellular matrix production via p38, JNK, and NF-κB-signaling
pathways in nasal polyp-derived fibroblasts. Int Forum Allergy
Rhinol. 10:636–645. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang Y, Guan DX, Shi J, Gao H, Li JJ,
Zhao JS, Qiu L, Liu J, Li N, Guo WX, et al: All-trans retinoic acid
potentiates the chemotherapeutic effect of cisplatin by inducing
differentiation of tumor initiating cells in liver cancer. J
Hepatol. 59:1255–1263. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Abu Lila AS and Ishida T: Liposomal
delivery systems: Design optimization and current applications.
Biol Pharm Bull. 40:1–10. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li L and Wang H: Heterogeneity of liver
cancer and personalized therapy. Cancer Lett. 379:191–197. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu T and Dai Y: Tumor microenvironment and
therapeutic response. Cancer Lett. 387:61–68. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Han S, Wang W, Wang S, Yang T, Zhang G,
Wang D, Ju R, Lu Y, Wang H and Wang L: Tumor microenvironment
remodeling and tumor therapy based on M2-like tumor associated
macrophage-targeting nano-complexes. Theranostics. 11:2892–2916.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hui L and Chen Y: Tumor microenvironment:
Sanctuary of the devil. Cancer Lett. 368:7–13. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mohr R, Özdirik B, Lambrecht J, Demir M,
Eschrich J, Geisler L, Hellberg T, Loosen SH, Luedde T, Tacke F, et
al: From liver cirrhosis to cancer: The role of Micro-RNAs in
hepatocarcinogenesis. Int J Mol Sci. 22:14922021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Brodt P: Role of the microenvironment in
liver metastasis: From pre- to prometastatic niches. Clin Cancer
Res. 22:5971–5982. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Muppala S: Significance of the tumor
microenvironment in liver cancer progression. Crit Rev Oncog.
25:1–9. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kahn AM, Blenman KRM, Sonis ST and
Lustberg MB: Strategies to mitigate the toxicity of cancer
therapeutics. Adv Cancer Res. 155:215–244. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou F, Teng F, Deng P, Meng N, Song Z and
Feng R: Recent progress of nano-drug delivery system for liver
cancer treatment. Anticancer Agents Med Chem. 17:1884–1897. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sun D, Zhang J, Wang L, Yu Z, O'Driscoll
CM and Guo J: Nanodelivery of immunogenic cell death-inducers for
cancer immunotherapy. Drug Discov Today. 26:651–662. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sudha PN and Rose MH: Beneficial effects
of hyaluronic acid. Adv Food Nutr Res. 72:137–176. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Vasvani S, Kulkarni P and Rawtani D:
Hyaluronic acid: A review on its biology, aspects of drug delivery,
route of administrations and a special emphasis on its approved
marketed products and recent clinical studies. Int J Biol Macromol.
151:1012–1029. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lokeshwar VB, Mirza S and Jordan A:
Targeting hyaluronic acid family for cancer chemoprevention and
therapy. Adv Cancer Res. 123:35–65. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Benitez A, Yates TJ, Lopez LE, Cerwinka
WH, Bakkar A and Lokeshwar VB: Targeting hyaluronidase for cancer
therapy: Antitumor activity of sulfated hyaluronic acid in prostate
cancer cells. Cancer Res. 71:4085–4095. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jordan AR, Lokeshwar SD, Lopez LE, Hennig
M, Chipollini J, Yates T, Hupe MC, Merseburger AS, Shiedlin A,
Cerwinka WH, et al: Antitumor activity of sulfated hyaluronic acid
fragments in pre-clinical models of bladder cancer. Oncotarget.
8:24262–24274. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhou X, He C, Liu M, Chen Q, Zhang L, Xu
X, Xu H, Qian Y, Yu F, Wu Y, et al: Self-assembly of hyaluronic
acid-mediated tumor-targeting theranostic nanoparticles. Biomater
Sci. 9:2221–2229. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bai Y, Liu CP, Chen D, Liu CF, Zhuo LH, Li
H, Wang C, Bu HT and Tian W: β-Cyclodextrin-modified hyaluronic
acid-based supramolecular self-assemblies for pH- and
esterase-dual-responsive drug delivery. Carbohydr Polym.
246:1166542020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cai J, Fu J, Li R, Zhang F, Ling G and
Zhang P: A potential carrier for anti-tumor targeted
delivery-hyaluronic acid nanoparticles. Carbohydr Polym.
208:356–364. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lei C, Liu XR, Chen QB, Li Y, Zhou JL,
Zhou LY and Zou T: Hyaluronic acid and albumin based nanoparticles
for drug delivery. J Control Release. 331:416–433. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Du W, Yang X, He S, Wang J, Guo Y, Kou B,
Jiang Y, Bian P, Li B and Yin L: Novel hyaluronic acid
oligosaccharide-loaded and CD44v6-targeting oxaliplatin
nanoparticles for the treatment of colorectal cancer. Drug Deliv.
28:920–929. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lai H, Ding X, Ye J, Deng J and Cui S:
pH-responsive hyaluronic acid-based nanoparticles for targeted
curcumin delivery and enhanced cancer therapy. Colloids Surf B
Biointerfaces. 198:1114552021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nieto C, Vega MA and Martín Del Valle E:
Nature-inspired nanoparticles as paclitaxel targeted carrier for
the treatment of HER2-positive breast cancer. Cancers (Basel).
13:25262021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Silva EL, Carneiro G, Caetano PA, Goulart
G, Ferreira Costa D, de Souza-Fagundes EM, Gomes DA and Ferreira
LA: Nanostructured lipid carriers loaded with tributyrin as an
alternative to improve anticancer activity of all-trans retinoic
acid. Expert Rev Anticancer Ther. 15:247–256. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xu ZC, Shen HX, Chen C, Ma L, Li WZ, Wang
L and Geng ZM: Neuropilin-1 promotes primary liver cancer
progression by potentiating the activity of hepatic stellate cells.
Oncol Lett. 15:2245–2251. 2018.PubMed/NCBI
|
|
35
|
Justus CR, Leffler N, Ruiz-Echevarria M
and Yang LV: In vitro cell migration and invasion assays. J Vis
Exp. 88:510462014.
|
|
36
|
Bourebaba N and Marycz K: Hepatic stellate
cells role in the course of metabolic disorders development-A
molecular overview. Pharmacol Res. 170:1057392021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gu L, Zhang F, Wu J and Zhuge Y:
Nanotechnology in drug delivery for liver fibrosis. Front Mol
Biosci. 8:8043962022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hellerbrand C: Hepatic stellate cells-the
pericytes in the liver. Pflugers Arch. 465:775–778. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Iwahasi S, Rui F, Morine Y, Yamada S,
Saito YU, Ikemoto T, Imura S and Shimada M: Hepatic stellate cells
contribute to the tumor malignancy of hepatocellular carcinoma
through the IL-6 pathway. Anticancer Res. 40:743–749. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Maeda H, Wu J, Sawa T, Matsumura Y and
Hori K: Tumor vascular permeability and the EPR effect in
macromolecular therapeutics: A review. J Control Release.
65:271–284. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shi Y, van der Meel R, Chen X and Lammers
T: The EPR effect and beyond: Strategies to improve tumor targeting
and cancer nanomedicine treatment efficacy. Theranostics.
10:7921–7924. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li Z, Wang F, Li Y, Wang X, Lu Q, Wang D,
Qi C, Li C, Li Z, Lian B, et al: Combined anti-hepatocellular
carcinoma therapy inhibit drug-resistance and metastasis via
targeting ‘substance P-hepatic stellate cells-hepatocellular
carcinoma’ axis. Biomaterials. 276:1210032021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Meng Q, Luo X, Chen J, Wang D, Chen E,
Zhang W, Zhang G, Zhou W, Xu J and Song Z: Unmasking
carcinoma-associated fibroblasts: Key transformation player within
the tumor microenvironment. Biochim Biophys Acta Rev Cancer.
1874:1884432020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fiori ME, Di Franco S, Villanova L, Bianca
P, Stassi G and De Maria R: Cancer-associated fibroblasts as
abettors of tumor progression at the crossroads of EMT and therapy
resistance. Mol Cancer. 18:702019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li T, Kang G, Wang T and Huang H: Tumor
angiogenesis and anti-angiogenic gene therapy for cancer. Oncol
Lett. 16:687–702. 2018.PubMed/NCBI
|
|
46
|
Bhat SM, Badiger VA, Vasishta S,
Chakraborty J, Prasad S, Ghosh S and Joshi MB: 3D tumor
angiogenesis models: Recent advances and challenges. J Cancer Res
Clin Oncol. 147:3477–3494. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang Y, Xu Z, Guo S, Zhang L, Sharma A,
Robertson GP and Huang L: Intravenous delivery of siRNA targeting
CD47 effectively inhibits melanoma tumor growth and lung
metastasis. Mol Ther. 21:1919–1929. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen Q, Bai H, Wu W, Huang G, Li Y, Wu M,
Tang G and Ping Y: Bioengineering bacterial vesicle-coated
polymeric nanomedicine for enhanced cancer immunotherapy and
metastasis prevention. Nano Lett. 20:11–21. 2020. View Article : Google Scholar : PubMed/NCBI
|