Introduction
Adriamycin is an anthracycline antitumor drug that
has been widely used in the therapy of various tumors including
breast cancer, bladder cancer, lymphoma, and acute lymphocytic
leukemia. Although its clinical therapeutic efficacy is well
established, its use is limited owing to various side effects,
including irreversible cardiotoxicity (1). Exploring the pathophysiological
process and developing novel therapeutic agents to antagonize the
toxic effect of Adriamycin is necessary for tumor treatment, along
with the prevention of heart disease.
Adriamycin inhibits DNA and RNA biosynthesis via DNA
intercalation, further inducing a series of cytotoxic reactions
(2). Mitochondrial dysfunction,
redox imbalance, apoptosis, dysregulation of autophagy and the
inflammatory response are involved in the pathophysiological
process of Adriamycin organ damage (3); however, not all tissues and organs
respond to Adriamycin in the same way and they may display varying
degrees of lesions (4). Adriamycin
cardiotoxicity is characterized by cardiomyocyte cell death through
necrosis or apoptosis (5), and
previous studies have suggested that cardiomyocyte atrophy is
central to the pathogenesis of Adriamycin-induced cardiotoxicity
(6,7). This process is largely attributed to
the dysregulation of autophagy and the ubiquitin-proteasome system
(8,9). Despite extensive studies, the initial
mechanisms leading to the progression of Adriamycin-induced
cardiotoxicity remain unclear.
Artemisinin is extracted from a well-known Chinese
medicinal herb Artemisia annua L., also known as qinghao
(10). Artemether is a derivative
of artemisinin and is a commonly administered antimalarial drug
(11). Artemisinin-based drugs
possess remarkable biological activities, such as antifungal,
antibacterial, anti-HIV, anticancer and antidiabetic properties
(12,13). Previous studies by the authors have
indicated that the therapeutic mechanisms of artemether are closely
related to the regulation of mitochondrial function and redox
status (14–18). Cardiomyocytes are rich in
mitochondria and redox imbalance plays an important role in
Adriamycin-induced cardiotoxicity (19); therefore, the present study aimed
to explore the therapeutic effect of artemether on Adriamycin
cardiotoxicity and investigate the underlying mechanisms.
Materials and methods
Animals and Adriamycin cardiotoxicity
model
Male BALB/c mice (20–25 g, 8 weeks old, n=24, the
success rate of mouse modeling was about 80%) were purchased from
the Laboratory Animal Center of Southern Medical University
(Guangzhou, China) and housed at room temperature 20–23°C, 12 h
light/dark cycle, relative humidity 50–60%. All mice had free
access to water and food. An Adriamycin cardiotoxicity mouse model
was established by a single injection of Adriamycin (10.4 mg/kg;
MilliporeSigma) through the tail vein. Adriamycin was dissolved in
normal saline with a final concentration ~1.74 mg/ml. The total
injection volume was about 120–150 µl for a dose of 10.4 mg/kg.
After 2 weeks, the mice were randomly divided into the Adriamycin
cardiotoxicity (AC) and AC + artemether (AC + Art) groups. Normal
control group mice (mice that were not administered Adriamycin)
were injected with an equal volume of saline solution. Control and
AC group mice were fed a regular standard diet. Mice in the AC +
Art group were fed a medicated diet containing 0.3 g/kg artemether
(Chengdu ConBon Bio-tech Co., Ltd.). The intervention time lasted
for 2 weeks. Animal studies were approved (approval no.
20200331002) and performed under the guidelines of The
Institutional Animal Care and Use Committee at Guangzhou University
of Chinese Medicine (Guangzhou, China).
Tissue preparation
At the end of treatment, the mice (18–29 g) were
anesthetized with isoflurane (~3% for anesthetic induction and 1–2%
for anesthetic maintenance), before drawing blood (~400 µl) from
the orbital sinus vein. Afterwards, the mice were euthanized by
cervical dislocation. Mouse death was verified by cessation of
heartbeat, respiratory arrest and pupil dilation. The heart tissues
were then isolated and weighed. Considering that tibial length is a
more reliable reference for assessing organ changes when body
weight fluctuates (20). The tibia
length (left and right) was measured using a vernier caliper and
heart weight/tibia length (average of left and right) was
calculated. Sections of the heart tissues (4 µm) were fixed in 10%
formalin (at room temperature for 48 h) for pathological
examination and immunohistochemical staining. The remaining tissues
were immediately frozen in liquid nitrogen and stored at −80°C for
future analysis. Paraffin sections of the heart tissues were
stained with hematoxylin and eosin (H&E) at room temperature (1
min for hematoxylin and 2 min for eosin) to observe the
pathological changes. The serum lactate dehydrogenase (LDH) level
was determined by using an automatic biochemical analyzer (Roche
Diagnostics).
Immunohistochemical staining
Immunohistochemical staining was performed as
previously described (16).
Briefly, cardiac sections were deparaffinized using two sequential
5 min washes in fresh xylene and rehydrated in a descending ethanol
series. After antigen retrieval using citrate buffer, the sections
were incubated overnight at 4°C with primary antibodies against
connexin 43 (Cx43, 1:100; cat. no. 3512), N-cadherin (1:100; cat.
no. 14215), Bax (1:100; cat. no. 2772; all from Cell Signaling
Technology, Inc.), Bcl-2 (1:100; cat. no. SAB4500003;
Sigma-Aldrich; Merck KGaA) and Optic Atrophy 1 (OPA1, 1:100; cat.
no. 612606; BD Biosciences). The sections were then washed and
incubated at room temperature with ready-to-use horseradish
peroxidase-conjugated secondary antibodies without dilution for 15
min (cat. no. KIT-5020; Fuzhou MaiXin Biotech Co, Ltd.). The
chromogenic reagent used was 3,3′-diaminobenzidine, and the
sections were counterstained using hematoxylin.
Immunofluorescent staining
After incubation with Cx43 (1:50) and N-cadherin
(1:50) primary antibodies, Alexa Fluor-conjugated secondary
antibodies (1:500; Invitrogen; Thermo Fisher Scientific, Inc; cat.
no. A-11037; Goat anti-Rabbit Secondary Antibody, Alexa Fluor™ 594;
and cat. no. A-11029; Goat anti-Mouse Secondary Antibody, Alexa
Fluor™ 488) were incubated for 1 h at room temperature. The tissues
were visualized and images were captured using a confocal
microscope (LSM710; Carl Zeiss AG).
Enzyme-linked immunosorbent assay
Serum B-type natriuretic peptide (BNP) levels were
determined using an enzyme-linked immunosorbent assay (cat. no.
SEKM-0151; Beijing Solarbio Science & Technology Co., Ltd.),
according to the manufacturer's instructions.
Serum H2O2
determination
Serum H2O2 levels were
detected using Amplex UltraRed reagent (cat. no. A36006;
Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol.
Immunoblot analysis
Heart tissues were homogenized and prepared in
sample loading buffer (cat. no. 1610747, Bio-Rad Laboratories,
Inc.). The protein concentrations were determined using Bradford
Dye Reagent (cat. no. 500-0205, Bio-Rad Laboratories, Inc.). The
same mass of protein (~15 µl/45 ug) was loaded per lane. The
homogenates were separated using sodium dodecyl
sulfate-polyacrylamide gel (10%) electrophoresis and subsequently
electro-transferred to polyvinylidene fluoride membranes
(MilliporeSigma). After blocking in 5% non-fat milk for 1 h at room
temperature, the membranes were incubated overnight at 4°C with the
following primary antibodies: LC3B (1:500; cat. no. L7543;
Sigma-Aldrich; Merck KGaA), Beclin-1 (1:500; cat. no. 3495; Cell
Signaling Technology, Inc.), PTEN-induced kinase 1 (PINK1; 1:500;
cat. no. GTX59847; GeneTex, Inc.), p62 (1:500; cat. no. 18420;
Proteintech Group, Inc.), BNIP3L/Nix (1:500; cat. no. 12396; Cell
Signaling Technology, Inc.), Sestrin2 (SESN2; 1:500; cat. no.
GTX64418; GeneTex, Inc.), Bax (1:500; cat. no. 2772; Cell Signaling
Technology, Inc.), Bcl-2 (1:500; cat. no. SAB4500003;
Sigma-Aldrich; Merck KGaA), OPA1 (1:500; cat. no. 612606; BD
Biosciences), copper chaperone for superoxide dismutase (CCS;
1:500; cat. no. 22802; Proteintech Group, Inc.), superoxide
dismutase 1 (SOD1; 1:500; cat. no. 37385), SOD2 (1:1,000; cat. no.
13141) and Vinculin (1:500; cat. no. 13901; all from Cell Signaling
Technology, Inc.). After incubating with the corresponding
horseradish peroxidase-conjugated secondary antibodies (1:2,000;
cat. nos. 65-6120 or 62-6520; Invitrogen; Thermo Fisher Scientific,
Inc.) at room temperature for 1 h, the images were obtained using a
ChemiDoc Imaging System (Bio-Rad Laboratories, Inc.) and analyzed
using ImageJ software (Version 1.8.0; National Institutes of
Health).
Reverse transcription-quantitative
(RT-q)PCR
Total RNA from the heart tissues was purified using
an RNA purification kit (cat. no. 12183025; Invitrogen; Thermo
Fisher Scientific, Inc.). The first-strand cDNA was synthesized
using oligo(dT)12-18 primers (cat. no. 18418012;
Invitrogen; Thermo Fisher Scientific, Inc.), dNTP Mix (cat. no.
18427-013; Invitrogen), Ribonuclease Inhibitor (cat. no. 10777-019;
Invitrogen), DTT (cat. no. Y00147; Invitrogen), and M-MLV Reverse
Transcriptase (cat. no. 28025-013; Invitrogen). The reaction
process was under 37°C for 50 min and then was terminated at 70°C
for 15 min. qPCR was performed using SYBR green PCR master mix
(cat. no. 4367659; Applied Biosystems; Thermo Fisher Scientific,
Inc.) using the Applied Biosystems QuantStudio 5 platform and the
amplification conditions (95°C for 5 min followed by 45 cycles of
95°C for 15 s, 55°C for 15 s and 72°C for 20 s) were set as
previously described (16). Gene
specific primers (Table I) were
provided by Sangon Biotech Co., Ltd. The relative RNA expression
levels were calculated using 2−ΔΔCq (21) and normalized against the
housekeeping gene, 18S rRNA.
 | Table I.Sequences of the primers used for
reverse transcription-quantitative PCR. |
Table I.
Sequences of the primers used for
reverse transcription-quantitative PCR.
| Gene name | Primer sequence
(5′-3′) |
|---|
| Mouse BNP | F:
GGCCTCACAAAAGAACACCC |
|
| R:
TTCAGTGCGTTACAGCCCAA |
| Mouse Cx43 | F:
ACAGCGGTTGAGTCAGCTTG |
|
| R:
GAGAGATGGGGAAGGACTTGT |
| Mouse
N-cadherin | F:
AGCGCAGTCTTACCGAAGG |
|
| R:
TCGCTGCTTTCATACTGAACTTT |
| Mouse OPA1 | F:
TGGAAAATGGTTCGAGAGTCAG |
|
| R:
CATTCCGTCTCTAGGTTAAAGCG |
| Mouse 16S rRNA | F:
CCGCAAGGGAAAGATGAAAGAC |
|
| R:
TCGTTTGGTTTCGGGGTTTC |
| Mouse ND1 | F:
CTAGCAGAAACAAACCGGGC |
|
| R:
CCGGCTGCGTATTCTACGTT |
| Mouse CytoB | F:
GCCACCTTGACCCGATTCTTCGC |
|
| R:
TGAACGATTGCTAGGGCCGCG |
| Mouse 18S rRNA | F:
GAGGCCCTGTAATTGGAATGAG |
|
| R:
GCAGCAACTTTAATATACGCTATTGG |
Statistical analysis
Data are presented as the mean ± standard deviation.
One-way analysis of variance followed by least significant
difference post hoc test was used for data analysis using SPSS
software (Version 22.0; IBM Corp.). P<0.05 was considered to
indicate a statistically significant difference.
Results
Artemether prevents Adriamycin-induced
cardiac atrophy
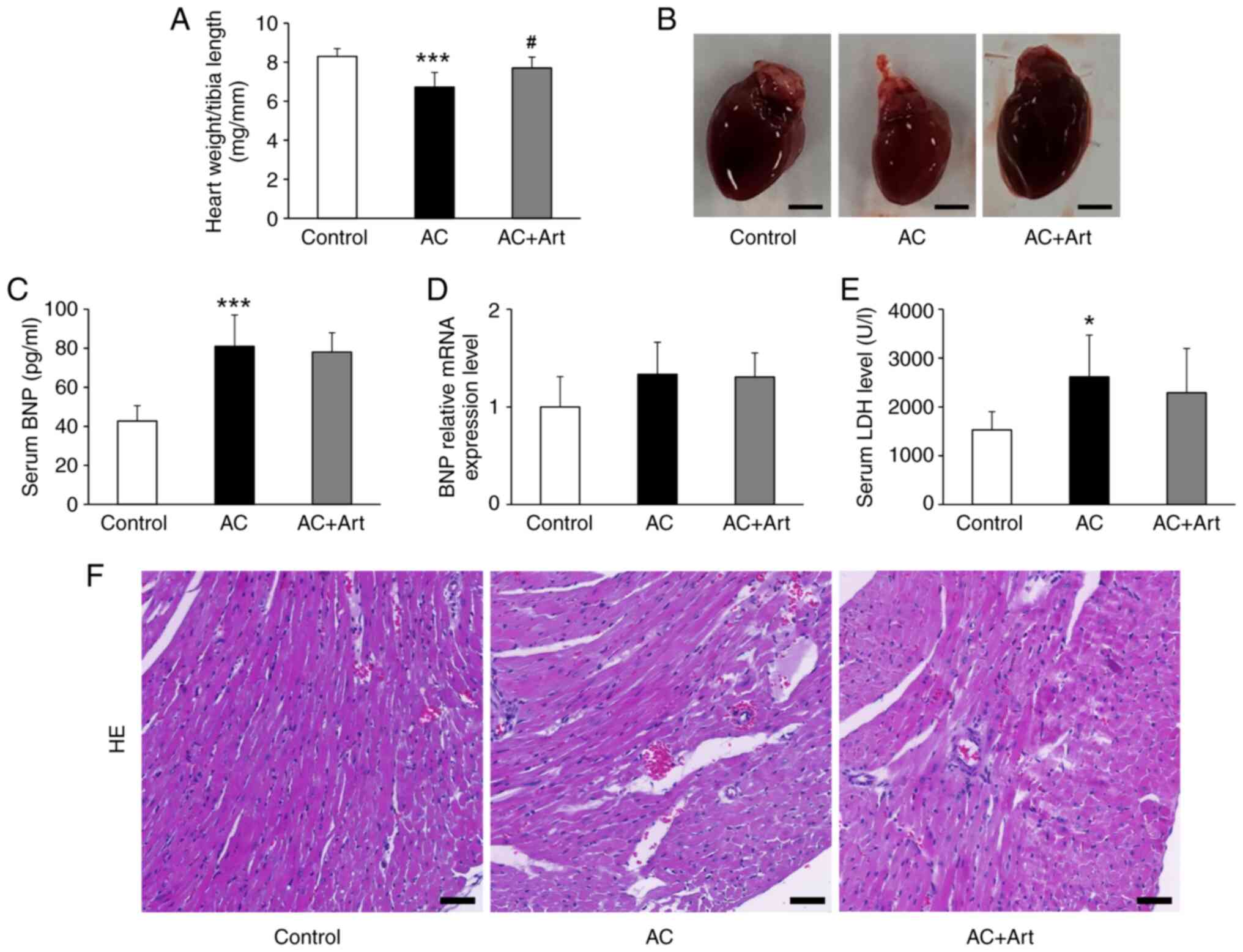
As shown in Fig. 1A and
B, a significant decrease in heart weight/tibia length was
observed in the AC group, and artemether treatment notably
prevented cardiac atrophy. Compared with the control group, the AC
group had significantly increased serum BNP and LDH levels
(Fig. 1C and E). Although BNP mRNA
expression in the heart tissue was also upregulated in the AC
group, the difference was not significant (Fig. 1D). Artemether therapy did not
affect BNP expression and secretion in the heart after Adriamycin
exposure (Fig. 1C and D). The
serum level of LDH was slightly reduced by artemether but not to
significant degree (Fig. 1E).
H&E staining revealed loosening or disruption of myocardial
fibers in the AC group, and these pathological changes were
attenuated by artemether therapy (Fig.
1F).
Artemether stabilizes the
intercellular junctions between ventricular myocytes
Cx43 and N-cadherin are the main constituents of the
gap and adherens junctions, respectively, in the myocardium
(22). They form the structural
basis for the synchronous coordination of myocardial fibers, which
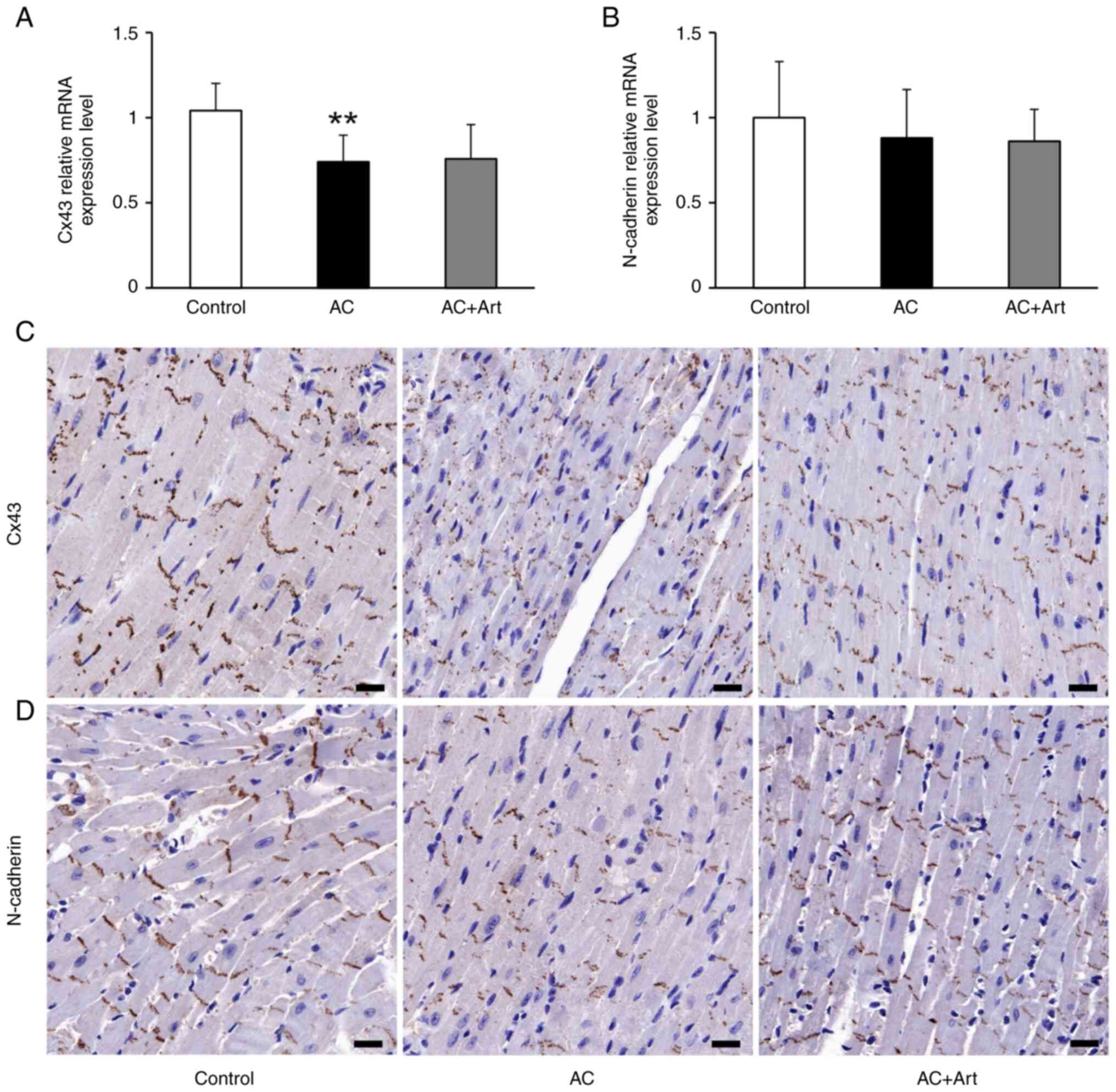
are essential for maintaining cardiac function (23). Compared with the control group, the
AC group had a significant decrease in Cx43 mRNA expression
(Fig. 2A). Immunohistochemical
staining demonstrated that Cx43 and N-cadherin were preferentially
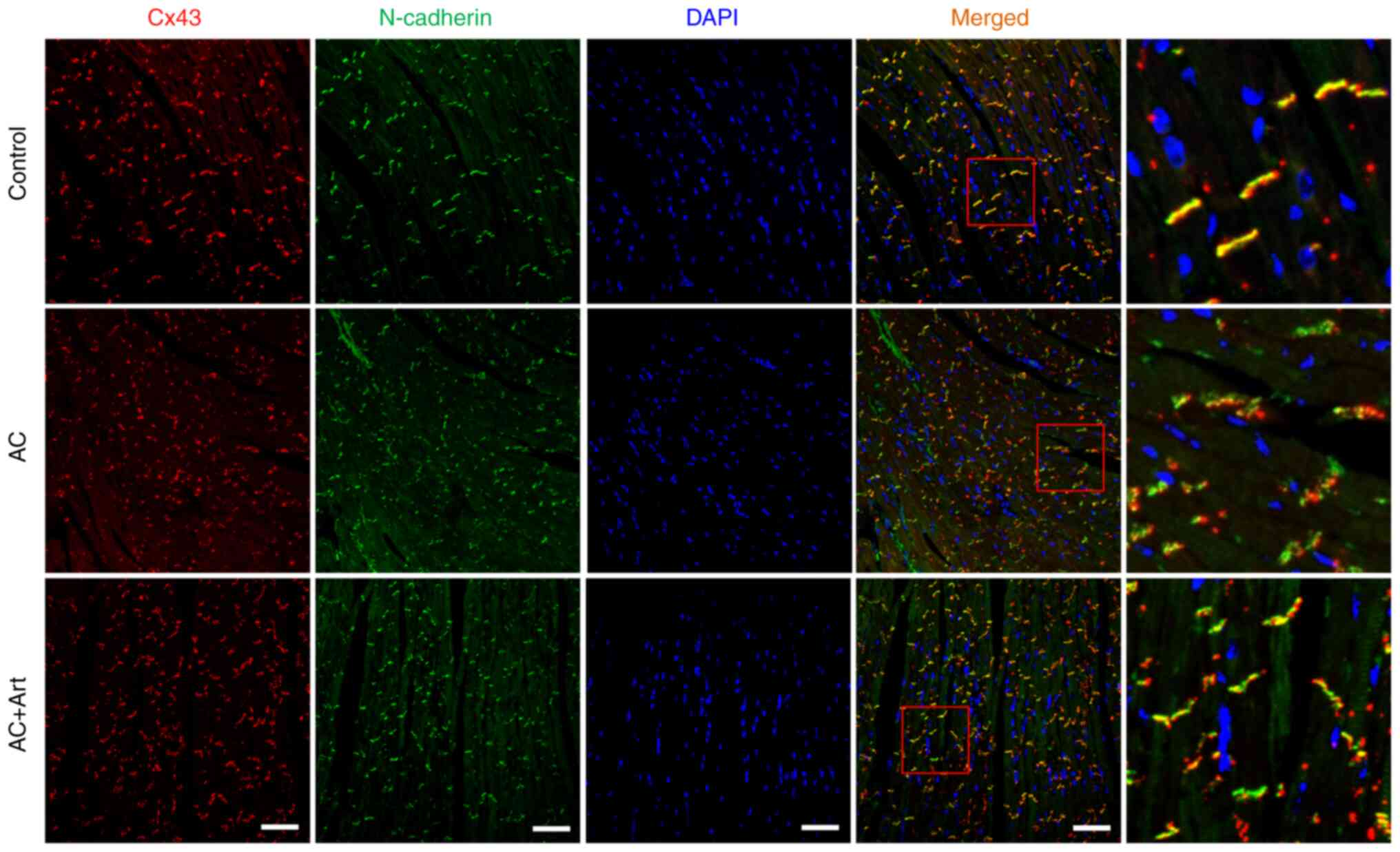
distributed in intercalated discs (Fig. 2C and D). As shown in Fig. 3, diminished colocalization of Cx43
and N-cadherin was observed at the intercalated discs in the AC
group. Although artemether treatment did not affect the expression
of Cx43 and N-cadherin (Fig. 2A and
B), it notably restored and stabilized their intercombination
(Figs. 2C and D and 3).
Artemether regulates myocardial cell
autophagy
Autophagy is a cellular protein degradation pathway,
which plays an important role in cardiac dysfunction and atrophy
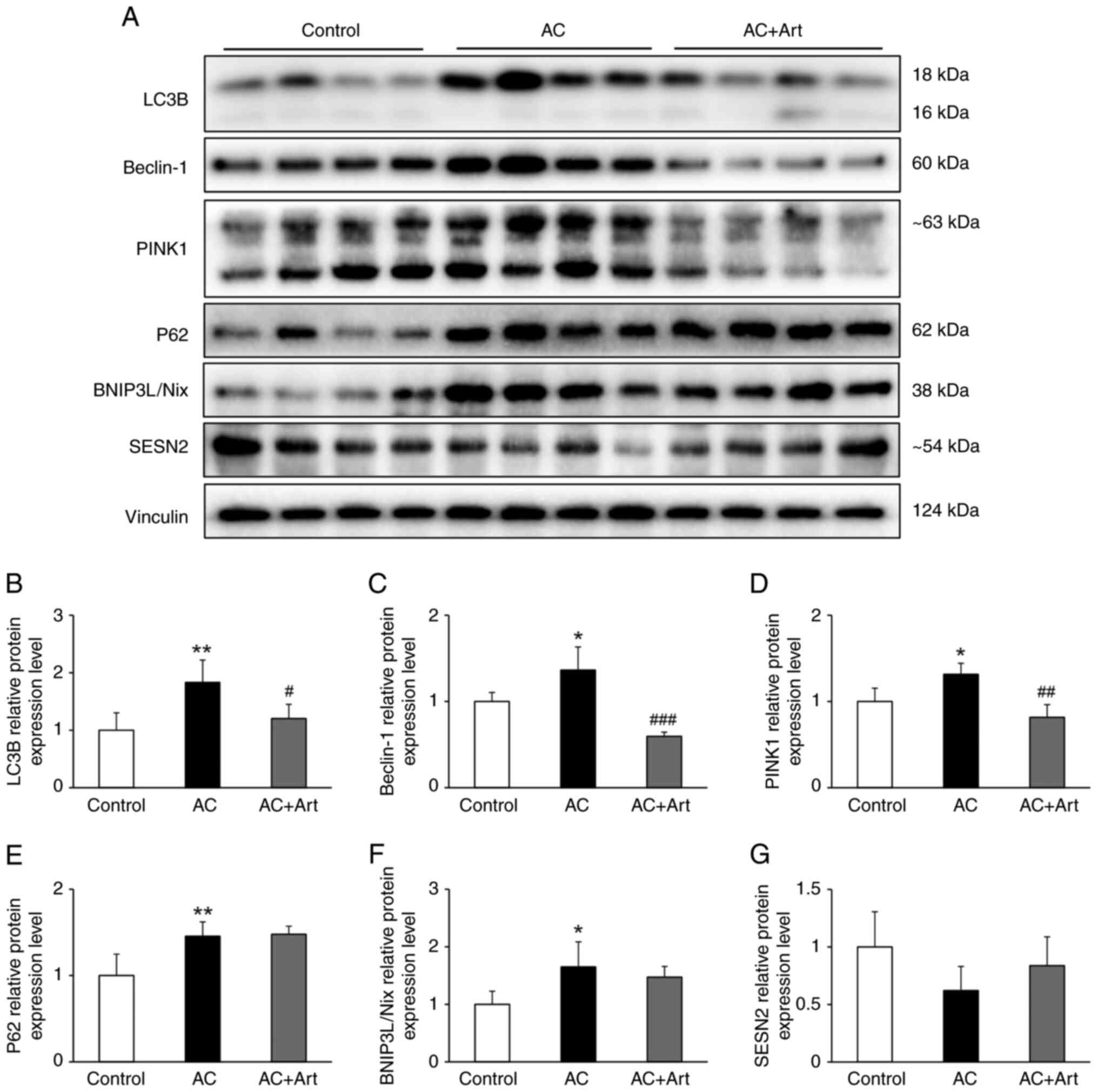
(24). In the present study,
significantly increased LC3B, Beclin-1, p62, BNIP3L/Nix and PINK1
protein levels were detected in the AC group, which indicated an
activation of autophagy following Adriamycin exposure (Fig. 4A-F). A marginal and insignificant
decline in SESN2 was observed in the AC group (Fig. 4A and G). After treatment with
artemether, the levels of LC3B, Beclin-1 and PINK1 were
significantly decreased, while the levels of p62 and BNIP3L/Nix
were unchanged; a marginal and insignificant increase in the level
of SESN2 was also observed (Fig.
4A-G).
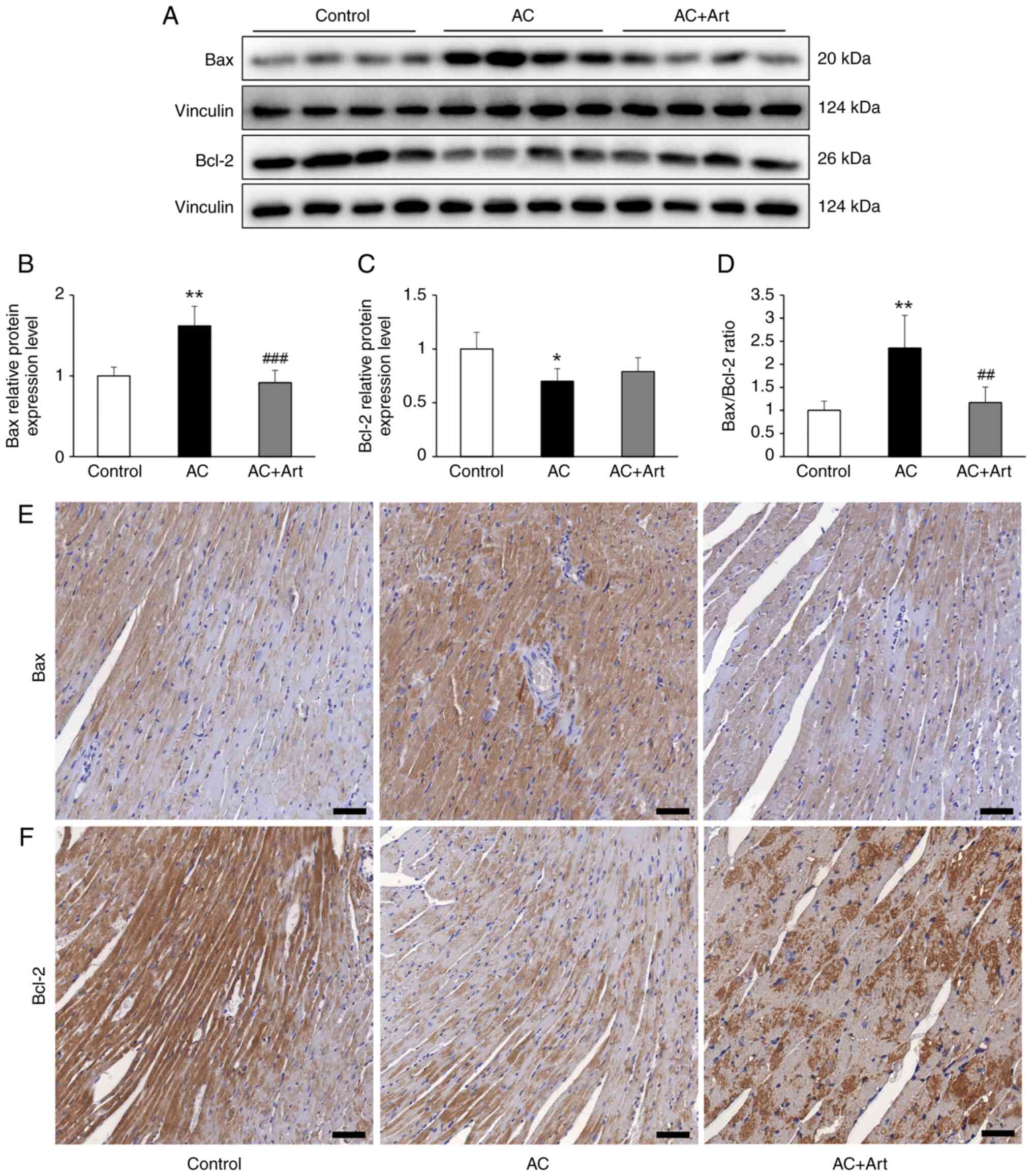
Artemether restores the unbalanced
ratio of Bax and Bcl-2 in myocardial cells
The Bax/Bcl-2 ratio is closely related to
mitochondrial function and the autophagy pathway (25,26);
therefore, Bax and Bcl-2 protein levels in heart tissue were
measured in the present study. As revealed in Fig. 5A-D, significantly increased Bax,
decreased Bcl-2 and an abnormally elevated Bax/Bcl-2 ratio was
observed in the AC group, compared with the control group. Although
artemether treatment did not significantly increase the Bcl-2
protein levels, it did significantly decrease the Bax levels and
the Bax/Bcl-2 ratio (Fig. 5A-D).
As indicated by immunohistochemical analysis, Bax and Bcl-2 were
primarily located in the myocardial cells and the expression levels
were in line with the immunoblotting results (Fig. 5E and F).
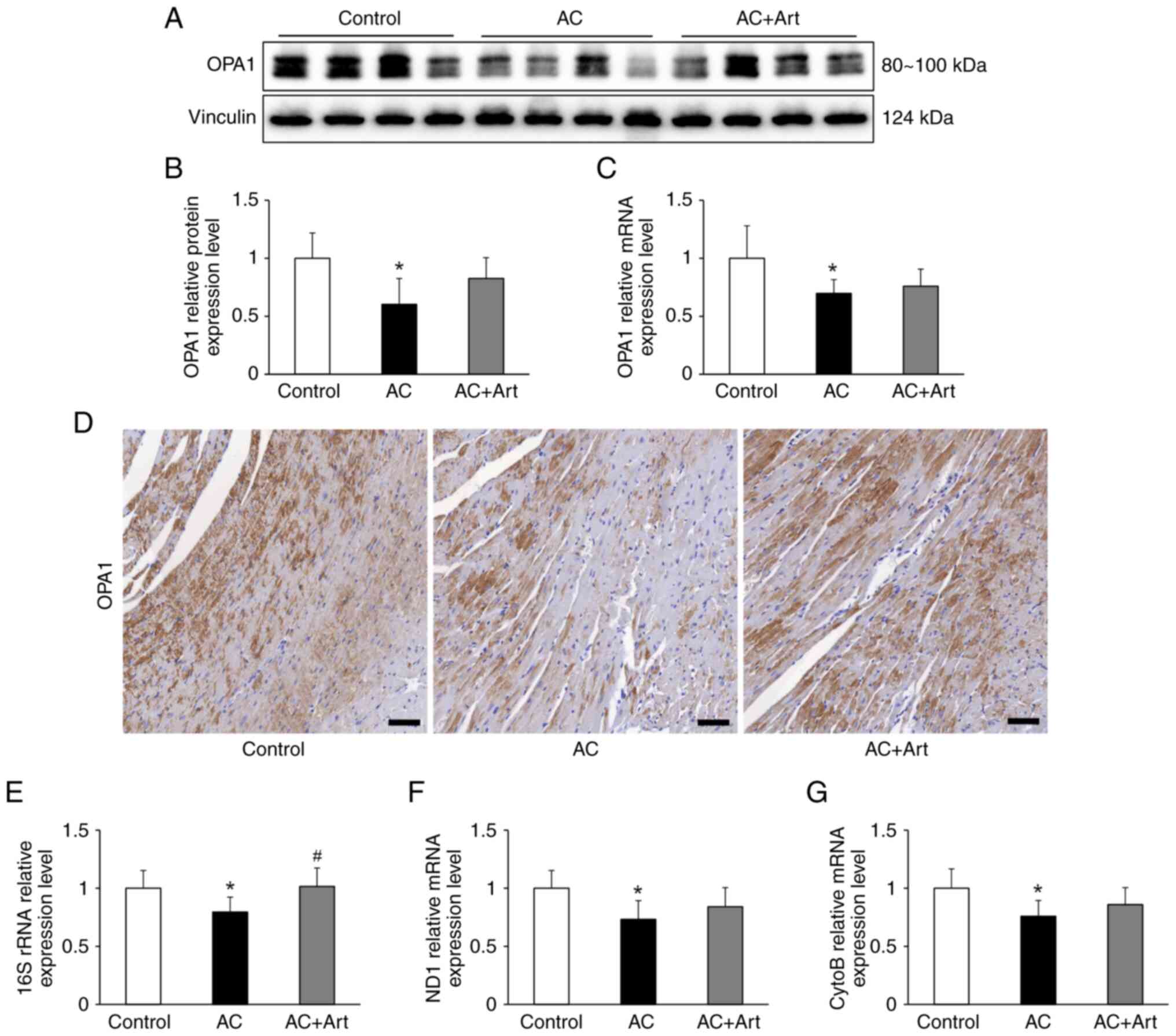
Artemether affects the levels of
mitochondria constituents in myocardial cells
OPA1 is essential for mitochondrial cristae
formation and its loss disrupts the mitochondrial inner membrane
structure and integrity, leading to mitochondrial dysfunction
(27). After Adriamycin exposure,
an expected decrease in the protein and mRNA levels of OPA1 were
observed in the myocardial cell, and these levels insignificantly
increased upon treatment with artemether (Fig. 6A-D). The mitochondrial DNA (mtDNA)
encoded mitochondrial constituents, including 16S rRNA, NADH
dehydrogenase 1 (ND1) and cytochrome b (CytoB), were significantly
decreased in the AC group (Fig.
6E-G). The level of 16S rRNA was significantly increased by
artemether treatment, and the ND1 and CytoB mRNA levels were also
increased by artemether, but the difference was not significant
(Fig. 6E-G).
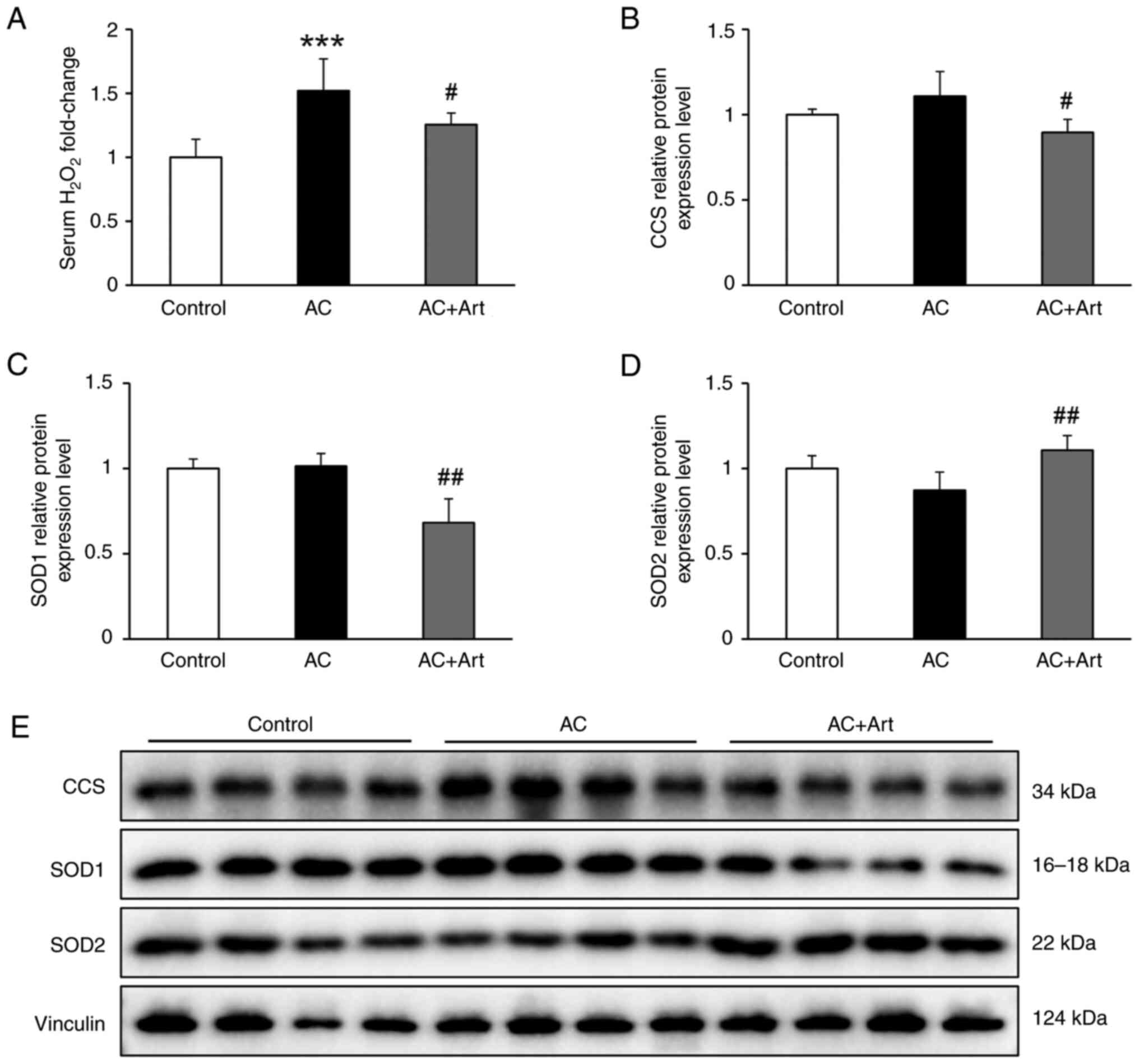
Artemether ameliorates cardiac redox
imbalance
Mitochondria are the main source of reactive oxygen
species (ROS), and its dysfunction is the primary cause of redox
imbalance in the body (28).
Compared with the control group, the AC groups exhibited
significantly elevated serum H2O2 levels,
which were significantly decreased by artemether treatment
(Fig. 7A). As demonstrated in
Fig. 7B-E, slightly increased CCS,
decreased SOD2 and unaltered SOD1 levels were detected in the AC
group, compared with the control group. After artemether therapy,
CCS and SOD1 levels were significantly decreased, and SOD2 levels
were significantly increased (Fig.
7B-E).
Discussion
In the present study, it was demonstrated that
artemether ameliorated Adriamycin-induced cardiac atrophy and the
mechanisms may be associated with regulating mitochondrial function
and autophagy.
Consistent with previous studies (9,29),
in the present study it was demonstrated that cardiac atrophy
occurred 4 weeks after Adriamycin injection. Significantly elevated
serum BNP levels and pathological alterations also reflected
cardiac dysfunction resulting from Adriamycin exposure. Artemether
treatment prevented cardiac atrophy and attenuated pathological
lesions; however, it did not affect BNP expression. BNP is produced
primarily by cardiac ventricular myocytes in response to altered
ventricular wall stress. Subsequently, it is secreted into the
blood and exerts a wide range of biological effects, including
diuresis, natriuresis, vasodilation, and smooth muscle relaxation
(30); therefore, it was
hypothesized that elevated serum BNP may be a compensatory response
to cardiac dysfunction. The integrity of cell membrane structure
and intercellular connections is essential for maintaining cell
function (31). Cardiac
intercalated discs are highly specialized structures that play a
key role in the coordination and synchronization of myocardial
contraction (22). In the present
study, in addition to the expected pathological alterations, such
as myocardial fiber disruption and cardiomyocyte interstitium
widening, the cardiac intercalated discs were also destroyed, which
manifested as decreased Cx43 expression and abnormal colocalization
of Cx43 and N-cadherin, although there was no significant
difference in N-cadherin mRNA levels between the control and AC
groups; however, N-cadherin immunohistochemical staining appeared
to be lower in the AC group than the control group. It was
hypothesized that this inconsistency suggests that N-cadherin may
be regulated at both the transcriptional and translational levels.
Artemether intervention did not affect Cx43 and N-cadherin mRNA
expression levels; however, it notably recovered and stabilized
their intercombination.
Cardiomyocyte atrophy, rather than cell death, is
the primary determinant of the subacute decrease in heart weight
after Adriamycin exposure (6,9). To
explore the pathophysiological mechanism of myocardial atrophy in
the present study, autophagy pathway-associated proteins in the
myocardial tissue were analyzed. The results suggested that
autophagy-related protein degradation occurred in the
cardiomyocytes, and artemether treatment markedly regulated the
pathway. As a major pathway responsible for the recycling of
intracellular substances, autophagy can be activated by a variety
of stress stimuli, including mitochondrial dysfunction (32). Bax and Bcl-2 belong to the Bcl-2
protein family, which regulates cell autophagy, mitochondrial
bioenergetic metabolism and redox homeostasis; their biological
effect occurs by translocation between the cytoplasm and the outer
mitochondrial membrane (25,26).
In the present study, the AC group had a significantly increased
Bax/Bcl-2 ratio, indicating compromised mitochondria in the
myocardial cell. OPA1 is an inner mitochondrial membrane protein
that helps to regulate mitochondrial fusion and cristae morphology
(27). In the present study,
significantly decreased OPA1 protein and mRNA levels also indicated
dysfunctional mitochondria in the myocardial cell. In addition, the
mtDNA encoded 16S rRNA, ND1 and CytoB were all decreased in the AC
group. Collectively, this evidence indicated that Adriamycin
exposure induced mitochondrial damage and activated the cellular
autophagy pathway. These mitochondrial alterations improved to
varying degrees by artemether treatment in the present study. It
has been reported that Adriamycin-induced cardiac atrophy is
mediated by the striated muscle-specific ubiquitin ligase, muscle
ring-finger protein-1, and activation of the ubiquitin-proteasome
system is associated with ROS production (9,33);
therefore, it was hypothesized that the ubiquitin-proteasome system
may also be the therapeutic target of artemether, however, this
needs further investigation.
Mitochondria are the principal generators of
cellular superoxide radicals. Considering that the myocardial cell
is rich in mitochondria, it is reasonable to hypothesize that the
significantly elevated serum H2O2 levels
observed in the present study are, at least partly, due to damaged
mitochondria in the heart. SOD2 and SOD1 are mitochondrial and
cytoplasmic superoxide free radical scavenging systems,
respectively (34). CCS is a
critical component of the redox system and is located in the
cytosol and intermembrane space of mitochondria (35). The loss of SOD2 correlates with the
overexpression of SOD1 and their mutual transformation is closely
associated with cellular redox imbalance and tumorigenesis
(36,37). Although Adriamycin did not
significantly alter their protein levels in the present study, the
SOD1/SOD2 ratio was altered to some extent; therefore, treatment
with artemether remodeled the intracellular antioxidant system.
In summary, the present study demonstrated that
artemether prevented Adriamycin-induced cardiac atrophy by
regulating mitochondrial function and the autophagy pathway. These
findings have potential clinical implications for preventing heart
disease or cardiotoxicity induced by chemotherapy drugs.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 82004156), the Shenzhen Science and
Technology Project (grant no. JCYJ20190812183603627) and the
Shenzhen Fund for Guangdong Provincial High level Clinical Key
Specialties.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
PH and HS proposed the conception and original
design of the present study. WW performed the majority of the
experiments and analyzed most of the data. XY participated in
animal tissue processing and sectioning. YD performed some of the
immunoblotting experiments. TW participated in the animal
experiments. MS contributed to some of the pathological
experiments. PH and WW wrote the manuscript. PH and HS confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
All animal procedures were approved (approval no.
20200331002) by the Animal Care and Use Committee of Guangzhou
University of Chinese Medicine (Guangzhou, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
van der Zanden SY, Qiao X and Neefjes J:
New insights into the activities and toxicities of the old
anticancer drug doxorubicin. FEBS J. 288:6095–6111. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tacar O, Sriamornsak P and Dass CR:
Doxorubicin: An update on anticancer molecular action, toxicity and
novel drug delivery systems. J Pharm Pharmacol. 65:157–170. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sritharan S and Sivalingam N: A
comprehensive review on time-tested anticancer drug doxorubicin.
Life Sci. 278:1195272021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pugazhendhi A, Edison TNJI, Velmurugan BK,
Jacob JA and Karuppusamy I: Toxicity of Doxorubicin (Dox) to
different experimental organ systems. Life Sci. 200:26–30. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Christidi E and Brunham LR: Regulated cell
death pathways in doxorubicin-induced cardiotoxicity. Cell Death
Dis. 12:3392021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen DS, Yan J and Yang PZ: Cardiomyocyte
Atrophy, an Underestimated Contributor in Doxorubicin-Induced
Cardiotoxicity. Front Cardiovasc Med. 9:8125782022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ferreira de Souza T, Quinaglia AC, Silva
T, Osorio Costa F, Shah R, Neilan TG, Velloso L, Nadruz W, Brenelli
F, Sposito AC, Matos-Souza JR, et al: Anthracycline therapy is
associated with cardiomyocyte atrophy and preclinical
manifestations of heart disease. JACC Cardiovasc Imaging.
11:1045–1055. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bartlett JJ, Trivedi PC and Pulinilkunnil
T: Autophagic dysregulation in doxorubicin cardiomyopathy. J Mol
Cell Cardiol. 104:1–8. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Willis MS, Parry TL, Brown DI, Mota RI,
Huang W, Beak JY, Sola M, Zhou C, Hicks ST, Caughey MC, et al:
Doxorubicin exposure causes subacute cardiac atrophy dependent on
the striated muscle-specific ubiquitin ligase MuRF1. Circ Heart
Fail. 12:e0052342019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ma N, Zhang Z, Liao F, Jiang T and Tu Y:
The birth of artemisinin. Pharmacol Ther. 216:1076582020.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Esu EB, Effa EE, Opie ON and Meremikwu MM:
Artemether for severe malaria. Cochrane Database Syst Rev.
6:CD0106782019.PubMed/NCBI
|
|
12
|
Wang Y, Wang Y, You F and Xue J: Novel use
for old drugs: The emerging role of artemisinin and its derivatives
in fibrosis. Pharmacol Res. 157:1048292020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dolivo D, Weathers P and Dominko T:
Artemisinin and artemisinin derivatives as anti-fibrotic
therapeutics. Acta Pharm Sin B. 11:322–339. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Han P, Wang Y, Zhan H, Weng W, Yu X, Ge N,
Wang W, Song G, Yi T, Li S, et al: Artemether ameliorates type 2
diabetic kidney disease by increasing mitochondrial pyruvate
carrier content in db/db mice. Am J Transl Res. 11:1389–1402.
2019.PubMed/NCBI
|
|
15
|
Wang Y, Han P, Wang M, Weng W, Zhan H, Yu
X, Yuan C, Shao M and Sun H: Artemether improves type 1 diabetic
kidney disease by regulating mitochondrial function. Am J Transl
Res. 11:3879–3889. 2019.PubMed/NCBI
|
|
16
|
Han P, Cai Y, Wang Y, Weng W, Chen Y, Wang
M, Zhan H, Yu X, Wang T, Shao M and Sun H: Artemether ameliorates
kidney injury by restoring redox imbalance and improving
mitochondrial function in Adriamycin nephropathy in mice. Sci Rep.
11:12662021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cheng X, Zhou P, Weng W, Sun Z, Liu H,
Chen Y, Cai Y, Yu X, Wang T, Shao M, et al: Artemether attenuates
renal tubular injury by targeting mitochondria in adriamycin
nephropathy mice. Am J Transl Res. 14:2002–2012. 2022.PubMed/NCBI
|
|
18
|
Weng W, Liu H, Sun Z, Zhou P, Yu X, Shao
M, Han P and Sun H: Combined treatment with niclosamide
ethanolamine and artemether combination improves type 1 diabetes
via the targeting of liver mitochondria. Exp Ther Med. 23:2392022.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wallace KB, Sardao VA and Oliveira PJ:
Mitochondrial determinants of doxorubicin-induced cardiomyopathy.
Circ Res. 126:926–941. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yin FC, Spurgeon HA, Rakusan K, Weisfeldt
ML and Lakatta EG: Use of tibial length to quantify cardiac
hypertrophy: Application in the aging rat. Am J Physiol.
243:H941–H947. 1982.PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao G, Qiu Y, Zhang HM and Yang D:
Intercalated discs: Cellular adhesion and signaling in heart health
and diseases. Heart Fail Rev. 24:115–132. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Leo-Macias A, Agullo-Pascual E and Delmar
M: The cardiac connexome: Non-canonical functions of connexin43 and
their role in cardiac arrhythmias. Semin Cell Dev Biol. 50:13–21.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Orogo AM and Gustafsson AB: Therapeutic
targeting of autophagy: Potential and concerns in treating
cardiovascular disease. Circ Res. 116:489–503. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gross A and Katz SG: Non-apoptotic
functions of BCL-2 family proteins. Cell Death Differ.
24:1348–1358. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chong SJF, Marchi S, Petroni G, Kroemer G,
Galluzzi L and Pervaiz S: Noncanonical cell fate regulation by
Bcl-2 Proteins. Trends Cell Biol. 30:537–555. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jang S and Javadov S: OPA1 regulates
respiratory supercomplexes assembly: The role of mitochondrial
swelling. Mitochondrion. 51:30–39. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dan Dunn J, Alvarez LA, Zhang X and
Soldati T: Reactive oxygen species and mitochondria: A nexus of
cellular homeostasis. Redox Biol. 6:472–485. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shimauchi T, Numaga-Tomita T, Ito T,
Nishimura A, Matsukane R, Oda S, Hoka S, Ide T, Koitabashi N,
Uchida K, et al: TRPC3-Nox2 complex mediates doxorubicin-induced
myocardial atrophy. JCI Insight. 2:e933582017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nash PL: Brain type natriuretic peptide.
Neonatal Netw. 27:343–346. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Belardi B, Son S, Felce JH, Dustin ML and
Fletcher DA: Cell-cell interfaces as specialized compartments
directing cell function. Nat Rev Mol Cell Biol. 21:750–764. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dikic I and Elazar Z: Mechanism and
medical implications of mammalian autophagy. Nat Rev Mol Cell Biol.
19:349–364. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Montalvo RN, Doerr V, Min K, Szeto HH and
Smuder AJ: Doxorubicin-induced oxidative stress differentially
regulates proteolytic signaling in cardiac and skeletal muscle. Am
J Physiol Regul Integr Comp Physiol. 318:R227–R233. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang Y, Branicky R, Noe A and Hekimi S:
Superoxide dismutases: Dual roles in controlling ROS damage and
regulating ROS signaling. J Cell Biol. 217:1915–1928. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Boyd SD, Ullrich MS, Skopp A and Winkler
DD: Copper Sources for Sod1 Activation. Antioxidants (Basel).
9:5002020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Papa L, Hahn M, Marsh EL, Evans BS and
Germain D: SOD2 to SOD1 switch in breast cancer. J Biol Chem.
289:5412–5416. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chang YC, Fong Y, Tsai EM, Chang YG, Chou
HL, Wu CY, Teng YN, Liu TC, Yuan SS and Chiu CC: Exogenous
C8-Ceramide induces apoptosis by overproduction of ROS
and the switch of superoxide dismutases SOD1 to SOD2 in human lung
cancer cells. Int J Mol Sci. 19:30102018. View Article : Google Scholar : PubMed/NCBI
|