Introduction
Chronic kidney disease (CKD) affects millions of
individuals globally (1).
Mortality due to cardiovascular complications in patients with CKD
is markedly higher than that in matched individuals from the
general population (2).
Cardiovascular disease (CVD) is the leading cause of death among
CKD patients (3). In addition to
accelerated atherosclerotic and vascular calcification,
CKD-associated cardiac injury is also a vital cardiovascular
complication of CKD, which is characterized by left ventricular
hypertrophy (LVH) and diastolic dysfunction (4). Growing evidence suggests that uremic
toxins serve an important role in the development of CKD-associated
cardiac injury (5–8).
Indole-3 acetic acid (IAA) is a protein-bound uremic
solute from tryptophan metabolism (9). The serum level of IAA is increased in
patients with CKD compared with healthy individuals (10). In patients with uremia, IAA cannot
be effectively removed by conventional dialysis and causes side
effects, such as cardiovascular toxicity (11). Mortality and cardiovascular events
are related to higher serum IAA in CKD patients; thus, IAA has been
reported to predict these outcomes in CKD patients (10). However, the toxic effects of IAA on
the heart and the underlying mechanisms of action remain
unclear.
Saikosaponin A (SSA) is a triterpenoid saponin
isolated from Bupleuri radix, a traditional medicinal herb
with numerous bioactive agents (12). It has been reported to have certain
pharmacological activities, such as anti-inflammatory and
antioxidant effects (13). A
previous study reported that SSA reduces pressure overload-induced
myocardial fibrosis (14),
indicating that SSA has a protective effect on the heart. Moreover,
SSA inhibits lead-induced kidney injury (15).
Tripartite motif-containing protein 16 (Trim16), a
member of the Trim family, has E3 ubiquitin ligase activity and
serves an important role in certain diseases, such as pathological
cardiac hypertrophy (16) and
breast cancer (17). Receptor
interacting protein kinase 2 (RIP2) belongs to the tyrosine
kinase-like family (18). RIP2
overexpression aggravates myocardial infarction-related cardiac
remodeling (19). Nevertheless,
whether SSA can help protect against CKD-associated cardiac injury,
an important uremic cardiovascular complication, is still unknown.
The present study investigated the protective effect of SSA against
cardiac damage induced by IAA and explored the underlying mechanism
and the roles of Trim16 and RIP2 in this process.
Materials and methods
Reagents and antibodies
DMEM, fetal bovine serum (FBS), trypsin and
collagenase II were purchased from Gibco (Thermo Fisher Scientific,
Inc.). Bromodeoxyuridine (BrdU) was purchased from Sigma-Aldrich
(Merck KGaA, cat. no. 19-160). Rabbit anti-Trim 16 antibodies (cat.
no. ab72129) and rabbit troponin antibodies (cat. no. ab209813)
were purchased from Abcam. Rabbit anti-total p38 (t-p38) antibodies
(cat. no. 8690), anti-phosphorylated p38 (p-p38) antibodies (cat.
no. 4511) and K48-linked ubiquitin rabbit antibodies (cat. no.
4289) were purchased from Cell Signaling Technology, Inc. Rabbit
anti-RIP 2 antibodies were purchased from Wuhan Sanying
Biotechnology (cat. no. 15366-1-AP). Anti-tubulin antibodies (cat.
no. 80762-1-RR) and anti-GAPDH antibodies (cat. no. 60004-1-Ig)
were purchased from Wuhan Sanying Biotechnology. SSA and IAA were
purchased from Med Chem Express (cat. no. HY-N0246) and
Sigma-Aldrich (Merck KGaA; cat. no. 6505-45-9), respectively.
Co-immunoprecipitation experiments were performed using a Pierce
Co-Immunoprecipitation Kit (Thermo Fisher Scientific, Inc.; cat.
no. 26149). PCR primers were purchased from Nanjing Ruizhen
Biotechnology Co., Ltd. Trim16-specific and nonspecific small
interfering RNAs (siRNAs) were purchased from Shanghai GenePharma
Co., Ltd. Primary cell siRNA transfection reagent was purchased
from Baidai Biology (cat. no. 11016).
Primary culture of neonatal
cardiomyocytes from mice
Primary cardiomyocytes were isolated from neonatal
mice 24–72 h post-birth obtained from the Animal Center of Gannan
Medical College, as previously described (20). The left ventricular tissue was cut
into small pieces (1 mm3) using scissors and digested
with digestion buffer (0.08% trypsin and 0.06% collagenase II
dissolved in Hanks' Balanced Salt Solution) at 37°C. After
terminating digestion with complete DMEM (containing 10% FBS), the
cardiomyocytes were cultured in DMEM with 10% FBS and 0.1 mM BrdU
for 48 h. Cell culture medium was then replaced with complete DMEM
(containing 10% FBS). Cardiomyocytes were identified via
immunofluorescence with troponin. Briefly, cardiomyocytes were
fixed in 4% paraformaldehyde for 10 min at room temperature and
were washed five times with PBS for 10 min. After incubation with
0.2% Triton X-100 (Jiangsu KeyGEN BioTECH Corp., Ltd.) and blocking
with 1% bovine serum albumin (Jiangsu KeyGEN BioTECH Corp., Ltd.)
at room temperature, cardiomyocytes were incubated with
anti-troponin antibodies (1:200; cat. no. ab209813; Abcam)
overnight at 4°C. The samples were then washed three times with
PBS, followed by incubation with fluorescein isothiocyanate
(FITC)-conjugated secondary antibodies (1:1,000; cat. no. ab7086;
Abcam) at room temperature for 1 h. To visualize the nuclei of the
cells, the cells were counterstained with
4,6-diamidino-2-phenylindole (DAPI) for 15 min at 37°C. Images were
captured with a fluorescence microscope system (Zeiss GmbH).
Animal treatment
Male C57BL/6J mice (n=24; age, 8 weeks; weight,
23–25 g) were bred from the Animal Center of Gannan Medical
College. All mice were housed in the animal facility of Gannan
Medical College at 19–21°C under a 12 h light/dark cycle with free
access to food and water. Eight-week-old mice were randomly
assigned to the following experimental groups: Control (n=8),
IAA-treated (n=8) (2.4 mg/kg/24 h IAA by oral gavage for 16 weeks)
and IAA + SSA-treated (n=8) (2.4 mg/kg/24 h IAA by oral gavage and
40 mg/kg/24 h SSA through intraperitoneal injection for 16
weeks).
The mice were sacrificed at 16 weeks after the
beginning of treatment by cervical dislocation after
anesthetization with intraperitoneal injection of pentobarbital (50
mg/kg). For analysis, the heart of each animal was harvested.
Reverse transcription-quantitative PCR
(RT-qPCR) analysis
Total RNA was extracted from the mouse
cardiomyocytes and whole hearts using Trizol (Takara Bio, Inc.).
The cDNA was prepared from 1 µg total RNA using the SuperScript
First-Strand Synthesis system for RT-PCR (Invitrogen; Thermo Fisher
Scientific, Inc.). qPCR was performed on a 10-µl reaction mixture
containing 1 µl cDNA, 0.2 µl forward primer, 0.2 µl reverse primer,
3.8 µl distilled water and 4.8 µl EX Taq (Takara Bio, Inc.) using a
real-time PCR detection system (Roche Diagnostics GmbH). The PCR
program was as follows: Pre-denaturation at 94°C for 1 min;
followed by 30 cycles of denaturation at 94°C for 30 sec, annealing
at 60°C for 30 sec and extension at 72°C for 1 min. A final
extension step was performed at 72°C for 3 min. The relative
expression of atrial natriuretic peptide (ANP), brain natriuretic
peptide (BNP) and β-myosin heavy chain (β-MHC) mRNA was analyzed
using the 2−ΔΔCq method (21) and normalized to GAPDH. Based on the
level of the control group, the results are presented as the fold
increase. The primers used for qPCR were as follows: ANP forward
(F), 5′-GGAGGAGAAGATGCCGGTAGA-3′ and reverse (R),
5′-GCTTCCTCAGTCTGCTCACTCA-3′; BNP F, 5′-AAGCTGCTGGAGCTGATAAGA-3′
and R, 5′-GTTACAGCCCAAACGACTGAC-3′; β-MHC F,
5′-GTGCCAAGGGCCTGAATGAG-3′ and R, 5′-GCAAAGGCTCCAGGTCTGA-3′; and
GAPDH F, 5′-CCAAGGTCATCCATGACAACT-3′ and R,
5′-GGGCCATCCACAGTCTTCT-3′.
siRNA transfection
A Trim16-specific siRNA (siTrim16) and a nonspecific
siRNA (siCntl) were purchased from GenePharma (Shanghai GenePharma
Co., Ltd.). According to manufacturer's instructions 100 nmol/l
siRNAs were transfected into cardiomyocytes with Primary Cell siRNA
Transfection Reagent (cat. no. 11016; Baidai Biology) at room
temperature. Briefly, cells (5×106 cells/ml) were seeded
into six-well plates. siRNA (100 nM) was diluted in 400 µl
serum-free culture medium, and 4 µl Primary Cell siRNA Transfection
Reagent was added to it. Cells were incubated with the transfection
complexes for 20 min at room temperature before being mixed with
1.6 ml fresh serum-free culture medium. After 8 h, the medium was
replaced by fresh culture medium containing 10% FBS. Subsequently,
cardiomyocytes were treated with IAA (50 µmol/l) or SSA (30 µmol/l)
for 48 h and used for assessment. The siRNA sequences used were as
follows: TRIM16 sense, 5′-AGUAAUUCACCAUGCAGGUUU-3′ and antisense,
5′-UCUCCCUCCUGCAUUUGUGUU-3′; and control (siCntl) sense,
5′-UUCUCAGAACGUGUCACGUTT-3′ and antisense,
5′-ACGUGACAAGUUCGGAGAATT-3′.
Immunoblotting
Total protein was extracted from neonatal left
ventricular heart sample tissues and cardiomyocytes using RIPA
buffer (cat. no. KGP10100; Jiangsu KeyGEN BioTECH Corp., Ltd.). The
protein concentration was detected using the BCA protein
quantitative kit (cat. no. P0012; Beyotime Institute of
Biotechnology). Protein (20 µg/lane) was separated by SDS-PAGE on
10% gels and was transferred to PVDF membranes (cat. no. FFP32;
Beyotime Institute of Biotechnology). The membranes were blocked
with 5% skimmed milk at room temperature for 30 min and incubated
with primary antibodies against Trim16 (1:1,000; cat. no. ab72129;
Abcam), t-p38 (1:1,000; cat. no. 8690; Cell Signaling Technology,
Inc.), p-p38 (1:1,000; cat. no. 4511; Cell Signaling Technology,
Inc.) and RIP2 (1:1,000; cat. no. 15366-1-AP; Wuhan Sanying
Biotechnology) at 4°C overnight. The membrane was then incubated
with goat anti-rabbit secondary antibodies (1:5,000; cat. no.
ZB2301; OriGene Technologies, Inc.). The intensity of the bands was
assessed using a chemiluminescence kit (cat. no. 32209; Thermo
Fisher Scientific, Inc.) and ImageJ software (version 1.5.3;
National Institutes of Health).
Coimmunoprecipitation experiments
Coimmunoprecipitation experiments were performed
utilizing a coimmunoprecipitation kit (cat. no. 26149; Thermo
Fisher Scientific, Inc.). Total proteins from the cardiomyocytes
were isolated using RIPA lysis buffer, and quantified using the BCA
kit. For immunoprecipitation, 500 µg protein was incubated with 2
µg appropriate antibodies, including RIP2 antibodies (cat. no.
15366-1-AP; Wuhan Sanying Biotechnology) or IgG negative control
antibodies (Beyotime Institute of Biotechnology; no. A7016)
overnight at 4°C. Subsequently, 40 µl Protein G/A agarose beads
(Invitrogen; Thermo Fisher Scientific, Inc.) were added to the cell
lysate and incubated for 2 h at room temperature. After beads were
washed with PBS three times, precipitated proteins eluted from the
beads (1 µg/µl) were resuspended in SDS-PAGE loading buffer, and
boiled for 5 min. Finally, western blot analysis was used to
measure the immunoprecipitation products as aforementioned.
Hematoxylin and eosin (H&E)
staining
The hearts from mice were fixed in 10%
paraformaldehyde for 48 h at room temperature. H&E staining was
performed according to routine protocols. Briefly, for H&E
staining, the heart slices were fixed using a graded alcohol series
(100, 95, 85 and 70%) for 5 min and hydrated by immersion in 1%
hydrochloric acid alcohol for 30 sec at room temperature. The
slices were then stained using hematoxylin for 5 min and rinsed in
water for 1 min at room temperature. The slices were subsequently
stained with 0.5% eosin for 3 min. Finally, sections were covered
using a coverslip and imaged under a light microscope (Zeiss
GmbH).
Echocardiography and Doppler
analysis
Echocardiography was performed using a
high-resolution ultrasound imaging system (Vevo 2100, VisualSonics,
Inc.) to assess cardiac structure and function. The mouse was fixed
in the supine position and anesthetized through inhalation of 2%
isoflurane/100% oxygen. Left ventricular end-diastolic anterior
wall depth (LVAWd), left ventricular end-systolic anterior wall
depth (LVAWs), left ventricular end-diastolic posterior wall depth
(LVPWd), left ventricular end-systolic posterior wall depth (LVPWs)
and left ventricular diastolic function indicators, such as the
ratio of left ventricular transmitral early peak flow velocity to
left ventricular transmitral late peak flow velocity (E/A ratio)
(22), were recorded.
Serum biochemistry analysis
The mice were anesthetized via an intraperitoneal
injection of pentobarbital (50 mg/kg) and were sacrificed by
exsanguination performed after removal of the eyeball. Blood
collected from the retro-orbital vein was centrifuged at 157 × g
for 5 min at 4°C, and the serum was collected and stored at −80°C.
Serum creatinine (Cr) and blood urea nitrogen (BUN) levels were
measured by Roche automatic biochemical analysis. Levels of BUN and
Cr were evaluated by urease-glutamate dehydrogenase and enzymatic
methods, respectively. BUN assay kits (cat. no. OSR6234) and Cr
assay kits (batch no. 20220912) were provided by Beckman Coulter,
Inc. and Shanghai KHB Co., Ltd., respectively.
Statistical analysis
SPSS statistical software (version 20.0, IMB Corp.)
was used for data analysis. The differences between groups were
determined by one-way analysis of variance (ANOVA) and Tukey's post
hoc test was used following ANOVA. Values are presented as the mean
± standard deviation and P<0.05 was considered to indicate a
statistically significant difference.
Results
SSA alleviates cardiomyocyte
hypertrophy induced by IAA
Cultured mouse cardiomyocytes were treated with
various concentrations of IAA. Compared with the control group, the
mRNA expression levels of ANP, BNP and β-MHC in the 10 and 50
µmol/l IAA-treated groups were significantly increased (Fig. 1A). Furthermore, SSA treatment
inhibited cardiomyocyte hypertrophy induced by IAA. Compared with
those in the 50 µmol/l IAA group, the mRNA expression levels of
ANP, BNP and β-MHC in the 30 µmol/l SSA-treated groups were
significantly decreased (Fig.
1B).
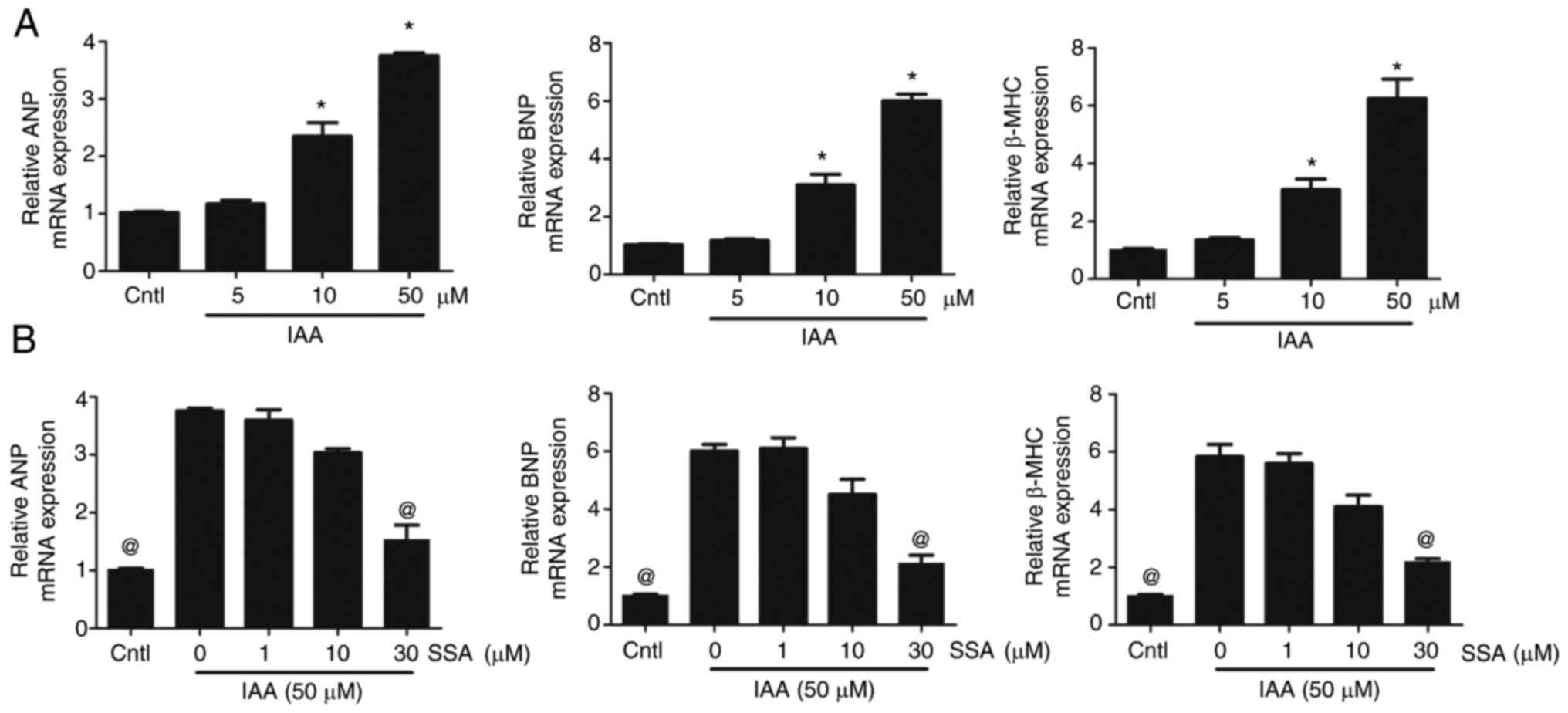 | Figure 1.SSA inhibits IAA-induced
cardiomyocyte hypertrophy. (A) Cardiomyocytes were treated with
different concentrations of IAA (5, 10 and 50 µmol/l) for 48 h and
mRNA expression levels of ANP, BNP and β-MHC were analyzed by qPCR.
(B) Cardiomyocytes were pretreated with SSA (1, 10 and 30 µmol/l)
for 1 h then exposed to IAA (50 µmol/l) for 48 h and mRNA
expression levels of ANP, BNP and β-MHC were analyzed by qPCR.
*P<0.01 vs. Cntl, @P<0.01 vs. 50 µmol/l IAA group.
SSA, saikosaponin A; IAA, indole-3 acetic acid; ANP, atrial
natriuretic peptide; BNP, brain natriuretic peptide; β-MHC,
β-myosin heavy chain; Cntl, control; qPCR, quantitative PCR. |
SSA inhibits downregulation of Trim16
expression and upregulation of RIP2 expression induced by IAA
Cardiomyocytes were transfected with (siCntl) or
siTrim16. Compared with the untransfected control and siCntl
groups, the protein expression level of Trim16 in the siTrim16
group was significantly decreased (Fig. 2A). Cardiomyocytes were treated with
either IAA (50 µmol/l) or IAA + SSA (50 µmol/l IAA + 30 µmol/l
SSA). Compared with the control group, the protein expression level
of Trim16 in the IAA-only group was significantly decreased, and
SSA treatment significantly blocked this decrease (Fig. 2B). Cardiomyocytes were treated with
siCntl, IAA (50 µmol/l IAA + siCntl), IAA + SSA (50 µmol/l IAA + 30
µmol/l SSA + siCntl) or IAA + SSA + siTrim16 (50 µmol/l IAA + 30
µmol/l SSA + siTrim16). Compared with the control siCntl group, the
protein expression level of RIP2 in the IAA-treated group was
significantly upregulated, but SSA treatment significantly
inhibited this IAA-induced RIP2 upregulation. Notably, Trim16
knockdown significantly blocked the inhibitory effect of SSA on
RIP2 upregulation (Fig. 2C).
SSA alleviates cardiomyocyte
hypertrophy induced by IAA and silencing of Trim16 blocks the
anti-hypertrophic effect of SSA
Cardiomyocytes were treated with siCntl, IAA (50
µmol/l IAA + siCntl), IAA + SSA (50 µmol/l IAA + 30 µmol/l SSA +
siCntl) or and IAA + SSA + siTrim16 (50 µmol/l IAA + 30 µmol/l SSA
+ siTrim16). Compared with the control group, the mRNA expression
levels of ANP, BNP and β-MHC in the IAA-treated group were
significantly increased. The mRNA expression levels of ANP, BNP and
β-MHC in the IAA + SSA-treated group were significantly
downregulated compared with that in the IAA-only group. Trim16
knockdown significantly reduced the inhibitory effect of SSA on the
expression of ANP, BNP and β-MHC (Fig.
3A). Immunofluorescence analysis of the morphological
alterations in cardiomyocytes demonstrated that SSA inhibited
cardiomyocyte hypertrophy induced by IAA. Silencing Trim16 blocked
the antihypertrophic effect of SSA (Fig. 3B). The phosphorylation of p38 was
significantly increased in the IAA-treated group compared with that
in the control group, however, SSA treatment significantly
decreased the phosphorylation of p38. Moreover, the effect of SSA
was significantly blocked by silencing Trim16 (Fig. 3C).
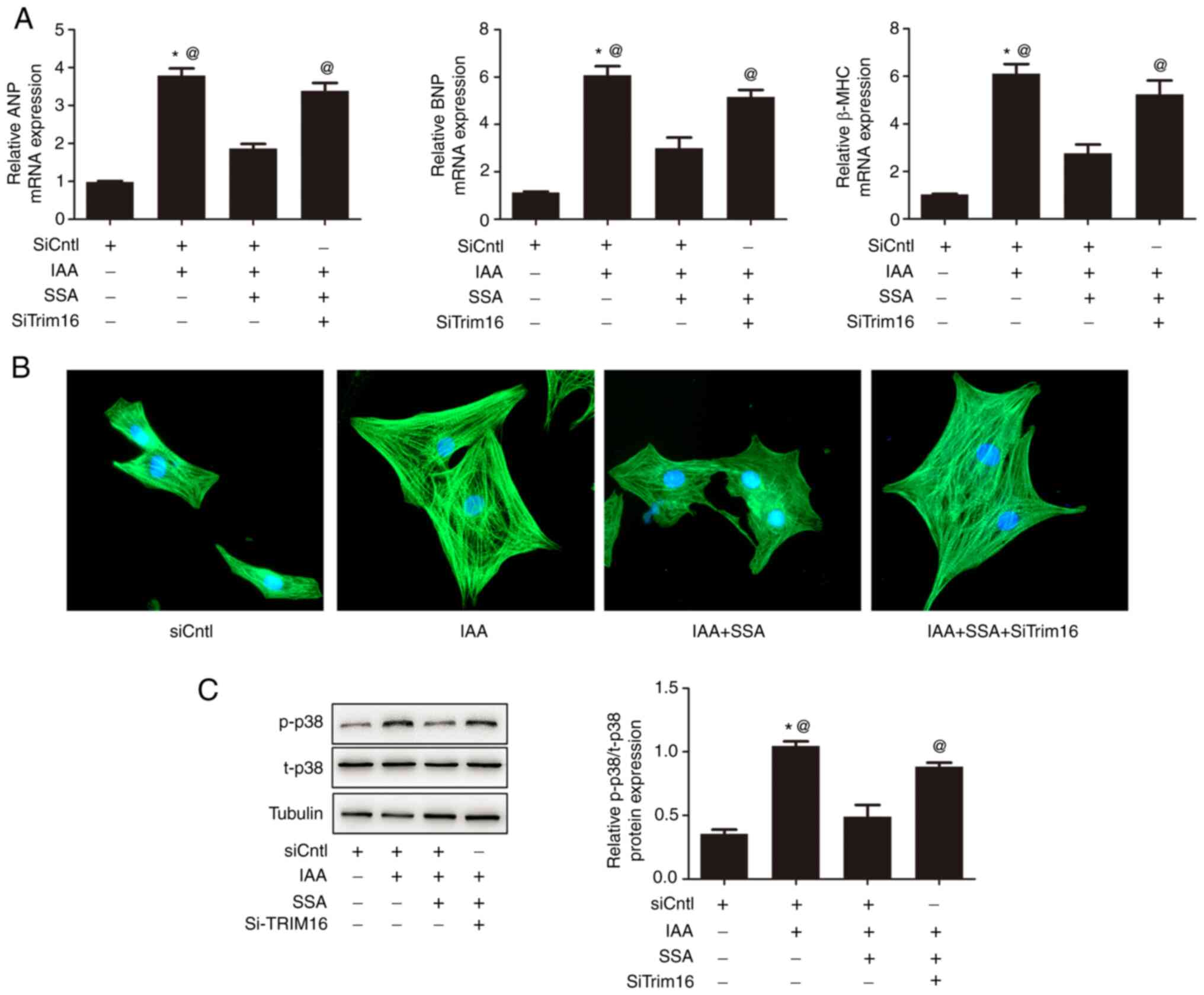 | Figure 3.Silencing of Trim16 blocks the
inhibitory effect of SSA on cardiomyocyte hypertrophy induced by
IAA. Cardiomyocytes were transfected with siTrim16 or scrambled
siRNA (siCntl), treated with SSA (30 µmol/l) for 1 h then treated
with IAA (50 µmol/l) for 48 h. (A) mRNA expression of ANP, BNP and
β-MHC were measured by quantitative PCR. (B) Cell size was observed
by immunofluorescence using a troponin antibody (magnification,
×400). (C) Expression of p-p38 and t-p38 was measured by western
blotting. *P<0.01 vs. siCntl, @P<0.01 vs. IAA +
SSA-treated group. SSA, saikosaponin A; IAA, indole-3 acetic acid;
Trim16, tripartite motif-containing protein 16; ANP, atrial
natriuretic peptide; BNP, brain natriuretic peptide; β-MHC,
β-myosin heavy chain; p, phosphorylated; t, total. |
IAA inhibits K48 ubiquitination of
RIP2 in cardiomyocytes and SSA-induced RIP2 ubiquitination in a
Trim16-dependent manner
Cardiomyocytes were treated with siCntl, IAA (50
µmol/l IAA + siCntl), IAA + SSA (50 µmol/l IAA + 30 µmol/l SSA +
siCntl) or IAA + SSA + siTrim16 (50 µmol/l IAA + 30 µmol/l SSA +
siTrim16). Compared with the control group, K48 ubiquitination of
RIP2 in cardiomyocytes in the IAA-treated group was significantly
decreased. SSA significantly increased the K48 ubiquitination of
RIP2. Furthermore, this effect of SSA was significantly reduced by
silencing Trim16 (Fig. 4).
 | Figure 4.SSA alleviates the inhibitory effect
of IAA on the K48 ubiquitination of RIP2 in cardiomyocytes. (A)
Cardiomyocytes were transfected with siTrim16 or scrambled siRNA
(siCntl), treated with SSA (30 µmol/l) for 1 h, then incubated with
IAA (50 µmol/l) for 48 h. K48 ubiquitination of RIP2 was measured
by immunoprecipitation. (B) Expression of RIP2 was measured by
western blotting *P<0.01 vs. IAA-treated group,
@P<0.05 vs. IAA + SSA-treated group. SSA,
saikosaponin A; IAA, indole-3 acetic acid; RIP2, receptor
interacting protein kinase 2; p, phosphorylated; t, total; IP,
immunoprecipitation; IB, immunoblotting; si, short interfering
RNA. |
SSA alleviates structural and
functional abnormalities of the heart induced by IAA in mice
Male C57BL/6J mice were divided into control,
IAA-treated and IAA + SSA-treated groups. Compared with those in
the control group, the levels of BUN and Cr in the IAA-treated
group were significantly increased 16 weeks after IAA
administration. Treatment of mice with SSA did not decrease the
IAA-mediated increase in BUN and Cr levels (Fig. 5A). Compared with the control group,
mRNA expression levels of ANP, BNP and β-MHC in the heart were
significantly increased in the IAA-treated group (Fig. 5B). Administration of SSA
significantly reduced the expression of ANP, BNP and β-MHC
(Fig. 5B). In addition, an
increase in cardiac hypertrophy in IAA-treated mice compared with
that in the control group was observed using H&E staining.
Treatment with SSA alleviated the cardiac hypertrophy observed in
IAA-treated mice (Fig. 5C).
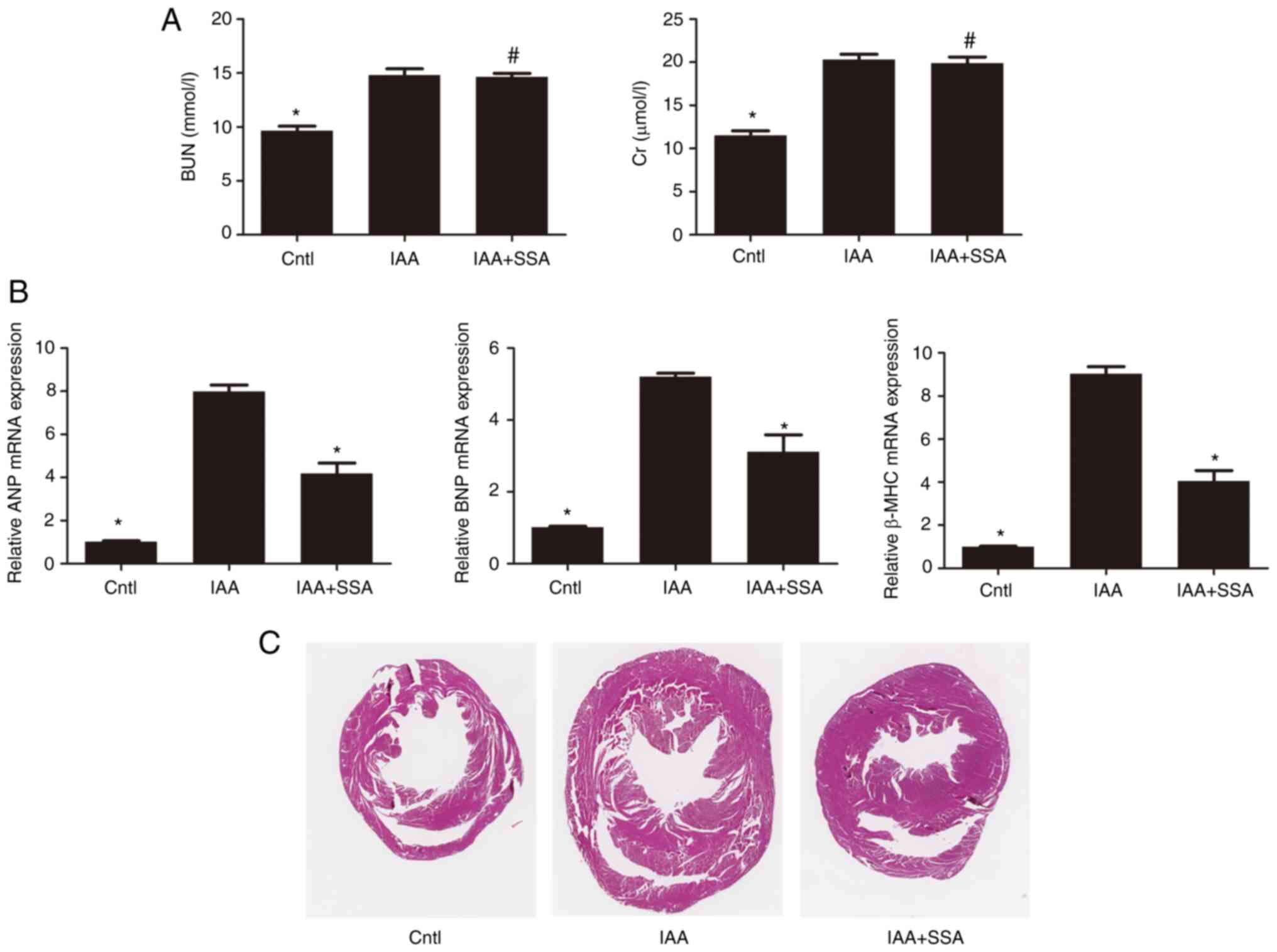 | Figure 5.SSA treatment ameliorates IAA-induced
cardiac hypertrophy in mice. (A) Serum BUN and Cr levels in the
control (n=8), IAA (n=8) and IAA + SSA (n=8) groups of mice were
analyzed by urease-glutamate dehydrogenase and enzymatic methods,
respectively. (B) The mRNA expression of ANP, BNP and β-MHC in the
myocardial tissues of control, IAA- IAA + SSA-treated groups of
mice were analyzed by quantitative PCR. (C) Representative
micrographs (magnification, 10×) of hematoxylin and eosin-stained
transverse sections from hearts of control, IAA- and IAA +
SSA-treated mice. *P<0.01 vs. IAA-treated group;
#P<0.01 vs. Cntl. SSA, Saikosaponin A; IAA, Indole-3
acetic acid; ANP, Atrial natriuretic peptide; BNP, Brain
natriuretic peptide; β-MHC, β-myosin heavy chain; Cntl, control;
BUN, blood urea nitrogen; Cr, serum creatinine. |
Echocardiography demonstrated that the LVPWs, LVPWd,
LVAWs and LVAWd in the IAA-treated group of mice were significantly
higher than those in the control group (Fig. 6A). Treatment with SSA significantly
decreased the LVPWs, LVPWd, LVAWs and LVAWd in IAA-treated mice
(Fig. 6A). Analysis of the
Doppler-derived mitral flow velocities demonstrated that there was
a significant reduction in the E/A ratio in the IAA-treated mice.
Such an alteration is always accompanied by diastolic relaxation
abnormalities (10), which
indicated that IAA treatment impaired cardiac diastolic function.
However, SSA treatment significantly ameliorated cardiac diastolic
function in IAA-treated mice (Fig.
6B).
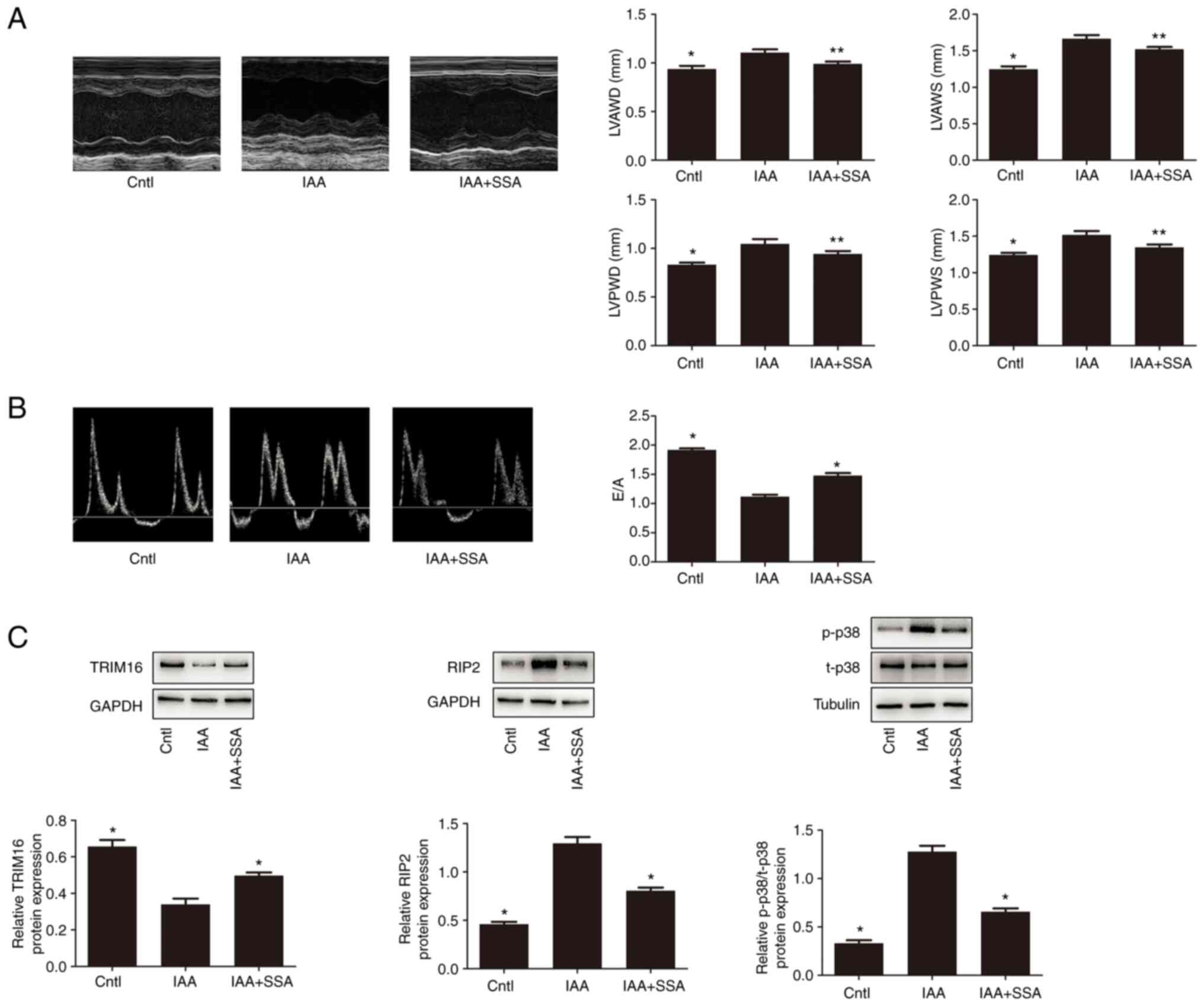 | Figure 6.SSA treatment ameliorates structural
and functional abnormalities of the heart in echocardiography of
mice treated with IAA, and also regulates expression of Trim16,
RIP2 and p-p38 in the heart. (A) Representative M-mode
echocardiograms and the mean LVPWs, LVPWd, LVAWs and LVAWd in the
control, IAA- and IAA + SSA-treated groups. (B) Representative
M-mode echocardiograms and the mean E/A in the control, IAA- and
IAA + SSA-treated groups. (C) Expression of Trim16, RIP2, p-p38 and
t-p38 measured by western blotting. *P<0.01, **P<0.05 vs.
IAA-treated group. SSA, saikosaponin A; IAA, indole-3 acetic acid;
Trim16, tripartite motif-containing protein 16; p, phosphorylated;
Cntl, control; RIP2, receptor interacting protein kinase 2; LVPWd,
left ventricular end-diastolic posterior wall depth; LVPWs, left
ventricular end-systolic posterior wall depth; LVAWd, left
ventricular end-diastolic anterior wall depth; LVAWs, left
ventricular end-systolic anterior wall depth. |
Effect of IAA and SSA on the
expression of Trim16, RIP2 and p38 in mice
Compared with the control group, the expression of
Trim16 in the heart in the IAA-treated mice was significantly
decreased; however, the expression of RIP2 and phosphorylation of
p38 in the IAA-treated group were significantly increased.
Intraperitoneal injection of SSA significantly inhibited the
downregulation of Trim16 expression, significantly upregulated RIP2
expression and significantly increased phosphorylation of p38 in
the heart of IAA-treated mice (Fig.
6C).
Discussion
CKD is a global health issue which has attracted
much attention (23). It is
estimated that 10–14% of the global population have CKD (4). Patel et al (4) reported a total of 37 and 2.6 million
patients with CKD in the USA and UK, respectively. As CKD
progresses, certain complications can occur, among which
cardiovascular complications are particularly important (24–26).
CKD-associated cardiac injury (also known as uremic cardiomyopathy)
is a widely prevalent cardiovascular disease in patients with CKD,
which accounts for ~50% of deaths due to CKD (27). CKD-associated cardiac injury leads
to left ventricular hypertrophy, left ventricular dilation and left
ventricular diastolic dysfunction. Severe CKD-associated cardiac
injury can lead to sudden cardiac death even in individuals without
cardiac symptoms (28).
Although previous studies have reported that
hypertension, volume overload, insulin resistance and
hyperphosphatemia serve important roles in CKD-associated cardiac
injury, the drivers and molecular mechanisms underlying
CKD-associated cardiac injury are still unclear. Since 2015,
mounting evidence (7,29) has indicated that uremic toxins have
a vital role in the development of CKD-associated cardiac
injury.
The uremic toxin indoxyl sulfate (IS) induces
cardiomyocyte hypertrophy in vitro and in vivo
(6,30). Moreover, our previous study
reported that another uremic toxin, p-cresyl sulfate (PCS), also
induced cardiomyocyte hypertrophy (5). In addition to IS and PCS, IAA is
another important uremic toxin (7,10).
IAA is a protein-bound uremic toxin that is mainly produced during
tryptophan metabolism by intestinal bacteria (31). Liabeuf et al (31) reported that serum IAA progressively
increased with CKD stage. Claro et al (32) demonstrated a negative correlation
between eGFR and IAA in patients with CKD in pre-dialysis. Dou
et al (10) reported that
mortality and the occurrence of cardiovascular events were
significantly higher in individuals with serum IAA levels >3.73
mmol/l compared with serum IAA levels <3.73 mmol/l in a study
that followed 120 patients with CKD over 966 days. Multivariate Cox
regression analysis has been reported to demonstrate that serum IAA
level can predict cardiovascular events and mortality after
adjusting for age, sex, cholesterol, systolic blood pressure,
smoking, C-reactive protein, serum phosphorus, body mass index,
albumin, diastolic blood pressure and history of cardiovascular
disease (10). Notably, when IS,
PCS and IAA were integrated into a multivariate Cox regression
model, only IAA predicted cardiovascular events and mortality,
which suggests that there was a close association between high
serum IAA and cardiovascular complications in CKD (10). Chinnappa et al (33) reported that IAA was closely
associated with peak cardiac power and aerobic exercise capacity in
patients with CKD.
Stockler-Pinto et al (34) reported that IAA stimulated
production of nuclear factor-kappa B (NF-κB) mRNA and decreased
nuclear E2-related factor 2 (Nrf2) expression in hemodialysis
patients, which indicated that IAA triggered inflammation and
oxidative stress in these individuals. Bataille et al
(35) reported that there was no
apparent association of IAA with anemia parameters in hemodialysis
patients. Therefore, further in-depth studies on the role of IAA in
complications of CKD should be performed. Furthermore, IAA
activates the aryl hydrocarbon receptor (AhR)/p38MAPK/NF-κB
signaling pathway and upregulates the expression of the
proinflammatory enzyme cyclooxygenase-2 in endothelial cells in
vitro (10). IAA treatment
also increases production of reactive oxygen species in endothelial
cells (10). Addi et al
(36) reported that IAA induced
tissue factor expression in multiple types of endothelial cells,
such as human umbilical vein endothelial cells (HUVECs), aortic
endothelial cells and cardiac-derived microvascular cells. NF-κB
p50 subunit translocation induced by IAA serves a key role in this
process (36). Furthermore,
inhibition of the AhR/p38MAPK signaling pathway reduces tissue
factor expression upregulation in IAA-treated endothelial cells
(36). Previous clinical and basic
studies demonstrate that IAA has a damaging effect on the
cardiovascular system, however, few studies on IAA in
CKD-associated cardiac injury have been previously reported.
Recently, Hager et al (37)
reported that IAA prompted cardiac necrosis in rats. The present
study demonstrated that IAA upregulated the expression of ANP, BNP
and β-MHC in mouse cardiomyocytes and induced cardiomyocyte
hypertrophy in vitro. Moreover, cardiac hypertrophy,
decreased diastolic function and increased expression of ANP, BNP
and β-MHC were demonstrated to occur in mice treated with IAA.
Therefore, IAA could induce CKD-associated cardiac injury both
in vivo and in vitro.
SSA is a triterpenoid saponin isolated from R.
bupleuri (12) that exerts
numerous pharmacological effects involving antioxidative stress and
anti-inflammation (13). SSA
regulates the expression of bone morphogenetic protein 4 in hepatic
stellate cells (38). SSA also
attenuates liver inflammation and fibrosis induced by carbon
tetrachloride (39). Zhou et
al (40) reported that SSA
alleviated ulcerative colitis through an anti-inflammatory pathway.
Du et al (41) reported
that SSA alleviated lipopolysaccharide (LPS)-induced acute lung
injury in mice by reducing the expression of TNF-α and IL-1β.
Furthermore, SSA demonstrates protective effects against neuronal
damage induced by ischemia-reperfusion injury, and this mechanism
involves downregulation of Toll-like receptor 4 and NF-κB
expression in the brain (42).
There are few reports of the effect of SSA on cardiovascular
disease. Fu et al (43)
reported that SSA inhibited LPS-induced oxidative stress and
inflammation in HUVECs. He et al (44) reported that SSA attenuated
atherosclerosis by inhibiting the PI3K/Akt/NF-κB/NLRP3 signaling
pathway. A previous study reported that SSA alleviated pressure
overload-induced cardiac fibrosis (14). Zhang et al (45) reported that Saikosaponin D, another
similar triterpenoid saponin isolated from R. bupleuri,
efficiently protected cardiomyocytes from Doxorubicin-induced
cardiotoxicity by inhibiting excessive oxidative stress. In
addition, SSA has a protective effect on the kidney (15) and complications of chronic kidney
disease (46). Huang et al
demonstrated that SSA improved CKD-induced muscle atrophy by
reducing oxidative stress through the PI3K/AKT/Nrf2 pathway
(46). However, there has been
little research on whether SSA alleviates CKD-associated cardiac
injury. In the present study, administration of SSA inhibited
cardiomyocyte hypertrophy induced by IAA. This confirmed that SSA
reduced ANP, BNP and β-MHC expression in cardiomyocytes and reduced
the size of these cells, reversing the increased expression and
size of cells induced by IAA treatment.
Trim16, a member of the Trim family of proteins
(47), was first identified as an
estrogen-responsive B-box protein which possesses transcriptional
activity (48). The molecular
structure of Trim16 is different from other members of the Trim
family, as it lacks the RING finger domain (RING domain) (49). Trim16 has numerous functions,
including regulation of cell differentiation, the innate immune
response and tumorigenesis (50–53).
The majority of Trim family members have E3 ubiquitin ligase
activity and serve an important role in protein posttranslational
modification (47). Although
Trim16 lacks the RING domain, it has two B-box domains and has E3
ubiquitin ligase activity.
Certain Trim family proteins, such as Trim8, Trim24,
Trim32 and Trim72, have been reported to serve key roles in cardiac
hypertrophy and other cardiovascular diseases, which indicated that
the TRIM family serve a critical role in heart disease (54,55).
Although the role of the Trim family in cardiac development,
cardiomyopathy and other cardiac diseases has been widely reported,
the role of Trim16 in cardiac diseases is still unclear. Trim16
inhibits inflammation and oxidative stress, which are often closely
associated with cardiovascular disease (56,57).
A previous study (16) found that
Trim16 deficiency aggravated phenylephrine-induced cardiomyocyte
hypertrophy in vitro and transverse aortic
constriction-induced mouse cardiac hypertrophy in vivo,
whereas overexpression of Trim16 inhibited cardiac hypertrophy. The
underlying mechanism was reported to be Trim16-increased
ubiquitination of Scr kinase (16). However, the role of Trim16 in
CKD-associated cardiac injury is currently unknown. In the present
study, the expression of Trim16 was downregulated in hypertrophic
cardiomyocytes treated with IAA, and SSA alleviated cardiomyocyte
hypertrophy and upregulated Trim16 expression, which suggested that
Trim16 may be involved in CKD-associated cardiac injury.
RIP2 belongs to the tyrosine kinase-like family of
proteins (18). RIP2 is involved
in the transduction of multiple signaling pathways, such as the
IKK/NF-κB and MAPK/AP1 signaling pathways (18), which implied that RIP2 may serve an
important role in the occurrence and development of certain
diseases, such as myocardial ischemia and septic cardiomyopathy
(58,59). RIP2 overexpression aggravates
myocardial infarction-related cardiac remodeling, and its mechanism
is related to the activation of p38 phosphorylation (19). Previously (60), it was demonstrated that the
expression of RIP2 was significantly increased in cardiac cells in
patients with heart failure, mice with aortic banding
surgery-induced pressure overload and phenylephrine-treated
cardiomyocytes in vitro. Notably, RIP2 overexpression
aggravates pressure overload-induced cardiac remodeling (60). The expression and function of RIP2
in CKD-associated cardiac injury have not yet been confirmed. The
present study demonstrated that the expression of RIP2 and
phosphorylated p38 was upregulated in hypertrophic cardiomyocytes
treated with IAA. Furthermore, SSA inhibited the upregulation of
RIP2 expression and p38 phosphorylation. Humphries et al
reported that RIP2 can be modified by ubiquitination, which in turn
affects the signaling pathway function of RIP2 (18). Trim16 has E3 ubiquitin ligase
activity and RIP2 can be regulated by ubiquitination (49,61),
however, whether Trim16 can regulate RIP2 ubiquitination has not
been previously reported. The present study demonstrated that
upregulation of Trim16 promoted RIP2 K48 ubiquitination, which is
normally associated with protein degradation, which indicated that
Trim16 alleviated CKD-associated cardiac injury by increasing the
ubiquitination of RIP2 at K48 and promoting RIP2 degradation.
Moreover, the present study demonstrated that Trim16 knockdown
blocked the inhibitory effect of SSA on IAA-induced upregulation of
RIP2 expression and cardiomyocyte hypertrophy.
In the present study, an IAA-induced CKD-associated
cardiac injury mouse model was established. Mice were administered
IAA by oral gavage for 16 weeks and echocardiography analysis
demonstrated increased LVAWd, LVAWs, LVPWs and LVPWd in IAA-treated
mice. In addition, heart hypertrophy was also observed in
IAA-treated mice. Analysis of the Doppler-derived mitral flow
velocity demonstrated a reduction in the diastolic function of the
heart in IAA-treated mice. Administration of SSA improved cardiac
hypertrophy and diastolic dysfunction. Moreover, SSA inhibited
IAA-induced downregulation of TRIM16 expression, upregulation of
RIP2 expression and p38 phosphorylation in the hearts of
CKD-associated cardiac injury mice.
The present study had certain limitations. First,
the conclusions were not tested in transgenic mouse models. If the
toxicity of IAA is reduced in TRIM16-overexpressing and
RIP2-knockout mice, the evidence to support this mechanism would be
stronger. In addition, IAA impairs renal function, so the damaging
effect of IAA on the heart may be related to the deterioration of
renal function. If the concentration of other uremic toxins such as
IS and PCS in the serum of mice were measured, more rigorous
results would be obtained.
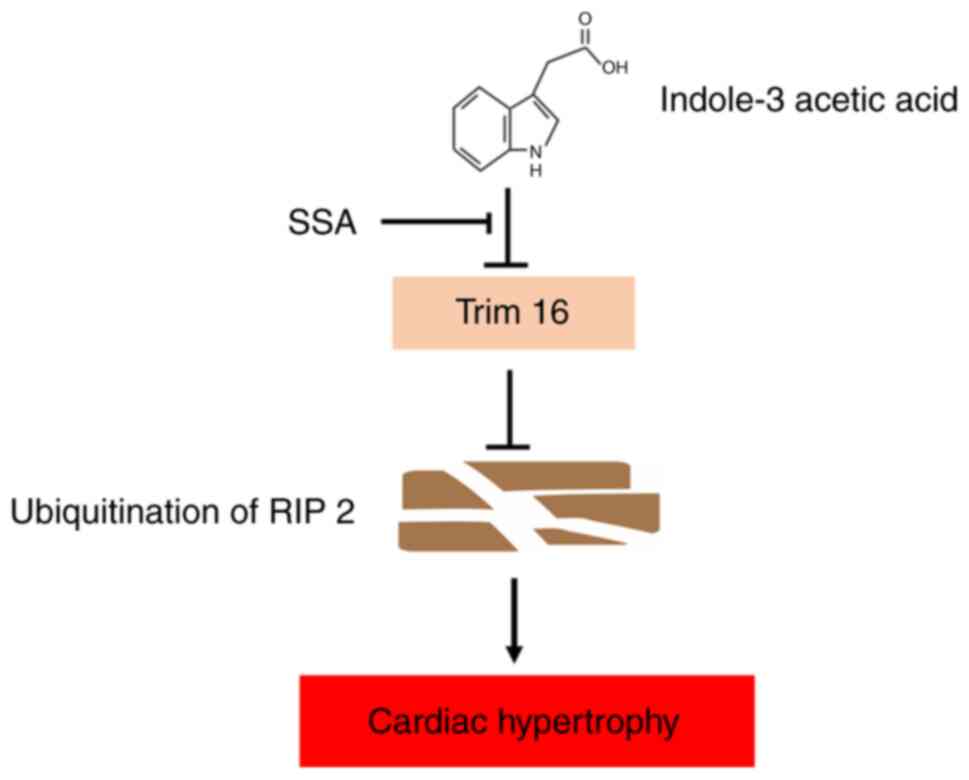
In summary, the present study demonstrated that the
uremic toxin IAA induced cardiomyocyte hypertrophy via regulation
of RIP2 ubiquitination mediated by TRIM16 and p38 phosphorylation
and that SSA antagonized the damaging effects of IAA (Fig. 7). The present study provided novel
information on the posttranslational modification of RIP2 by IAA
and SSA. As a result of these findings, new insights into
CKD-associated cardiac injury have been reported and have the
potential to contribute to the future development of treatments for
this disease.
Acknowledgements
Not applicable.
Funding
The present study was funded by the Green Yang golden phoenix
plan of Yangzhou (grant no. YZLYJFJH2021YXBS027), The Science and
Technology Plan Project of Jiangxi Province Health and Health
Commission (grant no. 202210913) and The Guiding Science and
Technology Plan Project of Ganzhou (grant no. GZ2021ZSF105).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CC and XC conceived and designed the experiments. XC
and XH performed the experiments. CC analyzed the data and wrote
the original draft. XC reviewed and edited the manuscript. CC and
XC confirm the authenticity of all the raw data. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
All animal protocols were approved by the Animal
Ethical and Welfare Committee of Gannan Medical College (approval
no. 2021092).
Patient consent for publication
Not applicable.
Authors' information
XC ORCID ID: 0000-0002-9206-4145
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lv JC and Zhang LX: Prevalence and disease
burden of chronic kidney disease. Adv Exp Med Biol. 1165:3–15.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Webster AC, Nagler EV, Morton RL and
Masson P: Chronic kidney disease. Lancet. 389:1238–1252. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Matsushita K, Ballew SH, Wang AY,
Kalyesubula R, Schaeffner E and Agarwal R: Epidemiology and risk of
cardiovascular disease in populations with chronic kidney disease.
Nat Rev Nephrol. 18:696–707. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Patel N, Yaqoob MM and Aksentijevic D:
Cardiac metabolic remodelling in chronic kidney disease. Nat Rev
Nephrol. 18:524–537. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen C, Xie C, Xiong Y, Wu H, Wu L, Zhu J,
Xing C and Mao H: Damage of uremic myocardium by p-cresyl sulfate
and the ameliorative effect of Klotho by regulating SIRT6
ubiquitination. Toxicol Lett. 367:19–31. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang K, Wang C, Nie L, Zhao X, Gu J, Guan
X, Wang S, Xiao T, Xu X, He T, et al: Klotho protects against
indoxyl sulphate-induced myocardial hypertrophy. J Am Soc Nephrol.
26:2434–2446. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Han H, Zhu J, Zhu Z, Ni J, Du R, Dai Y,
Chen Y, Wu Z, Lu L and Zhang R: p-Cresyl sulfate aggravates cardiac
dysfunction associated with chronic kidney disease by enhancing
apoptosis of cardiomyocytes. J Am Heart Assoc. 4:e0018522015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu CC, Hsieh MY, Hung SC, Kuo KL, Tsai TH,
Lai CL, Chen JW, Lin SJ, Huang PH and Tarng DC: Serum indoxyl
sulfate associates with postangioplasty thrombosis of dialysis
grafts. J Am Soc Nephrol. 27:1254–1264. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jourde-Chiche N, Dou L, Cerini C,
Dignat-George F, Vanholder R and Brunet P: Protein-bound
toxins-update 2009. Semin Dial. 22:334–339. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dou L, Sallée M, Cerini C, Poitevin S,
Gondouin B, Jourde-Chiche N, Fallague K, Brunet P, Calaf R, Dussol
B, et al: The cardiovascular effect of the uremic solute indole-3
acetic acid. J Am Soc Nephrol. 26:876–887. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shi Y, Tian H, Wang Y, Shen Y, Zhu Q and
Ding F: Improved dialysis removal of protein-bound uraemic toxins
with a combined displacement and adsorption technique. Blood Purif.
51:548–558. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shuai-Cheng W, Xiu-Ling C, Jian-Qing S,
Zong-Mei W, Zhen-Jiang Y and Lian-Tao L: Saikosaponin A protects
chickens against pullorum disease via modulation of cholesterol.
Poult Sci. 98:3539–3547. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhu Y, Chen X, Rao X, Zheng C and Peng X:
Saikosaponin a ameliorates l ipopolysaccharide and
d-galactosamine-induced liver injury via activating LXRα. Int
Immunopharmacol. 72:131–137. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu Y, Gao L, Zhao X, Guo S, Liu Y, Li R,
Liang C, Li L, Dong J, Li L and Yang H: Saikosaponin a protects
from pressure overload-induced cardiac fibrosis via inhibiting
fibroblast activation or endothelial cell endMT. Int J Biol Sci.
14:1923–1934. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Song Y, Sun H, Gao S, Tang K, Zhao Y, Xie
G and Gao H: Saikosaponin a attenuates lead-induced kidney injury
through activating Nrf2 signaling pathway. Comp Biochem Physiol C
Toxicol Pharmacol. 242:1089452021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu J, Li W, Deng KQ, Tian S, Liu H, Shi
H, Fang Q, Liu Z, Chen Z, Tian T, et al: The E3 ligase TRIM16 is a
key suppressor of pathological cardiac hypertrophy. Circ Res.
130:1586–1600. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Roshanazadeh MR, Adelipour M, Sanaei A,
Chenane H and Rashidi M: TRIM3 and TRIM16 as potential tumor
suppressors in breast cancer patients. BMC Res Notes. 15:3122022.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Humphries F, Yang S, Wang B and Moynagh
PN: RIP kinases: Key decision makers in cell death and innate
immunity. Cell Death Differ. 22:225–236. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jacquet S, Nishino Y, Kumphune S, Sicard
P, Clark JE, Kobayashi KS, Flavell RA, Eickhoff J, Cotton M and
Marber MS: The role of RIP2 in p38 MAPK activation in the stressed
heart. J Biol Chem. 283:11964–1171. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ravi V, Jain A, Taneja A, Chatterjee K and
Sundaresan NR: Isolation and culture of neonatal murine primary
cardiomyocytes. Curr Protoc. 1:e1962021.PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Oh JK, Appleton CP, Hatle LK, Nishimura
RA, Seward JB and Tajik AJ: The noninvasive assessment of left
ventricular diastolic function with two-dimensional and Doppler
echocardiography. J Am Soc Echocardiogr. 10:246–270. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Glassock RJ, Warnock DG and Delanaye P:
The global burden of chronic kidney disease: Estimates, variability
and pitfalls. Nat Rev Nephrol. 13:104–114. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen TK, Knicely DH and Grams ME: Chronic
kidney disease diagnosis and management: A review. JAMA.
322:1294–1304. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Thomas R, Kanso A and Sedor JR: Chronic
kidney disease and its complications. Prim Care. 35:329–344. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
House AA, Wanner C, Sarnak MJ, Piña IL,
McIntyre CW, Komenda P, Kasiske BL, Deswal A, deFilippi CR, Cleland
JGF, et al: Heart failure in chronic kidney disease: Conclusions
from a kidney disease: Improving global outcomes (KDIGO)
controversies conference. Kidney Int. 95:1304–1317. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen C, Xie C, Wu H, Wu L, Zhu J, Mao H
and Xing C: Uraemic cardiomyopathy in different mouse models. Front
Med (Lausanne). 8:6905172021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cao XS, Chen J, Zou JZ, Zhong YH, Teng J,
Ji J, Chen ZW, Liu ZH, Shen B, Nie YX, et al: Association of
indoxyl sulfate with heart failure among patients on hemodialysis.
Clin J Am Soc Nephrol. 10:111–119. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yamaguchi K, Yisireyili M, Goto S, Cheng
XW, Nakayama T, Matsushita T, Niwa T, Murohara T and Takeshita K:
Indoxyl sulfate activates NLRP3 inflammasome to induce cardiac
contractile dysfunction accompanied by myocardial fibrosis and
hypertrophy. Cardiovasc Toxicol. 22:365–377. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fernandez-Prado R, Esteras R, Perez-Gomez
MV, Gracia-Iguacel C, Gonzalez-Parra E, Sanz AB, Ortiz A and
Sanchez-Niño MD: Nutrients turned into toxins: Microbiota
modulation of nutrient properties in chronic kidney disease.
Nutrients. 9:4892017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liabeuf S, Laville SM, Glorieux G,
Cheddani L, Brazier F, Beauport DT, Valholder R, Choukroun G and
Massy ZA: Difference in profiles of the gut-derived tryptophan
metabolite indole acetic acid between transplanted and
non-transplanted patients with chronic kidney disease. Int J Mol
Sci. 21:20312020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Claro LM, Moreno-Amaral AN, Gadotti AC,
Dolenga CJ, Nakao LS, Azevedo MLV, de Noronha L, Olandoski M, de
Moraes TP, Stinghen AEM and Pécoits-Filho R: The impact of uremic
toxicity induced inflammatory response on the cardiovascular burden
in chronic kidney disease. Toxins (Basel). 10:3842018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chinnappa S, Tu YK, Yeh YC, Glorieux G,
Vanholder R and Mooney A: Association between protein-bound uremic
toxins and asymptomatic cardiac dysfunction in patients with
chronic kidney disease. Toxins (Basel). 10:5202018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Stockler-Pinto MB, Soulage CO, Borges NA,
Cardozo LFM, Dolenga CJ, Nakao LS, Pecoits-Filho R, Fouque D and
Mafra D: From bench to the hemodialysis clinic: Protein-bound
uremic toxins modulate NF-κB/Nrf2 expression. Int Urol Nephrol.
50:347–354. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bataille S, Pelletier M, Sallée M, Berland
Y, McKay N, Duval A, Gentile S, Mouelhi Y, Brunet P and Burtey S:
Indole 3-acetic acid, indoxyl sulfate and paracresyl-sulfate do not
influence anemia parameters in hemodialysis patients. BMC Nephrol.
18:2512017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Addi T, Poitevin S, McKay N, El Mecherfi
KE, Kheroua O, Jourde-Chiche N, de Macedo A, Gondouin B, Cerini C,
Brunet P, et al: Mechanisms of tissue factor induction by the
uremic toxin indole-3 acetic acid through aryl hydrocarbon
receptor/nuclear factor-kappa B signaling pathway in human
endothelial cells. Arch Toxicol. 93:121–136. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hager THI: Assessment toxic effects of
exposure to 3-indoleacetic acid via hemato-biochemical, hormonal,
and histopathological screening in rats. Environ Sci Pollut Res
Int. 29:90703–90718. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang X, Wang Q, Burczynski FJ, Kong W and
Gong Y: Saikosaponin A of Bupleurum chinense (Chaihu) elevates bone
morphogenetic protein 4 (BMP-4) during hepatic stellate cell
activation. Phytomedicine. 20:1330–1335. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wu SJ, Tam KW, Tsai YH, Chang CC and Chao
JC: Curcumin and saikosaponin a inhibit chemical-induced liver
inflammation and fibrosis in rats. Am J Chin Med. 38:99–111. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhou F, Wang N, Yang L, Zhang LC, Meng LJ
and Xia YC: Saikosaponin A protects against dextran sulfate
sodium-induced colitis in mice. Int Immunopharmacol. 72:454–458.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Du ZA, Sun MN and Hu ZS: Saikosaponin a
Ameliorates LPS-induced acute lung injury in mice. Inflammation.
41:193–198. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang X and Yang G: Saikosaponin A
attenuates neural injury caused by ischemia/reperfusion. Transl
Neurosci. 11:227–235. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fu Y, Hu X, Cao Y, Zhang Z and Zhang N:
Saikosaponin a inhibits lipopolysaccharide-oxidative stress and
inflammation in Human umbilical vein endothelial cells via
preventing TLR4 translocation into lipid rafts. Free Radic Biol
Med. 89:777–785. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
He D, Wang H, Xu L, Wang X, Peng K, Wang
L, Liu P and Qu P: Saikosaponin-a attenuates oxidized LDL uptake
and prompts cholesterol efflux in THP-1 cells. J Cardiovasc
Pharmacol. 67:510–518. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang YJ, Wu SS, Chen XM, Pi JK, Cheng YF,
Zhang Y, Wang XJ, Luo D, Zhou JH, Xu JY, et al: Saikosaponin D
alleviates DOX-induced cardiac injury in vivo and in vitro. J
Cardiovasc Pharmacol. 79:558–567. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Huang M, Yan Y, Deng Z, Zhou L, She M,
Yang Y, Zhang M and Wang D: Saikosaponin A and D attenuate skeletal
muscle atrophy in chronic kidney disease by reducing oxidative
stress through activation of PI3K/AKT/Nrf2 pathway. Phytomedicine.
114:1547662023. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ozato K, Shin DM, Chang TH and Morse HC
III: TRIM family proteins and their emerging roles in innate
immunity. Nat Rev Immunol. 8:849–860. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Spirina LV, Yunusova NV, Kondakova IV and
Tarasenko NV: Transcription factors Brn-3α and TRIM16 in cancers,
association with hormone reception. Heliyon. 5:e020902019.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bell JL, Malyukova A, Holien JK, Koach J,
Parker MW, Kavallaris M, Marshall GM and Cheung BB: TRIM16 acts as
an E3 ubiquitin ligase and can Heterodimerize with other TRIM
family members. PLoS One. 7:e374702012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Jena KK, Mehto S, Kolapalli SP, Nath P,
Sahu R, Chauhan NR, Sahoo PK, Dhar K, Das SK and Chauhan S and
Chauhan S: TRIM16 governs the biogenesis and disposal of
stress-induced protein aggregates to evade cytotoxicity:
Implication for neurodegeneration and cancer. Autophagy.
15:924–926. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kim PY, Tan O, Liu B, Trahair T, Liu T,
Haber M, Norris MD, Marshall GM and Cheung BB: High TDP43
expression is required for TRIM16-induced inhibition of cancer cell
growth and correlated with good prognosis of neuroblastoma and
breast cancer patients. Cancer Lett. 374:315–323. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Chauhan S, Kumar S, Jain A, Ponpuak M,
Mudd MH, Kimura T, Choi SW, Peters R, Mandell M, Bruun JA, et al:
TRIMs and galectins globally cooperate and TRIM16 and galectin-3
co-direct autophagy in endomembrane damage homeostasis. Dev Cell.
39:13–27. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Di Rienzo M, Romagnoli A, Antonioli M,
Piacentini M and Fimia GM: TRIM proteins in autophagy: Selective
sensors in cell damage and innate immune responses. Cell Death
Differ. 27:887–902. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Borlepawar A, Rangrez AY, Bernt A,
Christen L, Sossalla S, Frank D and Frey N: TRIM24 protein promotes
and TRIM32 protein inhibits cardiomyocyte hypertrophy via
regulation of dysbindin protein levels. J Biol Chem.
292:10180–10196. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Chen L, Huang J, Ji Y, Zhang X, Wang P,
Deng K, Jiang X, Ma G and Li H: Tripartite motif 32 prevents
pathological cardiac hypertrophy. Clin Sci. 130:813–828. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhao Y, Liu H, Xi X, Chen S and Liu D:
TRIM16 protects human periodontal ligament stem cells from
oxidative stress-induced damage via activation of PICOT. Exp Cell
Res. 397:1123362020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Jena KK, Kolapalli SP, Mehto S, Nath P,
Das B, Sahoo PK, Ahad A, Syed GH, Raghav SK, Senapati S, et al:
TRIM16 controls assembly and degradation of protein aggregates by
modulating the p62-NRF2 axis and autophagy. EMBO J. 37:e983582018.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Andersson L, Täng MS, Lundqvist A, Lindbom
M, Mardani I, Fogelstrand P, Shahrouki P, Redfors B, Omerovic E,
Levin M, et al: Rip2 modifies VEGF-induced signalling and vascular
permeability in myocardial ischaemia. Cardiovasc Res. 107:478–486.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lin Z, Liao HH, Zhou ZY, Zhang N, Li WJ
and Tang QZ: RIP2 inhibition alleviates lipopolysaccharide-induced
septic cardiomyopathy via regulating TAK1 signaling. Eur J
Pharmacol. 947:1756792023. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Yang JJ, Zhang N, Zhou ZY, Ni J, Feng H,
Li WJ, Mou SQ, Wu HM, Deng W, Liao HH and Tang QZ:
Cardiomyocyte-specific RIP2 overexpression exacerbated pathologic
remodeling and contributed to spontaneous cardiac hypertrophy.
Front Cell Dev Biol. 9:6882382021. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yang S, Wang B, Humphries F, Jackson R,
Healy ME, Bergin R, Aviello G, Hall B, McNamara D, Darby T, et al:
Pellino3 ubiquitinates RIP2 and mediates Nod2-induced signaling and
protective effects in colitis. Nat Immunol. 14:927–936. 2013.
View Article : Google Scholar : PubMed/NCBI
|