Chronic kidney disease (CKD) poses a significant
challenge to healthcare systems, affecting an estimated 8–15% of
the global population (1,2). This condition is signified by the
gradual deterioration of kidney function over time, culminating in
end-stage renal disease, which requires treatment through dialysis
or kidney transplantation (3).
Fibrosis originates from kidney damage stemming from a range of
factors such as diabetes, hypertension, glomerular diseases,
toxins, infections and autoimmune conditions, resulting in
excessive accumulation of the extracellular matrix (ECM), which in
turn disrupts the normal tissue structure (4,5). At
present, no treatments have received approval to specifically
address the fibrotic process in an effort to prevent, arrest or
reverse CKD (6). The limited
efficacy observed in addressing kidney fibrosis underscores the
need for a comprehensive review of its foundational mechanisms and
the exploration of emerging therapeutic targets.
Transforming growth factor-β1 (TGF-β1) signaling is
intrinsically associated with the advancement of renal fibrosis
(RF) in CKD (5). Extensive animal
research underscores TGF-β1 as the key pathogenic driver,
propelling both glomerular and tubulointerstitial fibrosis
(5). TGF-β1 signaling is regulated
at several stages to ensure homeostasis (7). This encompasses TGF-β activation, the
creation, activation and breakdown of functional TGF-β receptor
complexes, the modulation of Smad both in activation and
degradation, and the assembly of Smad transcription complexes
(7). These complexes co-operate
with various DNA-binding transcription factors and coregulators at
gene regulatory sequences (8).
During these procedures, the hyperactivation of TGF-β1/Smad3
signaling may occur, which subsequently causes tubular dysfunction,
interstitial fibroblast proliferation, inflammation and augmented
ECM deposition (8). Reestablishing
the balance of TGF-β1 signaling offers a compelling antifibrotic
strategy to halt CKD progression (7). In this context, the present study
aimed to delve deeper into the complex functions of the TGF-β/Smad
pathway in detail, examining its influence on biological activities
such as renal tubular epithelial cell apoptosis, inflammation,
myofibroblast activation, cellular aging and its involvement in
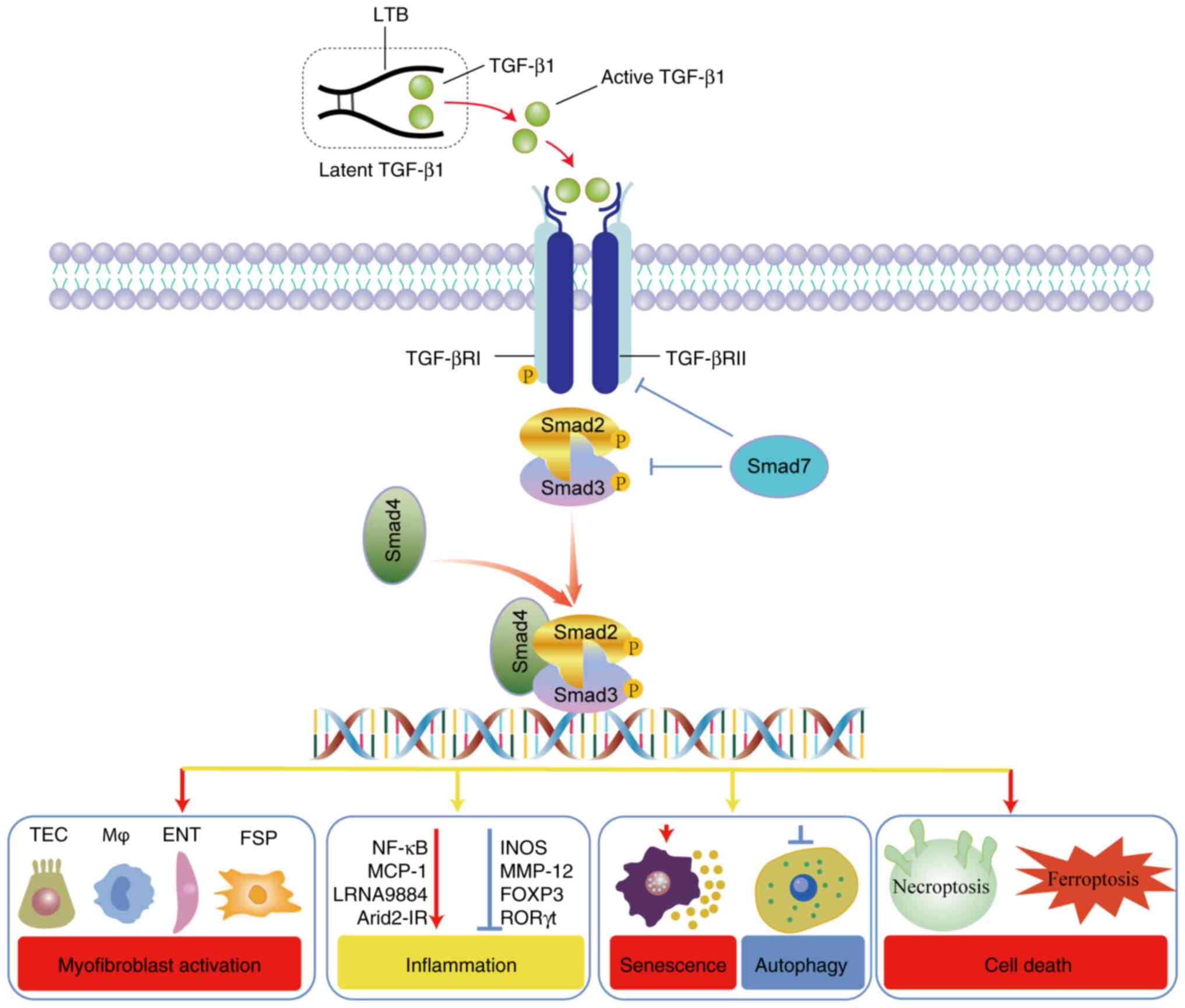
autophagy (Fig. 1). Of particular
emphasis in the present review, recent findings regarding the roles
of post-translational modifications (PTMs) including but not
limited to phosphorylation, ubiquitination, SUMOylation, and
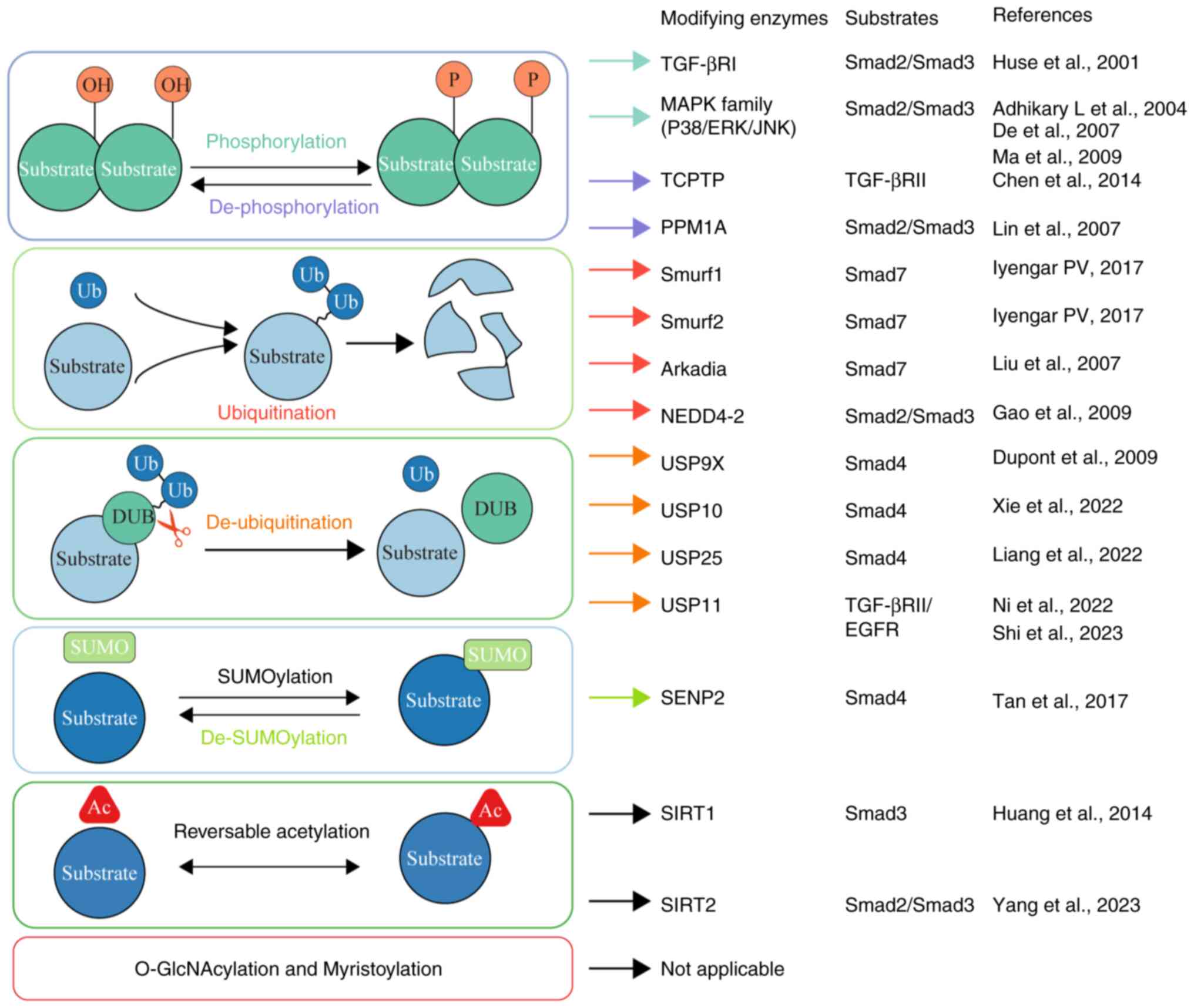
acetylation (Fig. 2) in
determining the strength and duration of TGF-β/Smad signaling were
comprehensively summarized.
TGF-β ligands comprise three distinct isoforms,
namely TGF-β1, TGF-β2 and TGF-β3 (9). These isoforms are ubiquitously
distributed across various cellular and tissue contexts, with
TGF-β1 being the most prevalent (10). TGF-β ligands initially emerge as
precursor proteins, which undergo a cleavage process at the
N-terminal region (11). The
cleavage process results in the formation of the latency-associated
peptide, which stays bound to the mature TGF-β homodimer at the
C-terminal (Fig. 1). This complex
association with the latent TGF-β-binding proteins ensures that
TGF-β remains inactive, commonly termed latent TGF-βs (12,13).
To become biologically active, these latent forms require the
intervention of certain environmental triggers such as specific
enzymes or an acidic milieu (5).
In the extracellular environment, associations with the ECM and
subsequent cleavage by various proteases, including plasmin and
specific matrix metalloproteinases, facilitate the liberation and
activation of the TGF-β ligands (11).
Upon engagement with the TGF-β1 ligand, the TGF-β
receptor (TGF-βR) II triggers the activation of TGF-βRI through
phosphorylation (5). This series
of activations subsequently culminates in the activation of Smad2/3
transcription factors through phosphorylation, commencing the
standard signaling process (5).
Integral to this cascade is Smad4, which associates with Smad2/3
after phosphorylation, directing the Smad2/3/4 complex towards the
nucleus (7) (Fig. 1). This migration to the nucleus is
a pivotal step for transcribing genes, which includes key genes
such as NADPH oxidase 4 (NOX4), connective tissue growth factor
(CTGF) and others [receptor interacting protein kinase 3 and
proto-oncogene tyrosine-protein kinase Src (Src)] involved in
tissue repair and cellular regulation (14–21).
Furthermore, Smad7, an inhibitory molecule, becomes active in
response to Smad3, and engages in competitive binding with TGF-βRI,
thereby hindering the phosphorylation process of Smad2 and Smad3
(22). The transcriptional
modulation orchestrated by Smad3-containing complexes is influenced
by an array of non-Smad co-activators, including the
transcriptional coactivator p300, activator protein 1 and
yes-associated protein, as well as co-suppressors such as SKI-like
proto-oncogene (SKI) and Ski-like oncoprotein N (SnoN) (23–26).
The transcriptional effects of TGF-β1 necessitate the involvement
of these accessory factors, driving structural alterations in
Smad2/3 to interact effectively with target motifs (8). In addition, TGF-β1 activates an array
of pathways independent of Smad signaling, which contribute to its
biological activities. These routes include TGF-β-activated kinase
1, phosphatidylinositol 3-kinase/Akt and Rho-like GTPase signaling
pathways (27,28).
Activation of myofibroblasts and the ensuing
accumulation of ECM are pivotal events in RF (29). The activated myofibroblast serves
as the key driver of RF, given its significant capacity to produce
the majority of the matrix (29).
While myofibroblasts are scarce under normal conditions, their
numbers surge in fibrotic kidneys (7,29).
Proposed precursors for myofibroblasts include pericytes, cells of
epithelial and endothelial origin, circulating cells derived from
bone marrow and local fibroblasts (30–35).
For epithelial cells, research has clarified the fibrosis-promoting
influence of TGF-β1, emphasizing the critical roles of key
molecules in the Smad signaling pathway, such as TGF-βRI, TGF-βRII
and Smad3, in epithelial-to-mesenchymal transition (EMT) (36–39).
Furthermore, a number of miRNAs and long non-coding RNAs (lncRNAs)
have been identified that are reliant on Smad3 function in
different capacities to regulate EMT (7,40–42).
For circulating bone marrow-derived cells, mounting evidence
indicates that macrophages originating from bone marrow can
directly transition into myofibroblasts (MMT) (43,44).
In fibrotic kidneys, the recruited Smad3-deficient macrophages do
not differentiate into myofibroblasts (44). Additionally, a series of factors
transcriptionally regulated by Smad3, such as Pou4f1, P2Y12 and
Src, have been proven to be involved in the MMT process (20,45–48).
This suggests that the progression of MMT is closely governed by
the TGF-β/Smad3 signaling pathway. For endothelium, in models of RF
triggered by either folic acid or unilateral ureteral obstruction,
the specific reduction of TGF-βRII levels in endothelial cells led
to a mitigation of fibrotic remodeling and an inhibition of
endothelial-to-mesenchymal transition (49). For fibroblasts, researchers
discovered that half of the myofibroblasts arise from the
proliferation of local resident fibroblasts, while an additional
35% originate from bone marrow fibroblasts (50). The TGF-β pathway appears
instrumental in this process, as evidenced by the fact that
specific deletion of TGF-βRII in α-smooth muscle actin (+) cells
leads to a marked decrease in fibroblast numbers (50). Furthermore, initiating the
conditional deletion of Smad2 in fibroblasts under the influence of
the fibroblast-specific protein 1 promoter, diminishes RF in
streptozotocin (STZ)-triggered diabetic nephropathy (DN) (51). In summary, the TGF-β/Smad pathway
plays a crucial role in guiding cellular dynamics and transitions
vital for RF.
Sterile inflammation, characterized by the
inflammatory response devoid of any infectious agents or specific
immunogens, serves as a key trigger for the development of RF
(52). TGF-β is instrumental in
the formation, balance, diversification and tolerance of immune
cells (53,54). Diminishment of TGF-β1 can result in
the hyperactivation of immunocytes and trigger the occurrence of
autoimmune diseases, a phenomenon noted in mice lacking either
TGF-β1 or its receptors. In such cases, excessive inflammatory
responses with massive lymphocyte and macrophage infiltration were
observed in many organs, primarily in the heart and lungs (55,56).
Consequently, broadly blocking upstream TGF-β signaling might
exacerbate renal inflammation.
Smad3 acts as a central regulator in renal
inflammation, uniquely modulating its dynamics by either inhibiting
or promoting the functions of macrophages and T cells (8). As a crucial effector molecule, Smad3
is involved in the TGF-β1-driven suppression of macrophage
activation, as demonstrated by its capability to inhibit the
regulatory actions of the inducible nitric oxide synthase and
matrix metalloproteinase-12 promoters within these cells (57). Furthermore, Smad3 has a critical
function in preserving the equilibrium between Treg and Th17 immune
responses. This is verified by the observation that a lack of Smad3
results in diminished forkhead box P3 induction, while
simultaneously enhancing Th17 cell generation by inhibition of
retinoid acid receptor-related orphan receptor γt transcriptional
activity within both controlled (in vitro) and living (in
vivo) contexts (58).
Conversely, Smad3 potentially facilitates macrophage recruitment
during renal inflammation through its chemotactic effects, as it
has been found to engage with macrophage chemotactic protein-1
(MCP-1), in turn amplifying the renal inflammation driven by
macrophages (59). Furthermore,
LRNA9884, a lncRNA that is transcriptionally regulated by Smad3 has
been shown to influence DN in db/db mice by stimulating the
synthesis of MCP-1, further amplifying renal inflammation (60). Additionally, in obstructed
nephropathy, AT-rich interaction domain 2 intronic transcript,
facilitated by TGF-β/Smad3, enhances the inflammation triggered by
IL-1β via the NF-κB pathway, without influencing the fibrosis
modulated by the TGF-β/Smad3 process (61). Notably, Smad7 serves as a negative
regulator of TGF-β/Smad signaling (62). Previous investigations have further
demonstrated that Smad7 functions as a key adjuster by stimulating
IκBα, a suppressor of NF-κB, consequently mitigating renal
inflammation (63,64). The observed phenomenon suggests
that a deficiency in Smad3 inhibits renal inflammation, which is
driven by NF-κB in the unilateral ureteral obstruction (UUO) model
(65). Presumably, the removal of
Smad3 inhibits the breakdown of Smad7 by E3 ubiquitin-protein
ligases, such as Smad ubiquitin regulatory factor (Smurf)1/Smurf2
(66). In summary, Smad3 has been
identified as a key controller in renal inflammation, orchestrating
various molecular interactions and pathways to either amplify or
mitigate inflammatory responses.
Cellular senescence describes the process wherein
cells lose their ability to replicate and permanently exit the cell
cycle after repeated duplications (67). These senescent cells resist
apoptosis and consistently release a diverse secretome, termed the
senescence-associated secretory phenotype, which includes
pro-inflammatory and pro-fibrotic mediators (67). In recent studies, cellular
senescence in renal tubular epithelial cells has been identified as
a primary contributor to the onset of RF, and consequently,
delaying this senescence presents an effective intervention to curb
RF and offers a crucial strategy for decelerating the progression
of CKD (67–69). A previous study indicates that the
TGF-β/Smad pathway promotes cellular senescence by reducing histone
4 lysine 20 tri-methylation through miR-29, impacting DNA repair
and genome stability (70).
Furthermore, a recent study has demonstrated that
ubiquitin-specific protease 11 (USP11) promotes cellular senescence
and fibrosis regulated by the Smad/P53 complex, by inhibiting the
ubiquitination of TGF-βRII (71).
In the context of the aging kidney, heightened stimulation of the
TGF-β/Smad3 pathway in podocyte-specific TGF-β overexpressing mice
leads to cell senescence through processes that involve
transference of p16 and initiation of p21 (72).
Autophagy, a preserved lysosomal pathway,
facilitates the degradation of cytoplasmic constituents (73). Yet, its role in kidney fibrosis,
whether protective or pathological, remains ambiguous (73). Moreover, the exact mechanisms and
signaling pathways that govern autophagy responses across various
kidney cell types and disease spectra require further elucidation
(73). Recent studies highlighted
the influence of Smad3 on autophagy and its prospective role as a
treatment focus for fibrotic diseases. A study has demonstrated
that Smad3 contributes to lysosomal depletion by inhibiting
transcription factor EB-mediated lysosome biogenesis, resulting in
impaired autophagy during the advancement of DN (74). Moreover, TGFβ, through an
epigenetic mechanism that involves the Smad3-mediated decrease of
histone acetyltransferase KAT8 (also termed as MYST1), activates
autophagy which promotes fibrotic diseases, including dermal and
pulmonary fibrosis, suggesting a potential therapeutic target
(75).
Preventing the death of renal tubular epithelial
cells is crucial in halting the progression of CKD (76). It is widely acknowledged that TGF-β
is known to facilitate cell death by the interruption of the cell
cycle at its G1 phase, orchestrated through the Smad pathway
(77–79). A previous study revealed that Smad3
contributes to acute kidney injury (AKI) by directly interacting
with cyclin-dependent kinase inhibitor proteins p21 and p27
(78). This interaction results in
the death of renal tubular epithelial cells (TECs) due to G1 phase
cell cycle arrest (79). Moreover,
a recent study has indicated that Smad3 can also transcriptionally
activate the receptor-interacting protein kinase 3/mixed lineage
kinase domain-like protein necroptosis pathway, subsequently
resulting in the death of TECs (21).
Ferroptosis, a regulated form of cell death induced
by oxidative stress and dependent on iron-mediated lipid
peroxidation, is intricately linked with numerous renal and
fibrotic diseases, including AKI, CKD and diabetic kidney diseases
(80,81). However, the precise mechanisms
driving RF through ferroptosis are yet to be fully understood.
Recently published studies demonstrated that Smad3 induces
ferroptosis in TECs, primarily through the modulation of NOX4 gene
transcription (18,82,83).
It also works in conjunction with activating transcription factor 3
(ATF3) to suppress the gene expression of solute carrier family 7
member 11 (SLC7A11), thereby modulating the ferroptosis process
(84). In light of these findings,
the present research group has made some interesting discoveries.
Preliminary investigations have revealed that active natural
compounds exhibit specificity towards key components involved in
the ferroptosis process. For example, tectorigenin has been found
to specifically target Smad3, regulating NOX4 and thereby
inhibiting the ferroptosis pathway, suggesting a potential
therapeutic role against RF (18).
Formononetin, on the other hand, suppresses the binding of the
SMAD3/ATF3 complex, promoting the expression of SLC7A11 and
highlighting its potential as a novel modulator for ferroptosis in
RF (84). Lastly, kaempferitrin
has demonstrated the ability to bind with NOX4, leading to an
improvement in tubular ferroptosis within the kidneys, proposing a
new approach to the treatment of RF through its impact on
ferroptosis (83). These promising
steps forward provide valuable insights into how natural compounds
can potentially be utilized in the modulation of ferroptosis and
treatment of RF.
Phosphorylation is a process occurring after protein
translation, characterized by the addition of a phosphate group to
certain amino acids in a protein, typically serine, threonine or
tyrosine residues (87). Normally,
Smad2 and Smad3, which are among the receptor-regulated Smads
(R-Smads), undergo activation via ligand-induced phosphorylation at
two serine residues within their carboxy-terminal SSXS motif,
mediated by TGF-βRI (88,89). Beyond the established TGF-β
signaling pathway involving Smad2 and Smad3, the TGF-β/Smad
signaling pathway is further influenced by multiple kinases,
providing further refinement, expansion or modulation of the
signaling output (90). The
mitogen-activated protein kinase (MAPK) family, comprising three
primary kinases including p38 MAPK, c-Jun N-terminal kinase (JNK)
and extracellular signal-regulated kinase (ERK), has been shown to
be activated in humans with both acute and CKD, as indicated by a
number of studies (91–93). In addition, through employing
pharmacological and genetic interventions, the blocking of p38
and/or JNK effectively mitigates RF across diverse animal studies
(94–96). Notably, each of the three MAPKs are
capable of phosphorylating specific sites within the linker regions
of both Smad2 and Smad3 (97).
These regions are situated in the interdomain space that separates
the mad homology domain 1 (MH1) and MH2 domains within a Smad
protein, subsequently altering the transcription of Smad3-dependent
genes (97). Hence, besides being
activated by TGF-β1, Smad3 activation can be triggered by various
stress-inducing agents such as angiotensin II (Ang II), advanced
end products (AGE) and C-reactive protein (CRP) through
MAPK-dependent pathways. Specifically, research reveals that Ang II
has the capability to directly stimulate Smad3, thereby promoting
the upregulation of both CTGF and collagen I via the AT1-ERK/p38
MAPK interaction route (98). In
addition, there is a strong link between AGEs and the expression of
CTGF, and EMT. Through the Smad3 pathway that functions
independently of TGF-β1, introducing AGEs induces CTGF expression
in TECs devoid of TGF-β1, evidenced by the swift activation of
Smad2/3, ERK1/2 and p38 (99).
Furthermore, CRP, reported as an inflammatory marker and mediator,
is known to regulate the fibrotic process, primarily by modulating
the CD32b-ERK/p38 MAPK pathway, subsequently activating Smad3
(100).
Protein dephosphorylation, mediated by protein
phosphatases, serves as a key control process that influences the
operation of a number of proteins within signal transduction routes
(87). Chen et al (101) discovered that loss of integrin
α1β1, a regulator for collagen synthesis, exacerbated RF in a UUO
model via a TGF-β/Smad-dependent manner. In terms of the mechanism,
integrin α1β1 promotes the recruitment of the phosphatase, T cell
protein tyrosine phosphatase (TCPTP) to TGF-βRII which results in
the dephosphorylation of tyrosine residues in the TGF-βRII
cytoplasmic tail, subsequently impairing TGF-βR-dependent fibrotic
signaling transmission (101). In
addition, protein phosphatase magnesium-dependent 1A (PPM1A)
facilitates the dephosphorylation of TGF-β-activated Smad2/3 within
their carboxy-terminal SSXS motif, subsequently promoting their
nuclear export (102). In
obstructive and aristolochic acid-induced nephropathy, a decrease
in PPM1A levels within the tubulointerstitium has been noted and
this diminution plays a role in enhancing Smad3 phosphorylation,
leading to subsequent RF (103).
In obstructive nephropathy, maxacalcitol, an analog of vitamin D,
enhances the function of the PPM1A/vitamin D receptor complex,
resulting in the dephosphorylation of Smad3, thereby reducing
tubulointerstitial fibrosis (104). Furthermore, PPM1A and PTEN
collaboratively work to diminish the phosphorylation of Smad3 and
the activation of genes associated with fibrosis (105).
In all organ tissues, intracellular proteins undergo
continuous turnover through degradation and synthesis (106). The ubiquitin-proteasome system
(UPS) primarily degrades intracellular proteins (106). This multi-enzyme process
covalently attaches ubiquitin to a substrate protein, which the
proteasome, a core proteolytic complex of the UPS, then recognizes
and degrades (106).
Deubiquitination, the counterpart to ubiquitination,
is a key cellular procedure encompassing the removal of ubiquitin
molecules that have been added to proteins (117,118). This dynamic interplay between
ubiquitination and deubiquitination ensures the precise regulation
of protein function, stability and interactions within the cell
(117). Deubiquitinating enzymes,
also known as deubiquitinases (DUBs), constitute a vast group of
proteases responsible for removing ubiquitin from proteins
(117). Moreover, they aid in the
creation of independent entities from freshly translated
polyubiquitins and repurpose ubiquitins after the breakdown of
polyubiquitinated protein substrates (119). While the control of TGF-β
signaling through ubiquitination has been widely studied in the
past decades, the function of deubiquitination steered by DUBs in
the TGF-β signaling pathway, especially in the context of CKD, has
only begun to gain attention recently.
PR-619, a comprehensive DUB inhibitor, mitigated
fibrosis in mice undergoing UUO and in rat kidney fibroblast cells,
namely NRK-49F cells, triggered by TGF-β1 (120). Furthermore, PR-619 demonstrates
an inhibitory effect on Smad4 levels, while it does not affect the
production of TGF-βR, Smad2 or Smad3 (120). This indicates that DUBs may
regulate fibrosis by modulating Smad4 (120). At present, members of the
ubiquitin specific proteases family (USPs), which have been
reported to regulate Smad4 deubiquitination, include USP9X
(121), USP10 (122), USP13 (123), USP17 (124) and USP25 (125). USPs are currently known as the
most extensive and predominant family of enzymes associated with
deubiquitination (117). In fact,
USP9X is documented to suppress fibrosis triggered by the
stimulation of AGEs in mesangial cells, as well as EMT in renal
tubular cells (126,127). USP10 has been reported to
counteract renal impairment caused by sepsis, primarily by reducing
apoptosis in TECs and mitigating oxidative stress (128). Furthermore, recent research has
confirmed that USP25 is instrumental in advancing hypertensive
renal disease (125). Knockout of
USP25 in mice has been shown to reduce kidney malfunctions and
fibrotic conditions (125). From
a mechanistic viewpoint, USP25 is associated with the regulation of
TGF-β signaling activation (125). Specifically, USP25 functions by
reducing Smad4 K63-linked polyubiquitination (125). For R-Smads and Smad7, although
some DUBs have been reported to regulate their ubiquitination
processes (129–133), their functions in CKD remain to
be further elucidated.
SUMOylation, a process where SUMO covalently
attaches to specific protein targets, plays a pivotal role in
modulating signal transduction through changes in subcellular
localization, influencing enzymatic activity and directing the
ubiquitin-mediated breakdown of its target substrates (137). This modification process is
driven by a series of enzymes requiring ATP, encompassing the E1
activator, the E2 conjugator known as Ubc9 and various E3 ligases
(138). Notably, the function of
SUMOylation in the TGF-β signaling pathway is attracting more and
more interest (139).
Within the scope of TGF-β signaling, the SUMOylation
process of TGF-βR1 amplifies its capability to interact with Smad3,
which promotes the phosphorylation of Smad3 (139). SENP2 counteracts this alteration,
and an upsurge in SENP2 expression curtails the EMT instigated by
TGF-β (143). The role of Smad4
SUMOylation in TGF-β transcription regulation remains contentious.
Some researchers posit a detrimental effect of Smad4 SUMOylation on
TGF-β signaling, highlighting that the K113R/K159R mutation
curtails the polyubiquitination of Smad4 (144). In the context of renal mesangial
cells under high glucose conditions, the SUMO2/3-driven SUMOylation
of Smad4 triggers the TGF-β/Smad pathway, subsequently elevating
fibronectin levels (145). These
contrasting perspectives might arise from distinct cellular
contexts (146). Conclusively,
adjusting the (de-)SUMOylation of the TGF-β/Smad pathway presents a
hopeful approach for CKD treatment.
Protein acetylation is recognized as a significant
and reversible post-translational modification, underscoring its
various cellular and physiological activities (147). Reversible acetylation is
orchestrated by two primary enzyme classes: Acetyltransferases
(KATs) and deacetylases (KDACs) (147). KATs enable the addition of acetyl
groups onto lysine residues and encompass the general control
non-derepressible 5, p300 and MYST families, along with other
unclassified KATs (147).
Although KATs primarily acetylate histones, enzymes such as p300
also influence the TGF-β/Smad pathway, and are also recognized for
enhancing TGF-β activity through the acetylation of Smad2 or Smad3
(148,149). Furthermore, the inhibition of
p300 with a novel FATp300 inhibitor, L002, mitigates RF caused by
hypertension and opposes fibrogenic responses in fibroblasts
(150).
Unlike KATs, KDACs are divided into two main
categories: Classical histone deacetylases, which are
Zn2+-dependent, and sirtuin deacetylases (SIRTs), which
rely on NAD+ (147).
In recent studies, seven mammalian counterparts of the yeast Sir2,
specifically labeled as SIRT1 to SIRT7, have been discerned
(151). Within the kidney, SIRT1
stands out as the predominant SIRTs under investigation. Primarily
localized in the nucleus, it orchestrates the acetylation patterns
of nucleosome histones and influences the dynamics of multiple
transcriptional regulators (151). As a result, SIRT1 plays a pivotal
role in cellular defense by mitigating processes such as apoptosis,
inflammation and fibrosis (151).
SIRT1 deficiency promotes Smad3 acetylation, subsequently
activating TGF-β signaling and exacerbating the progression of CKD
(152). Resveratrol intervention
facilitates the interaction between SIRT1 and Smad3, thereby
attenuating Smad3 acetylation (153). Moreover, elevating SIRT1 levels
in tubular cells impedes the transition from AKI to
tubulointerstitial fibrosis (151). This also curtails the subsequent
accumulation of matrix metalloproteinase-7 in the kidney through
the deacetylation of Smad4 (154). Hence, SIRT1 emerges as a
promising candidate for therapy in treating CKD (152). Unlike SIRT1, SIRT2 predominantly
resides in the cytoplasm and plays a role in hindering fibrosis
within renal tubules (155). The
specific removal of SIRT2 from TECs aggravates RF, while its
deliberate overexpression in these cells reduces RF (155). In terms of mechanism, SIRT2 forms
a direct association with Smad2 and Smad3, leading to their
deacetylation; this interaction subsequently mitigates the fibrotic
effects triggered by TGF-β (155). This highlights the therapeutic
potential of SIRT2 in addressing fibrosis. In the context of CKD,
there are limited reports on the regulatory roles of other SIRT
family members, specifically SIRT3-7, concerning the TGF-β/Smad
pathway. To sum up, the complex equilibrium and interaction between
these enzymes underscore their potential as therapeutic targets in
CKD and fibrotic conditions.
O-GlcNAcylation, a fluctuating and reversible type
of PTM, entails the addition of a β-D-N-acetylglucosamine (GlcNAc)
molecule onto serine or threonine residues within proteins
(156). This modification,
predominantly occurring within the cytoplasm and nucleus, is
distinct from the conventional N- and O-glycosylations that take
place in the endoplasmic reticulum and Golgi apparatus (156). The enzymes O-GlcNAc transferase
and O-GlcNAcase are key in controlling O-GlcNAcylation, which is
vital for numerous cellular functions such as signal transduction
and transcription, as well as maintaining protein stability
(156,157). Due to its significance, changes
in O-GlcNAcylation are linked to various diseases including
diabetes, neurodegenerative conditions and cancer (158–160).
A previous study has indicated that an increase in
O-GlcNAcylation levels in kidney tissues following UUO, and that
glucosamine-driven O-GlcNAcylation can promote fibrosis in the
renal parenchymal cell, HK2 TECs (161). Notably, a previous study observed
that Smad4 has been identified as a newly discovered protein
undergoing O-GlcNAc modification in human lung cancer cells. In
this context, O-GlcNAc hinders the linkage between Smad4 and
GSK-3β, interactions that are crucial for the proteasomal breakdown
of Smad4 (162). Nevertheless,
the precise function of O-GlcNAcylation in fibrosis, along with its
interplay with the TGF-β1/Smad3 signaling pathway remains to be
further elucidated.
Myristoylation, a post-translational modification
process, involves covalently linking myristate (a 14-carbon fatty
acid) to the N-terminal glycine residue of the protein via an amide
bond (163). This modification is
critical in numerous protein signaling systems as it imparts
various effects such as modulating protein stability, facilitating
protein-protein interactions and enhancing subcellular localization
to organelles or the plasma membrane (163).
There is relatively scant research focusing on the
role of myristoylation in the progression of CKD. Notably, a
previous study noted that myristoylated TGF-βRI and TGF-βRII can
induce transcriptional activation of Smad2, suggesting a potential
role for myristoylation in the activation of the TGF-β pathway
(164). In addition,
myristoylation may exert an indirect influence on the regulation of
the TGF-β signaling pathway. For instance, the myristoylation of
PPM1A could enhance the phosphatase activity of PPM1A, as promoted
by the cellular senescence-inhibited gene, thereby inhibiting TGF-β
signaling further (165). This
suggests that the modulation of myristoylation states could
represent a novel strategy for managing TGF-β-driven processes in
CKD.
However, despite these insights, there remain
several considerable limitations that hinder a comprehensive
understanding. A complete grasp of individual PTMs and how each
modification precisely regulates this process is still lacking,
necessitating further exploration into each PTM to clarify their
specific roles and impacts on overall signaling. In addition, the
intricate and complex interactions among various PTMs and their
collective influence on TGF-β/Smad signaling presents a challenging
aspect that is not yet fully understood due to several reasons.
First, the crosstalk between different PTMs can be complex and
context-dependent, making it difficult to predict the net effect on
Smad signaling (87). Second, many
PTMs can target multiple proteins within the pathway, further
complicating the overall picture (136,155,166). Finally, technological limitations
can hinder our ability to comprehensively analyze these
interactions and their dynamic regulation within cells (167). Despite these challenges,
unraveling these complexities holds immense promise for developing
more targeted therapeutic strategies for fibrotic diseases. More
research is needed to elucidate these dynamic relationships and
create a more holistic view of the signaling network. Furthermore,
despite advances in proteomics techniques, current approaches are
limited in their ability to uncover disease-specific regulations in
patient-derived samples (168).
The development and application of more sophisticated tools could
enable a finer resolution of these regulatory intricacies,
providing invaluable insight into disease progression and response
to treatment (168). Moreover,
the current landscape of therapeutic strategies lacks
personalization, with most approaches not considering the unique
progression patterns of CKD in different individuals (167). This represents a significant gap
in effectively managing and treating CKD. Lastly, inefficiencies in
existing drug delivery systems, particularly those targeting the
kidneys, pose another challenge by reducing therapeutic efficacy
and leading to potential side effects (169). Existing kidney-targeting drug
delivery systems face challenges related to nanoparticle size.
Small nanoparticles (<6-8 nm) can pass through the glomerular
filtration barrier but are quickly cleared by urine, limiting their
use for sustained drug delivery (170). Larger nanoparticles (350–400 nm)
may accumulate in the kidneys but struggle with bioavailability and
filtration (170). Additionally,
the protein corona on nanoparticles can reduce targeting
efficiency, complicating effective treatment (170).
Emerging research horizons include finding ways to
translate cellular insights into physiologically relevant disease
models, elucidating the interconnections between diverse PTMs,
broadening the spectrum of known post-translational regulators and
leveraging advanced proteomics techniques to decode previously
concealed, disease-specific regulations in patient-derived samples.
A more detailed and stage-specific understanding of TGF-β/Smad
signaling regulation could pave the way for personalized
therapeutic strategies tailored to individual CKD progressions,
thus enhancing CKD treatment outcomes and preserving renal
architecture and functionality in patients.
Not applicable.
This study received financial support from the following
sources: National Natural Science Foundation of China (grant no.
82104665); The Science and Technology Department of Sichuan
Province (grant nos. 2023NSFSC1763 and 2022YFS0621); and The
Innovation Team Project of the Affiliated Traditional Chinese
Medicine Hospital of Southwest Medical University (grant no.
2022-CXTD-03).
Not applicable.
The manuscript was initially drafted by JL and YZ.
It was then edited and revised by JK, HS, LW and ND. All authors
read and approved the final version of the manuscript. Data
authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Webster AC, Nagler EV, Morton RL and
Masson P: Chronic kidney disease. Lancet. 389:1238–1252. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
GBD Chronic Kidney Disease Collaboration,
. Global, regional, and national burden of chronic kidney disease,
1990-2017: A systematic analysis for the Global Burden of Disease
Study 2017. Lancet. 395:709–733. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen TK, Knicely DH and Grams ME: Chronic
kidney disease diagnosis and management: A review. JAMA.
322:1294–1304. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu Y: Cellular and molecular mechanisms
of renal fibrosis. Nat Rev Nephrol. 7:684–696. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Meng X, Nikolic-Paterson DJ and Lan HY:
TGF-β: The master regulator of fibrosis. Nat Rev Nephrol.
12:325–338. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ruiz-Ortega M, Rayego-Mateos S, Lamas S,
Ortiz A and Rodrigues-Diez RR: Targeting the progression of chronic
kidney disease. Nat Rev Nephrol. 16:269–288. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gu YY, Liu XS, Huang XR, Yu XQ and Lan HY:
TGF-β in renal fibrosis: Triumphs and challenges. Future Med Chem.
12:853–866. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu W, Wang X, Yu X and Lan HY: Smad3
Signatures in Renal Inflammation and Fibrosis. Int J Biol Sci.
18:2795–2806. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Meng XM, Tang PM, Li J and Lan HY:
TGF-β/Smad signaling in renal fibrosis. Front Physiol. 6:822015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yu L, Border WA, Huang Y and Noble NA:
TGF-beta isoforms in renal fibrogenesis. Kidney Int. 64:844–856.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Weiss A and Attisano L: The TGFbeta
superfamily signaling pathway. Wiley Interdiscip Rev Dev Biol.
2:47–63. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Annes JP, Munger JS and Rifkin DB: Making
sense of latent TGFbeta activation. J Cell Sci. 116:217–224. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Robertson IB, Horiguchi M, Zilberberg L,
Dabovic B, Hadjiolova K and Rifkin DB: Latent TGF-β-binding
proteins. Matrix Biol. 47:44–53. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Macconi D, Remuzzi G and Benigni A: Key
fibrogenic mediators: Old players. Renin-angiotensin system. Kidney
Int. Suppl (2011):4:58–64. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Loeffler I and Wolf G: Transforming growth
factor-β and the progression of renal disease. Nephrol Dial
Transplant. 29 (Suppl 1):i37–i45. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Samarakoon R, Overstreet JM and Higgins
PJ: TGF-β signaling in tissue fibrosis: Redox controls, target
genes and therapeutic opportunities. Cell Signal. 25:264–268. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Samarakoon R, Overstreet JM, Higgins SP
and Higgins PJ: TGF-β1 → SMAD/p53/USF2 → PAI-1 transcriptional axis
in ureteral obstruction-induced renal fibrosis. Cell Tissue Res.
347:117–128. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li J, Yang J, Zhu B, Fan J, Hu Q and Wang
L: Tectorigenin protects against unilateral ureteral obstruction by
inhibiting Smad3-mediated ferroptosis and fibrosis. Phytother Res.
36:475–487. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang Q, Gao L, Hu XW, Wang JN, Zhang Y,
Dong YH, Lan HY and Meng XM: Smad3-Targeted Therapy Protects
against Cisplatin-Induced AKI by attenuating programmed cell death
and inflammation via a NOX4-dependent mechanism. Kidney Dis
(Basel). 7:372–390. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tang PM, Zhou S, Li CJ, Liao J, Xiao J,
Wang QM, Lian GY, Li J, Huang XR, To KF, et al: The proto-oncogene
tyrosine protein kinase Src is essential for
macrophage-myofibroblast transition during renal scarring. Kidney
Int. 93:173–187. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liang L, Wang W, Chen J, Wu W, Huang XR,
Wei B, Zhong Y, Ma RCW, Yu X and Lan HY: SARS-CoV-2 N protein
induces acute kidney injury in diabetic mice via the
Smad3-Ripk3/MLKL necroptosis pathway. Signal Transduct Target Ther.
8:1472023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hayashi H, Abdollah S, Qiu Y, Cai J, Xu
YY, Grinnell BW, Richardson MA, Topper JN, Gimbrone MA Jr, Wrana JL
and Falb D: The MAD-related protein Smad7 associates with the
TGFbeta receptor and functions as an antagonist of TGFbeta
signaling. Cell. 89:1165–1173. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang Y, Feng XH and Derynck R: Smad3 and
Smad4 cooperate with c-Jun/c-Fos to mediate TGF-beta-induced
transcription. Nature. 394:909–913. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Samarakoon R, Dobberfuhl AD, Cooley C,
Overstreet JM, Patel S, Goldschmeding R, Meldrum KK and Higgins PJ:
Induction of renal fibrotic genes by TGF-β1 requires EGFR
activation, p53 and reactive oxygen species. Cell Signal.
25:2198–2209. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Samarakoon R, Higgins SP, Higgins CE and
Higgins PJ: TGF-beta1-induced plasminogen activator inhibitor-1
expression in vascular smooth muscle cells requires
pp60(c-src)/EGFR(Y845) and Rho/ROCK signaling. J Mol Cell Cardiol.
44:527–538. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Meng XM, Chung ACK and Lan HY: Role of the
TGF-β/BMP-7/Smad pathways in renal diseases. Clin Sci (Lond).
124:243–254. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang YE: Non-Smad signaling pathways of
the TGF-β Family. Cold Spring Harb Perspect Biol. 9:a0221292017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim SI and Choi ME: TGF-β-activated
kinase-1: New insights into the mechanism of TGF-β signaling and
kidney disease. Kidney Res Clin Pract. 31:94–105. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Humphreys BD: Mechanisms of Renal
Fibrosis. Annu Rev Physiol. 80:309–326. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lin SL, Kisseleva T, Brenner DA and
Duffield JS: Pericytes and perivascular fibroblasts are the primary
source of collagen-producing cells in obstructive fibrosis of the
kidney. Am J Pathol. 173:1617–1627. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nakamura J, Sato Y, Kitai Y, Wajima S,
Yamamoto S, Oguchi A, Yamada R, Kaneko K, Kondo M, Uchino E, et al:
Myofibroblasts acquire retinoic acid-producing ability during
fibroblast-to-myofibroblast transition following kidney injury.
Kidney Int. 95:526–539. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen YT, Chang FC, Wu CF, Chou YH, Hsu HL,
Chiang WC, Shen J, Chen YM, Wu KD, Tsai TJ, et al: Platelet-derived
growth factor receptor signaling activates pericyte-myofibroblast
transition in obstructive and post-ischemic kidney fibrosis. Kidney
Int. 80:1170–1181. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kramann R, Schneider RK, DiRocco DP,
Machado F, Fleig S, Bondzie PA, Henderson JM, Ebert BL and
Humphreys BD: Perivascular Gli1+ progenitors are key contributors
to injury-induced organ fibrosis. Cell Stem Cell. 16:51–66. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li J, Qu X and Bertram JF:
Endothelial-myofibroblast transition contributes to the early
development of diabetic renal interstitial fibrosis in
streptozotocin-induced diabetic mice. Am J Pathol. 175:1380–1388.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Meng X, Jin J and Lan HY: Driving role of
macrophages in transition from acute kidney injury to chronic
kidney disease. Chin Med J (Engl). 135:757–766. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zeisberg M and Kalluri R: The role of
epithelial-to-mesenchymal transition in renal fibrosis. J Mol Med
(Berl). 82:175–181. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kim KK, Sheppard D and Chapman HA: TGF-β1
signaling and tissue fibrosis. Cold Spring Harb Perspect Biol.
10:a0222932018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Meng XM, Huang XR, Chung AC, Qin W, Shao
X, Igarashi P, Ju W, Bottinger EP and Lan HY: Smad2 protects
against TGF-beta/Smad3-mediated renal fibrosis. J Am Soc Nephrol.
21:1477–1487. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sato M, Muragaki Y, Saika S, Roberts AB
and Ooshima A: Targeted disruption of TGF-beta1/Smad3 signaling
protects against renal tubulointerstitial fibrosis induced by
unilateral ureteral obstruction. J Clin Invest. 112:1486–1494.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zarjou A, Yang S, Abraham E, Agarwal A and
Liu G: Identification of a microRNA signature in renal fibrosis:
Role of miR-21. Am J Physiol Renal Physiol. 301:F793–F801. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhong X, Chung AC, Chen HY, Meng XM and
Lan HY: Smad3-mediated upregulation of miR-21 promotes renal
fibrosis. J Am Soc Nephrol. 22:1668–1681. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang B, Komers R, Carew R, Winbanks CE, Xu
B, Herman-Edelstein M, Koh P, Thomas M, Jandeleit-Dahm K,
Gregorevic P, et al: Suppression of microRNA-29 expression by
TGF-β1 promotes collagen expression and renal fibrosis. J Am Soc
Nephrol. 23:252–265. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Meng XM, Wang S, Huang XR, Yang C, Xiao J,
Zhang Y, To KF, Nikolic-Paterson DJ and Lan HY: Inflammatory
macrophages can transdifferentiate into myofibroblasts during renal
fibrosis. Cell Death Dis. 7:e24952016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang YY, Jiang H, Pan J, Huang XR, Wang
YC, Huang HF, To KF, Nikolic-Paterson DJ, Lan HY and Chen JH:
Macrophage-to-Myofibroblast transition contributes to interstitial
fibrosis in chronic renal allograft injury. J Am Soc Nephrol.
28:2053–2067. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wei J, Xu Z and Yan X: The role of the
macrophage-to-myofibroblast transition in renal fibrosis. Front
Immunol. 13:9343772022. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tang PM, Zhang YY, Xiao J, Tang PC, Chung
JY, Li J, Xue VW, Huang XR, Chong CC, Ng CF, et al: Neural
transcription factor Pou4f1 promotes renal fibrosis via
macrophage-myofibroblast transition. Proc Natl Acad Sci USA.
117:20741–20752. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang S, Meng XM, Ng YY, Ma FY, Zhou S,
Zhang Y, Yang C, Huang XR, Xiao J, Wang YY, et al: TGF-β/Smad3
signalling regulates the transition of bone marrow-derived
macrophages into myofibroblasts during tissue fibrosis. Oncotarget.
7:8809–8822. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen J, Tang Y, Zhong Y, Wei B, Huang XR,
Tang PM, Xu A and Lan HY: P2Y12 inhibitor clopidogrel inhibits
renal fibrosis by blocking macrophage-to-myofibroblast transition.
Mol Ther. 30:3017–3033. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Xavier S, Vasko R, Matsumoto K, Zullo JA,
Chen R, Maizel J, Chander PN and Goligorsky MS: Curtailing
endothelial TGF-β signaling is sufficient to reduce
endothelial-mesenchymal transition and fibrosis in CKD. J Am Soc
Nephrol. 26:817–829. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
LeBleu VS, Taduri G, O'Connell J, Teng Y,
Cooke VG, Woda C, Sugimoto H and Kalluri R: Origin and function of
myofibroblasts in kidney fibrosis. Nat Med. 19:1047–1053. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Loeffler I, Liebisch M, Allert S, Kunisch
E, Kinne RW and Wolf G: FSP1-specific SMAD2 knockout in renal
tubular, endothelial, and interstitial cells reduces fibrosis and
epithelial-to-mesenchymal transition in murine STZ-induced diabetic
nephropathy. Cell Tissue Res. 372:115–133. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lv W, Booz GW, Wang Y, Fan F and Roman RJ:
Inflammation and renal fibrosis: Recent developments on key
signaling molecules as potential therapeutic targets. Eur J
Pharmacol. 820:65–76. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Li MO and Flavell RA: TGF-beta: A master
of all T cell trades. Cell. 134:392–404. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Li MO, Wan YY, Sanjabi S, Robertson AKL
and Flavell RA: Transforming growth factor-beta regulation of
immune responses. Annu Rev Immunol. 24:99–146. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kulkarni AB, Huh CG, Becker D, Geiser A,
Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM and Karlsson S:
Transforming growth factor beta 1 null mutation in mice causes
excessive inflammatory response and early death. Proc Natl Acad Sci
USA. 90:770–774. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yaswen L, Kulkarni AB, Fredrickson T,
Mittleman B, Schiffman R, Payne S, Longenecker G, Mozes E and
Karlsson S: Autoimmune manifestations in the transforming growth
factor-beta 1 knockout mouse. Blood. 87:1439–1445. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Werner F, Jain MK, Feinberg MW, Sibinga
NE, Pellacani A, Wiesel P, Chin MT, Topper JN, Perrella MA and Lee
ME: Transforming growth factor-beta 1 inhibition of macrophage
activation is mediated via Smad3. J Biol Chem. 275:36653–36658.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Martinez GJ, Zhang Z, Chung Y, Reynolds
JM, Lin X, Jetten AM, Feng XH and Dong C: Smad3 differentially
regulates the induction of regulatory and inflammatory T cell
differentiation. J Biol Chem. 284:35283–35286. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zhang F, Tsai S, Kato K, Yamanouchi D,
Wang C, Rafii S, Liu B and Kent KC: Transforming growth factor-beta
promotes recruitment of bone marrow cells and bone marrow-derived
mesenchymal stem cells through stimulation of MCP-1 production in
vascular smooth muscle cells. J Biol Chem. 284:17564–17574. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhang YY, Tang PM, Tang PC, Xiao J, Huang
XR, Yu C, Ma RCW and Lan HY: LRNA9884, a Novel Smad3-Dependent long
noncoding rna, promotes diabetic kidney injury in db/db Mice via
Enhancing MCP-1-Dependent renal inflammation. Diabetes.
68:1485–1498. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zhou Q, Huang XR, Yu J, Yu X and Lan HY:
Long Noncoding RNA Arid2-IR Is a Novel Therapeutic Target for Renal
Inflammation. Mol Ther. 23:1034–1043. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wang W, Huang XR, Li AG, Liu F, Li JH,
Truong LD, Wang XJ and Lan HY: Signaling mechanism of TGF-beta1 in
prevention of renal inflammation: Role of Smad7. J Am Soc Nephrol.
16:1371–1383. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lan HY: Smad7 as a therapeutic agent for
chronic kidney diseases. Front Biosci. 13:4984–4992. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Chung AC, Huang XR, Zhou L, Heuchel R, Lai
KN and Lan HY: Disruption of the Smad7 gene promotes renal fibrosis
and inflammation in unilateral ureteral obstruction (UUO) in mice.
Nephrol Dial Transplant. 24:1443–1454. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
You YK, Wu WF, Huang XR, Li HD, Ren YP,
Zeng JC, Chen H and Lan HY: Deletion of Smad3 protects against
C-reactive protein-induced renal fibrosis and inflammation in
obstructive nephropathy. Int J Biol Sci. 17:3911–3922. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Liu Z, Huang XR, Chen HY, Fung E, Liu J
and Lan HY: Deletion of angiotensin-converting enzyme-2 promotes
hypertensive nephropathy by targeting Smad7 for ubiquitin
degradation. Hypertension. 70:822–830. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Wang Y, Wang Y, Yang M and Ma X:
Implication of cellular senescence in the progression of chronic
kidney disease and the treatment potencies. Biomed Pharmacother.
135:1111912021. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zhang JQ, Li YY, Zhang XY, Tian ZH, Liu C,
Wang ST and Zhang FR: Cellular senescence of renal tubular
epithelial cells in renal fibrosis. Front Endocrinol (Lausanne).
14:10856052023. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Li C, Shen Y, Huang L, Liu C and Wang J:
Senolytic therapy ameliorates renal fibrosis postacute kidney
injury by alleviating renal senescence. FASEB J.
35:e212292021.PubMed/NCBI
|
|
70
|
Lyu G, Guan Y, Zhang C, Zong L, Sun L,
Huang X, Huang L, Zhang L, Tian XL, Zhou Z and Tao W: TGF-β
signaling alters H4K20me3 status via miR-29 and contributes to
cellular senescence and cardiac aging. Nat Commun. 9:25602018.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Ni JY, Wang X, Xie HY, Yang NH, Li JY, Sun
XA, Guo HJ, Zhou L, Zhang W, Liu J and Lu LM: Deubiquitinating
enzyme USP11 promotes renal tubular cell senescence and fibrosis
via inhibiting the ubiquitin degradation of TGF-β receptor II. Acta
Pharmacol Sin. 44:584–595. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Ueda S, Tominaga T, Ochi A, Sakurai A,
Nishimura K, Shibata E, Wakino S, Tamaki M and Nagai K: TGF-β1 is
involved in senescence-related pathways in glomerular endothelial
cells via p16 translocation and p21 induction. Sci Rep.
11:216432021. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Tang C, Livingston MJ, Liu Z and Dong Z:
Autophagy in kidney homeostasis and disease. Nat Rev Nephrol.
16:489–508. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Yang C, Chen XC, Li ZH, Wu HL, Jing KP,
Huang XR, Ye L, Wei B, Lan HY and Liu HF: SMAD3 promotes autophagy
dysregulation by triggering lysosome depletion in tubular
epithelial cells in diabetic nephropathy. Autophagy. 17:2325–2344.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Zehender A, Li YN, Lin NY, Stefanica A,
Nüchel J, Chen CW, Hsu HH, Zhu H, Ding X, Huang J, et al: TGFβ
promotes fibrosis by MYST1-dependent epigenetic regulation of
autophagy. Nat Commun. 12:44042021. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Sanz AB, Sanchez-Niño MD, Ramos AM and
Ortiz A: Regulated cell death pathways in kidney disease. Nat Rev
Nephrol. 19:281–299. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Massagué J, Blain SW and Lo RS: TGFbeta
signaling in growth control, cancer, and heritable disorders. Cell.
103:295–309. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Wang W, Chen J, Hu D, Pan P, Liang L, Wu
W, Tang Y, Huang XR, Yu X, Wu J and Lan HY: SARS-CoV-2 N protein
induces acute kidney injury via Smad3-Dependent G1 cell cycle
arrest mechanism. Adv Sci (Weinh). 9:e21032482022. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Fu S, Tang Y, Huang XR, Feng M, Xu AP and
Lan HY: Smad7 protects against acute kidney injury by rescuing
tubular epithelial cells from the G1 cell cycle arrest. Clin Sci
(Lond). 131:1955–1969. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Zhang HY, Cheng M, Zhang L and Wang YP:
Ferroptosis and renal fibrosis: A new target for the future
(Review). Exp Ther Med. 25:132022. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Wang J, Wang Y, Liu Y, Cai X, Huang X, Fu
W, Wang L, Qiu L, Li J and Sun L: Ferroptosis, a new target for
treatment of renal injury and fibrosis in a 5/6 nephrectomy-induced
CKD rat model. Cell Death Discov. 8:1272022. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Wang JN, Yang Q, Yang C, Cai YT, Xing T,
Gao L, Wang F, Chen X, Liu XQ, He XY, et al: Smad3 promotes AKI
sensitivity in diabetic mice via interaction with p53 and induction
of NOX4-dependent ROS production. Redox Biol. 32:1014792020.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Li J, Yang J, Xian Q, Su H, Ni Y and Wang
L: Kaempferitrin attenuates unilateral ureteral obstruction-induced
renal inflammation and fibrosis in mice by inhibiting NOX4-mediated
tubular ferroptosis. Phytother Res. Mar 15–2024.(Epub ahead of
print). View Article : Google Scholar
|
|
84
|
Zhu B, Ni Y, Gong Y, Kang X, Guo H, Liu X,
Li J and Wang L: Formononetin ameliorates ferroptosis-associated
fibrosis in renal tubular epithelial cells and in mice with chronic
kidney disease by suppressing the Smad3/ATF3/SLC7A11 signaling.
Life Sci. 315:1213312023. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Streets A and Ong A: Post-translational
modifications of the polycystin proteins. Cell Signal.
72:1096442020. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Duan G and Walther D: The roles of
post-translational modifications in the context of protein
interaction networks. PLoS Comput Biol. 11:e10040492015. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Xu P, Liu J and Derynck R:
Post-translational regulation of TGF-β receptor and Smad signaling.
FEBS Lett. 586:1871–1884. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Huse M, Muir TW, Xu L, Chen YG, Kuriyan J
and Massagué J: The TGF beta receptor activation process: An
inhibitor- to substrate-binding switch. Mol Cell. 8:671–682. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Shi Y and Massagué J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Xu P, Lin X and Feng XH: Posttranslational
Regulation of Smads. Cold Spring Harb Perspect Biol. 8:a0220872016.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Adhikary L, Chow F, Nikolic-Paterson DJ,
Stambe C, Dowling J, Atkins RC and Tesch GH: Abnormal p38
mitogen-activated protein kinase signalling in human and
experimental diabetic nephropathy. Diabetologia. 47:1210–1222.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
De Borst MH, Prakash J, Melenhorst WB, van
den Heuvel MC, Kok RJ, Navis G and van Goor H: Glomerular and
tubular induction of the transcription factor c-Jun in human renal
disease. J Pathol. 213:219–228. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Ma FY, Sachchithananthan M, Flanc RS and
Nikolic-Paterson DJ: Mitogen activated protein kinases in renal
fibrosis. Front Biosci (Schol Ed). 1:171–187. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Stambe C, Atkins RC, Tesch GH, Masaki T,
Schreiner GF and Nikolic-Paterson DJ: The role of p38alpha
mitogen-activated protein kinase activation in renal fibrosis. J Am
Soc Nephrol. 15:370–379. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Ma FY, Flanc RS, Tesch GH, Bennett BL,
Friedman GC and Nikolic-Paterson DJ: Blockade of the c-Jun amino
terminal kinase prevents crescent formation and halts established
anti-GBM glomerulonephritis in the rat. Lab Invest. 89:470–484.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Müller R, Daniel C, Hugo C, Amann K,
Mielenz D, Endlich K, Braun T, van der Veen B, Heeringa P, Schett G
and Zwerina J: The mitogen-activated protein kinase p38α regulates
tubular damage in murine anti-glomerular basement membrane
nephritis. PLoS One. 8:e563162013. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Kamato D, Burch ML, Piva TJ, Rezaei HB,
Rostam MA, Xu S, Zheng W, Little PJ and Osman N: Transforming
growth factor-β signalling: Role and consequences of Smad linker
region phosphorylation. Cell Signal. 25:2017–2024. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Yang F, Chung ACK, Huang XR and Lan HY:
Angiotensin II induces connective tissue growth factor and collagen
I expression via transforming growth factor-beta-dependent and
-independent Smad pathways: The role of Smad3. Hypertension.
54:877–884. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Chung AC, Zhang H, Kong YZ, Tan JJ, Huang
XR, Kopp JB and Lan HY: Advanced glycation end-products induce
tubular CTGF via TGF-beta-independent Smad3 signaling. J Am Soc
Nephrol. 21:249–260. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
You YK, Huang XR, Chen HY, Lyu XF, Liu HF
and Lan HY: C-Reactive protein promotes diabetic kidney disease in
db/db Mice via the CD32b-Smad3-mTOR signaling pathway. Sci Rep.
6:267402016. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Chen X, Wang H, Liao HJ, Hu W, Gewin L,
Mernaugh G, Zhang S, Zhang ZY, Vega-Montoto L, Vanacore RM, et al:
Integrin-mediated type II TGF-β receptor tyrosine dephosphorylation
controls SMAD-dependent profibrotic signaling. J Clin Invest.
124:3295–3310. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Lin X, Duan X, Liang YY, Su Y, Wrighton
KH, Long J, Hu M, Davis CM, Wang J, Brunicardi F, et al: PPM1A
functions as a Smad phosphatase to terminate TGFbeta signaling.
Cell. 125:915–928. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Samarakoon R, Rehfuss A, Khakoo NS, Falke
LL, Dobberfuhl AD, Helo S, Overstreet JM, Goldschmeding R and
Higgins PJ: Loss of expression of protein phosphatase
magnesium-dependent 1A during kidney injury promotes fibrotic
maladaptive repair. FASEB J. 30:3308–3320. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Inoue K, Matsui I, Hamano T, Fujii N,
Shimomura A, Nakano C, Kusunoki Y, Takabatake Y, Hirata M,
Nishiyama A, et al: Maxacalcitol ameliorates tubulointerstitial
fibrosis in obstructed kidneys by recruiting PPM1A/VDR complex to
pSmad3. Lab Invest. 92:1686–1697. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Tang J, Goldschmeding R, Samarakoon R and
Higgins PJ: Protein phosphatase Mg2+/Mn2+ dependent-1A and PTEN
deregulation in renal fibrosis: Novel mechanisms and co-dependency
of expression. FASEB J. 34:2641–2656. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Meyer-Schwesinger C: The
ubiquitin-proteasome system in kidney physiology and disease. Nat
Rev Nephrol. 15:393–411. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Tan R, He W, Lin X, Kiss LP and Liu Y:
Smad ubiquitination regulatory factor-2 in the fibrotic kidney:
Regulation, target specificity, and functional implication. Am J
Physiol Renal Physiol. 294:F1076–F1083. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Iyengar PV: Regulation of ubiquitin
enzymes in the TGF-β Pathway. Int J Mol Sci. 18:8772017. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Bonni S, Wang HR, Causing CG, Kavsak P,
Stroschein SL, Luo K and Wrana JL: TGF-beta induces assembly of a
Smad2-Smurf2 ubiquitin ligase complex that targets SnoN for
degradation. Nat Cell Biol. 3:587–595. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Wang L, Zha H, Huang J and Shi L: Flavin
containing monooxygenase 2 regulates renal tubular cell fibrosis
and paracrine secretion via SMURF2 in AKI-CKD transformation. Int J
Mol Med. 52:1102023. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Liu FY, Li XZ, Peng YM, Liu H and Liu YH:
Arkadia-Smad7-mediated positive regulation of TGF-beta signaling in
a rat model of tubulointerstitial fibrosis. Am J Nephrol.
27:176–183. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Liu FY and Li XZ: The roles of Arkadia in
renal tubular epithelial to mesenchymal transition. Med Hypotheses.
67:1205–1207. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Wu W, Huang XR, You Y, Xue L, Wang XJ,
Meng X, Lin X, Shen J, Yu X, Lan HY and Chen H: Latent TGF-β1
protects against diabetic kidney disease via Arkadia/Smad7
signaling. Int J Biol Sci. 17:3583–3594. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Gao S, Alarcón C, Sapkota G, Rahman S,
Chen PY, Goerner N, Macias MJ, Erdjument-Bromage H, Tempst P and
Massagué J: Ubiquitin ligase Nedd4L targets activated Smad2/3 to
limit TGF-beta signaling. Mol Cell. 36:457–468. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Manning JA, Shah SS, Nikolic A, Henshall
TL, Khew-Goodall Y and Kumar S: The ubiquitin ligase NEDD4-2/NEDD4L
regulates both sodium homeostasis and fibrotic signaling to prevent
end-stage renal disease. Cell Death Dis. 12:3982021. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Henshall TL, Manning JA, Alfassy OS, Goel
P, Boase NA, Kawabe H and Kumar S: Deletion of Nedd4-2 results in
progressive kidney disease in mice. Cell Death Differ.
24:2150–2160. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Nijman SM, Luna-Vargas MP, Velds A,
Brummelkamp TR, Dirac AM, Sixma TK and Bernards R: A genomic and
functional inventory of deubiquitinating enzymes. Cell.
123:773–786. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Zhang J, Zhang X, Xie F, Zhang Z, van Dam
H, Zhang L and Zhou F: The regulation of TGF-β/SMAD signaling by
protein deubiquitination. Protein Cell. 5:503–517. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Komander D, Clague MJ and Urbé S: Breaking
the chains: structure and function of the deubiquitinases. Nat Rev
Mol Cell Biol. 10:550–563. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Soji K, Doi S, Nakashima A, Sasaki K, Doi
T and Masaki T: Deubiquitinase inhibitor PR-619 reduces Smad4
expression and suppresses renal fibrosis in mice with unilateral
ureteral obstruction. PLoS One. 13:e02024092018. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Dupont S, Mamidi A, Cordenonsi M,
Montagner M, Zacchigna L, Adorno M, Martello G, Stinchfield MJ,
Soligo S, Morsut L, et al: FAM/USP9×, a deubiquitinating enzyme
essential for TGFbeta signaling, controls Smad4 monoubiquitination.
Cell. 136:123–135. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Xie S, Xing Y, Shi W, Zhang M, Chen M,
Fang W, Liu S, Zhang T, Zeng X, Chen S, et al: Cardiac fibroblast
heat shock protein 47 aggravates cardiac fibrosis post myocardial
ischemia-reperfusion injury by encouraging ubiquitin specific
peptidase 10 dependent Smad4 deubiquitination. Acta Pharm Sin B.
12:4138–4153. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Liao X, Li Y, Liu J, Zhang Y, Tan J, Kass
DJ, Rojas M, Mallampalli RK, Zhao J and Zhao Y: Deubiquitinase
USP13 promotes extracellular matrix expression by stabilizing Smad4
in lung fibroblast cells. Transl Res. 223:15–24. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Song C, Liu W and Li J: USP17 is
upregulated in osteosarcoma and promotes cell proliferation,
metastasis, and epithelial-mesenchymal transition through
stabilizing SMAD4. Tumour Biol. 39:10104283177171382017. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Zhao Y, Chen X, Lin Y, Li Z, Su X, Fan S,
Chen Y, Wang X and Liang G: USP25 inhibits renal fibrosis by
regulating TGFβ-SMAD signaling pathway in Ang II-induced
hypertensive mice. Biochim Biophys Acta Mol Basis Dis.
1869:1667132023. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Sun XH, Xiao HM, Zhang M, Lin ZY, Yang Y,
Chen R, Liu PQ, Huang KP and Huang HQ: USP9X deubiquitinates
connexin43 to prevent high glucose-induced
epithelial-to-mesenchymal transition in NRK-52E cells. Biochem
Pharmacol. 188:1145622021. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Huang K and Zhao X: USP9X prevents
AGEs-induced upregulation of FN and TGF-β1 through activating
Nrf2-ARE pathway in rat glomerular mesangial cells. Exp Cell Res.
393:1121002020. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Gao F, Qian M, Liu G, Ao W, Dai D and Yin
C: USP10 alleviates sepsis-induced acute kidney injury by
regulating Sirt6-mediated Nrf2/ARE signaling pathway. J Inflamm
(Lond). 18:252021. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Huang Z, Shen S, Wang M, Li W, Wu G, Huang
W, Luo W and Liang G: Mouse endothelial OTUD1 promotes angiotensin
II-induced vascular remodeling by deubiquitinating SMAD3. EMBO Rep.
24:e561352023. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Huang YT, Cheng AC, Tang HC, Huang GC, Cai
L, Lin TH, Wu KJ, Tseng PH, Wang GG and Chen WY: USP7 facilitates
SMAD3 autoregulation to repress cancer progression in p53-deficient
lung cancer. Cell Death Dis. 12:8802021. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Wicks SJ, Haros K, Maillard M, Song L,
Cohen RE, Dijke PT and Chantry A: The deubiquitinating enzyme UCH37
interacts with Smads and regulates TGF-beta signalling. Oncogene.
24:8080–8084. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Tian Y, Liao F and Wu G, Chang D, Yang Y,
Dong X, Zhang Z, Zhang Y and Wu G: Ubiquitination and regulation of
Smad7 in the TGF-β1/Smad signaling of aristolochic acid
nephropathy. Toxicol Mech Methods. 25:645–652. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Zhao Y, Thornton AM, Kinney MC, Ma CA,
Spinner JJ, Fuss IJ, Shevach EM and Jain A: The Deubiquitinase CYLD
targets Smad7 protein to regulate transforming growth factor β
(TGF-β) signaling and the development of regulatory T cells. J Biol
Chem. 286:40520–40530. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Wang B, Xu X, Yang Z, Zhang L and Liu Y,
Ma A, Xu G, Tang M, Jing T, Wu L and Liu Y: POH1 contributes to
hyperactivation of TGF-β signaling and facilitates hepatocellular
carcinoma metastasis through deubiquitinating TGF-β receptors and
caveolin-1. EBioMedicine. 41:320–332. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Shi Y, Tao M, Chen H, Ma X, Wang Y, Hu Y,
Zhou X, Li J, Cui B, Qiu A, et al: Ubiquitin-specific protease 11
promotes partial epithelial-to-mesenchymal transition by
deubiquitinating the epidermal growth factor receptor during kidney
fibrosis. Kidney Int. 103:544–564. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Jacko AM, Nan L, Li S, Tan J, Zhao J, Kass
DJ and Zhao Y: De-ubiquitinating enzyme, USP11, promotes
transforming growth factor β-1 signaling through stabilization of
transforming growth factor β receptor II. Cell Death Dis.
7:e24742016. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Du C, Chen X, Su Q, Lu W, Wang Q, Yuan H,
Zhang Z, Wang X, Wu H and Qi Y: The function of SUMOylation and Its
critical roles in cardiovascular diseases and potential clinical
implications. Int J Mol Sci. 22:106182021. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Wang X, Liu T, Huang Y, Dai Y and Lin H:
Regulation of transforming growth factor-beta signalling by
SUMOylation and its role in fibrosis. Open Biol. 11:2100432021.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Kang JS, Saunier EF, Akhurst RJ and
Derynck R: The type I TGF-beta receptor is covalently modified and
regulated by sumoylation. Nat Cell Biol. 10:654–664. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Enserink JM: Sumo and the cellular stress
response. Cell Div. 10:42015. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Reverter D and Lima CD: A basis for SUMO
protease specificity provided by analysis of human Senp2 and a
Senp2-SUMO complex. Structure. 12:1519–1531. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Gong L and Yeh ETH: Characterization of a
family of nucleolar SUMO-specific proteases with preference for
SUMO-2 or SUMO-3. J Biol Chem. 281:15869–15877. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Tan M, Zhang D, Zhang E, Xu D, Liu Z, Qiu
J, Fan Y and Shen B: SENP2 suppresses epithelial-mesenchymal
transition of bladder cancer cells through deSUMOylation of
TGF-βRI. Mol Carcinog. 56:2332–2341. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Long J, Wang G, He D and Liu F: Repression
of Smad4 transcriptional activity by SUMO modification. Biochem J.
379((Pt 1)): 23–29. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Zhou X, Gao C, Huang W, Yang M, Chen G,
Jiang L, Gou F, Feng H, Ai N and Xu Y: High glucose induces
sumoylation of Smad4 via SUMO2/3 in mesangial cells. Biomed Res
Int. 2014:7826252014. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Liu P, Zhang J, Wang Y, Wang C, Qiu X and
Chen DQ: Natural products against renal fibrosis via modulation of
SUMOylation. Front Pharmacol. 13:8008102022. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Liu W, Yuan Q, Cao S, Wang G, Liu X, Xia
Y, Bian Y, Xu F and Chen Y: Review: Acetylation mechanisms and
targeted therapies in cardiac fibrosis. Pharmacol Res.
193:1068152023. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Bugyei-Twum A, Advani A, Advani SL, Zhang
Y, Thai K, Kelly DJ and Connelly KA: High glucose induces Smad
activation via the transcriptional coregulator p300 and contributes
to cardiac fibrosis and hypertrophy. Cardiovasc Diabetol.
13:892014. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Inoue Y, Itoh Y, Abe K, Okamoto T, Daitoku
H, Fukamizu A, Onozaki K and Hayashi H: Smad3 is acetylated by
p300/CBP to regulate its transactivation activity. Oncogene.
26:500–508. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Rai R, Verma SK, Kim D, Ramirez V, Lux E,
Li C, Sahoo S, Wilsbacher LD, Vaughan DE, Quaggin SE and Ghosh AK:
A novel acetyltransferase p300 inhibitor ameliorates
hypertension-associated cardio-renal fibrosis. Epigenetics.
12:1004–1013. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Morigi M, Perico L and Benigni A: Sirtuins
in renal health and disease. J Am Soc Nephrol. 29:1799–1809. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Huang XZ, Wen D, Zhang M, Xie Q, Ma L,
Guan Y, Ren Y, Chen J and Hao CM: Sirt1 activation ameliorates
renal fibrosis by inhibiting the TGF-β/Smad3 pathway. J Cell
Biochem. 115:996–1005. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Chen Q, Zeng Y, Yang X, Wu Y, Zhang S,
Huang S, Zhong Y and Chen M: Resveratrol ameliorates myocardial
fibrosis by regulating Sirt1/Smad3 deacetylation pathway in rat
model with dilated cardiomyopathy. BMC Cardiovasc Disord.
22:172022. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Simic P, Williams EO, Bell EL, Gong JJ,
Bonkowski M and Guarente L: SIRT1 suppresses the
epithelial-to-mesenchymal transition in cancer metastasis and organ
fibrosis. Cell Rep. 3:1175–1186. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Yang S, Yang G, Wang X, Xiang J, Kang L
and Liang Z: SIRT2 alleviated renal fibrosis by deacetylating SMAD2
and SMAD3 in renal tubular epithelial cells. Cell Death Dis.
14:6462023. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Ma J and Hart GW: O-GlcNAc profiling: From
proteins to proteomes. Clin Proteomics. 11:82014. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Harosh-Davidovich SB and Khalaila I:
O-GlcNAcylation affects β-catenin and E-cadherin expression, cell
motility and tumorigenicity of colorectal cancer. Exp Cell Res.
364:42–49. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
He XF, Hu X, Wen GJ, Wang Z and Lin WJ:
O-GlcNAcylation in cancer development and immunotherapy. Cancer
Lett. 566:2162582023. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Ma J and Hart GW: Protein O-GlcNAcylation
in diabetes and diabetic complications. Expert Rev Proteomics.
10:365–380. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Park J, Lai MKP, Arumugam TV and Jo DG:
O-GlcNAcylation as a therapeutic target for Alzheimer's disease.
Neuromolecular Med. 22:171–193. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Feng D, Sheng-Dong L, Tong W and Zhen-Xian
D: O-GlcNAcylation of RAF1 increases its stabilization and induces
the renal fibrosis. Biochim Biophys Acta Mol Basis Dis.
1866:1655562020. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Kim YJ, Kang MJ, Kim E, Kweon TH, Park YS,
Ji S, Yang WH, Yi EC and Cho JW: O-GlcNAc stabilizes SMAD4 by
inhibiting GSK-3β-mediated proteasomal degradation. Sci Rep.
10:199082020. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Yuan M, Song ZH, Ying MD, Zhu H, He QJ,
Yang B and Cao J: N-myristoylation: From cell biology to
translational medicine. Acta Pharmacol Sin. 41:1005–1015. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Stockwell BR and Schreiber SL:
TGF-beta-signaling with small molecule FKBP12 antagonists that bind
myristoylated FKBP12-TGF-beta type I receptor fusion proteins. Chem
Biol. 5:385–395. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Zhu F, Xie N, Jiang Z, Li G, Ma L and Tong
T: The cellular senescence-inhibited gene is essential for PPM1A
myristoylation to modulate transforming growth factor β signaling.
Mol Cell Biol. 38:e00414–18. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Al-Salihi MA, Herhaus L, Macartney T and
Sapkota GP: USP11 augments TGFβ signalling by deubiquitylating
ALK5. Open Biol. 2:1200632012. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Siwy J, Mischak H and Zürbig P: Proteomics
and personalized medicine: A focus on kidney disease. Expert Rev
Proteomics. 16:773–782. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Giudice G and Petsalaki E: Proteomics and
phosphoproteomics in precision medicine: Applications and
challenges. Brief Bioinform. 20:767–777. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Xu H, Wu T and Huang L: Therapeutic and
delivery strategies of phytoconstituents for renal fibrosis. Adv
Drug Deliv Rev. 177:1139112021. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Trac N, Ashraf A, Giblin J, Prakash S,
Mitragotri S and Chung EJ: Spotlight on genetic kidney diseases: A
call for drug delivery and nanomedicine solutions. ACS Nano.
17:6165–6177. 2023. View Article : Google Scholar : PubMed/NCBI
|