Introduction
A dental follicle (DF) is a connective tissue sac
that forms around nonerupting teeth during early tooth eruption,
which originates from the ectoderm of the cranial neural crest and
differentiates from cranial neural crest cells (1,2). DFs
can be the source of periodontal tissues, dominated by cementum,
alveolar bone and periodontal membrane, during tooth development
(3). In addition, DFs regulate
bone remodeling by affecting the activity of osteoblasts and
osteoclasts during tooth eruption (4,5).
Therefore, DFs serve an indispensable role in the tooth eruption
process. With an enhanced understanding of the DF, researchers have
increasingly focused on mesenchymal stem cells (MSCs) located
within the DF. MSCS associated with teeth, which have the ability
to differentiate, were first isolated from the DFs of human third
molars (6). The dental MSCs
(DMSCs) found in DFs exhibit similarities to other human MSCs and
are specifically referred to as DF progenitor/stem cells (DFPCs)
(7). DFPCs can differentiate into
cementocytes, osteoblasts, fibroblasts, adipocytes, chondrocytes,
neuron-like cells and periodontal ligament cells (5,8–10).
Previous research has demonstrated that proper differentiation and
functioning of DFPCs are crucial for the normal progression of
tooth eruption (11).
Tooth eruption refers to the migration of a tooth
from its developmental site within the jaw to its functional
position in the oral cavity, thus leading to occlusal contact with
the contralateral tooth (12).
Recent research has identified five stages of tooth eruption:
Pre-eruptive movement, intraosseous eruption, mucosal penetration,
pre-occlusal eruption and post-occlusal eruption (13). The resorption of alveolar bone at
the apex establishes an eruptive pathway, whereas alveolar bone
formation at the root facilitates tooth movement within the jaw
(14). Notably, the DF is involved
in the tooth eruption process. Studies have shown that different
parts of the DF have different functions; specifically, the crown
region is responsible for absorbing alveolar bone, whereas the root
region regulates alveolar bone formation (15,16).
Therefore, a normal DF is an essential factor for tooth
eruption.
Failed tooth eruptions include two conditions:
Delayed eruption and complete failed eruption (14,17).
Delayed eruption is defined as a tooth that deviates from the
average eruption time by >2 standard deviations (18). Complete failed eruption can be
categorized as primary retention, secondary retention or impaction
(19). Primary retention refers to
a tooth remaining embedded in the jaw without emerging into the
oral cavity (17), whereas
secondary retention occurs when teeth erupt but fail to establish
occlusion (20). Impaction is the
result of physical obstacles that exist in the path of eruption;
this barrier constitutes an independent factor, separate from the
eruption process itself (19).
Failed eruption of teeth is commonly attributed to
physical obstructions or disorders in the tooth eruption mechanism
itself (17,19,21).
The process of tooth eruption involves a complex array of
regulatory mechanisms and signaling pathways. DF cells (DFCs) serve
a crucial role in regulating signal transduction between
osteoblasts and osteoclasts, thereby governing alveolar bone
resorption and formation (5,22).
Additionally, a number of syndromes and systemic diseases have been
identified as causative factors for tooth eruption disorders
(23). Most of these diseases are
caused by genetic factors, with certain syndromes also contributing
to tooth eruption failure due to DF abnormalities.
A previous study on DFCs primarily focused on their
role in normal tooth eruption, with limited research concentrating
on abnormal eruptions (24). The
role of tooth eruption-related signaling pathways, such as Wnt and
TGF-β signaling pathways, in normal tooth eruption has been
extensively studied; however, there are few reviews on the
mechanisms underlying abnormal signaling pathways in DFCs that
result in failed tooth eruption (25,26).
Furthermore, numerous studies have summarized the abnormal tooth
eruption observed in various genetic diseases and systemic syndrome
(23,25,27–29);
however, the pathogenesis of these diseases involves multiple
causes, with some specifically implicating the DF in the aberrant
tooth eruption process. Notably, to the best of our knowledge,
there is currently no comprehensive summary available on diseases
in which the DF plays a role in pathogenesis. Therefore, the
present review aims to elucidate the signaling pathways associated
with abnormal DFCs during failed tooth eruption and to explore
their molecular impact on eruption mechanisms. Additionally, this
review aims to investigate known genetic diseases and syndromes
linked to abnormal tooth eruption, thus providing an overview of
their clinical manifestations and underlying causes while
emphasizing those involving DFCs. The atypical characteristics of
DFCs and factors contributing to tooth eruption failure within
these disease contexts are also highlighted.
Failure of tooth eruption caused by abnormal
signal transduction in DFCs
In recent years, significant insights have been
gained into the intricate mechanisms underlying tooth eruption. The
complex molecular interactions between cells involved in tooth
eruption and DFCs are widely acknowledged. To effectively treat
dental diseases characterized by abnormal tooth eruption, it is
imperative to understand the fundamental molecular mechanisms
within DFCs.
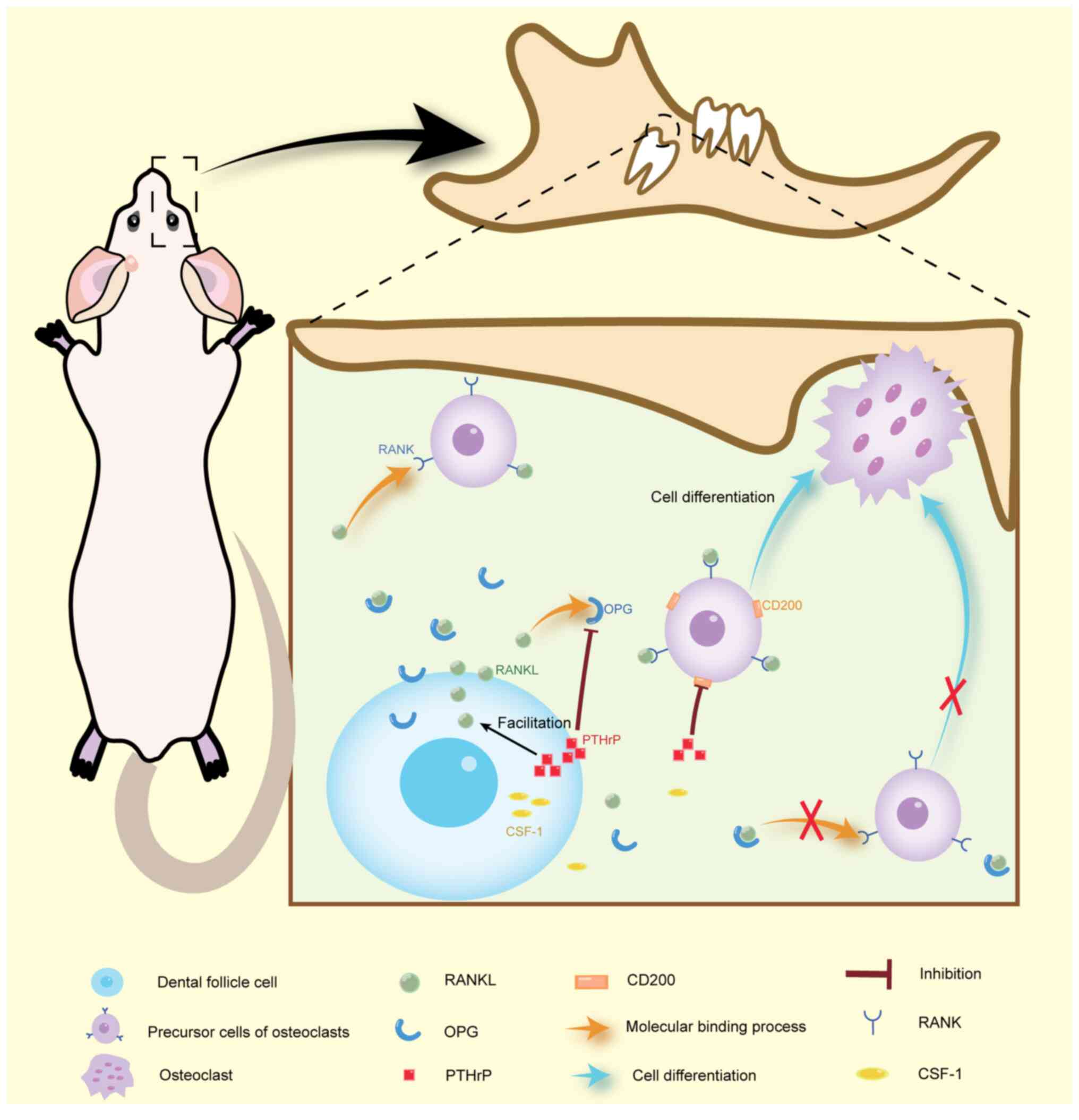
Abnormal parathyroid hormone
(PTH)-related protein (PTHrP)-PTHrP receptor (PPR) signaling
pathway activity in DFCs
PTHrP functions as a local autocrine/paracrine
factor capable of regulating cellular proliferation and
differentiation. It exerts regulatory control over
epithelial-mesenchymal interactions during organ development,
including those of the skin, hair follicles, mammary glands,
pancreas and developing teeth (30–34).
PTHrP has been reported to be highly expressed in DFCs and as a key
molecule necessary for tooth eruption (35). DFCs regulate both bone resorption
and formation around teeth, thereby promoting tooth eruption.
During this process, PTHrP is instrumental in promoting bone
resorption while inhibiting the osteogenesis of DFCs. Research has
demonstrated that DFCs, when treated with PTHrP and co-cultured,
display reduced expression of osteogenic-related genes, including
alkaline phosphatase (ALP), Runt-related gene 2 (RUNX2), bone
sialoprotein (BSP) and osteopontin (OPN) (36). Additionally, the Wnt/β-catenin
pathway serves as a key signaling pathway for tooth morphogenesis
(37), and its activation promotes
the stabilization and nuclear translocation of β-catenin (38). PTHrP inhibits the osteogenic
differentiation of co-cultured DFCs by suppressing activation of
the classical Wnt/β-catenin pathway, primarily through its impact
on phosphorylated (p)-GSK-3β. P-GSK-3β reduces the phosphorylation
of β-catenin, subsequently inducing nuclear translocation of
β-catenin (39). In PTHrP-treated
DFCs, the expression of p-GSK-3β has been shown to be reduced
(36).
PTHrP is also involved in regulating tooth root
development. Cementum covers the surface of the mineralized tissue
of the root, and its formation is crucial for root development.
Cementoblasts express the PTH/PTHrP receptor. PTHrP stimulation can
inhibit the expression of BSP and osteocalcin (OCN) in
cementoblasts in vitro, thereby blocking
cementoblast-mediated mineralization (40). In a previous study, after the
knockout of PTHrP in tooth tissues, including DFs, the surviving
mice exhibited tooth eruption failure and abnormal root formation
(41). By contrast, the injection
of PTHrP can accelerate tooth eruption and inhibit the osteogenesis
of DFCs (36). Additionally, PTHrP
signaling in the DFC may regulate osteoclast differentiation by
influencing the colony-stimulating factor 1 (CSF-1)-receptor
activator of NF-κB (RANK)-RANK ligand (RANKL)-osteoprotegerin (OPG)
pathway (11,42,43),
which is predominantly expressed by DFCs (35). CSF-1 and RANKL stimulate osteoclast
formation, whereas OPG inhibits it by competing with RANKL for
binding, thereby blocking its activity (44,45).
Osteoclasts serve a crucial role in alveolar bone resorption, thus
facilitating tooth eruption. PTHrP can promote bone resorption to
create a pathway for tooth eruption, whereas the expression of
RANKL and OPG serve as a key determinant of osteoclast activity
around the teeth (42). Studies
have demonstrated that PTHrP treatment increases the expression of
osteoclastogenic factors in DFC. Specifically, PTHrP has been shown
to elevate the expression of RANKL and reduce the expression of
OPG, thereby increasing the RANKL/OPG ratio in DFCs (36); this increased ratio promotes
osteoclast differentiation, thus accelerating the process of tooth
eruption. Previous studies have constructed RANKL-null mouse models
that exhibit impaired tooth eruption, thus suggesting that RANKL
plays an integral role in tooth eruption (46). PTHrP can also reduce
osteoclastogenesis through the downregulation of CD200, which is
closely related to RANKL (11,47).
Therefore, abnormal PTHrP expression in DFCs may be closely
associated with tooth eruption failure (Fig. 1).
PTH receptor-1 (PTH1R), which is also known as the
PTH/PPR (35), is a class B G
protein-coupled receptor composed of seven transmembrane helices
that is abundantly expressed in DFCs (48). PTH1R can interact with both PTH and
PTHrP (49,50). PPR can regulate the differentiation
of cementoblasts, as PPR-deficient progenitors have been shown to
exhibit both accelerated bone fibroblast differentiation and
upregulation of NFIC, leading to irregular cellular cementum
formation on the surface of roots that normally form acellular
cementum; this defect results in abnormal root development
(48). Mutations in PTH1R have
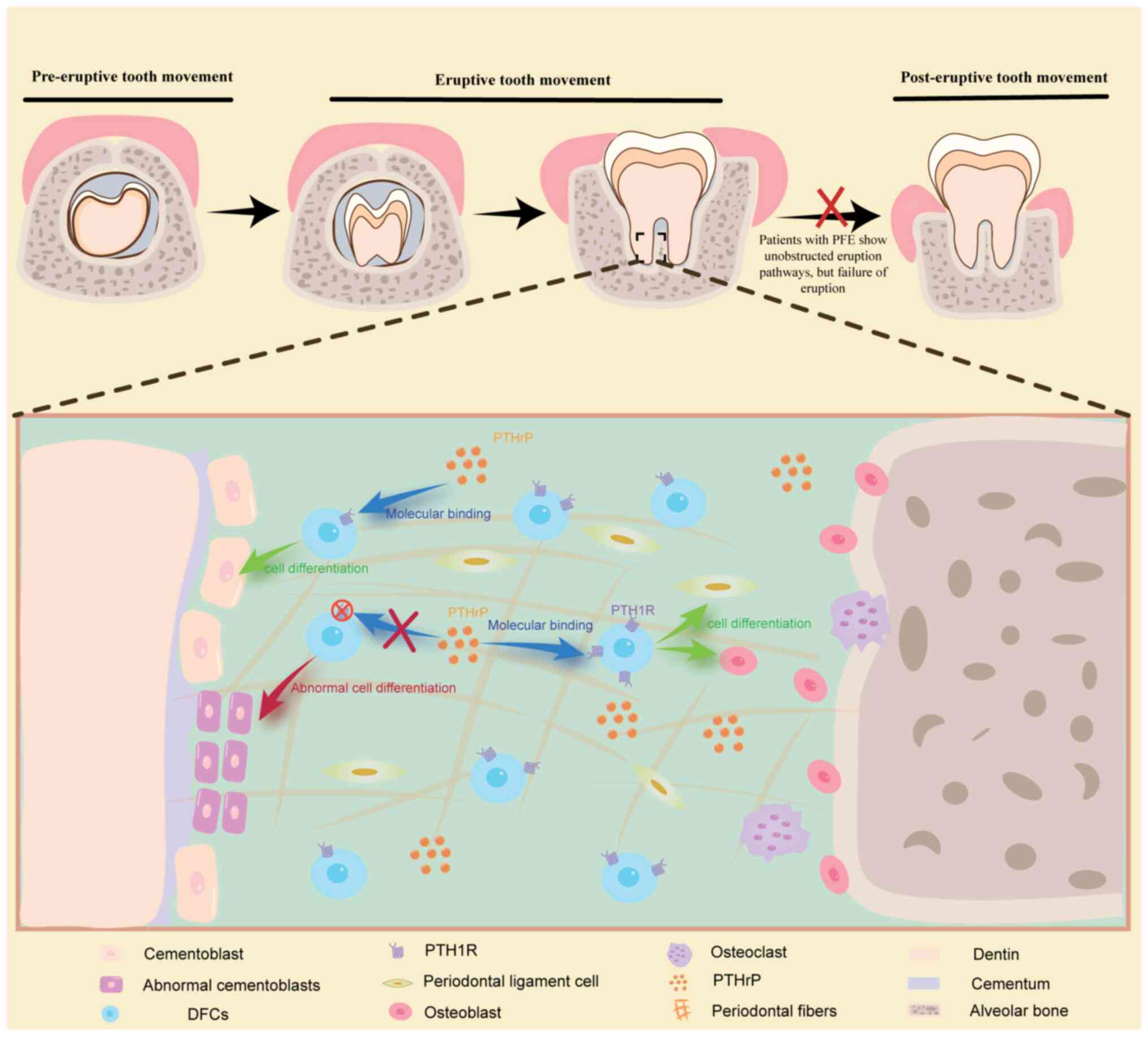
been associated with primary failure of eruption (PFE) (51–53).
PFE is characterized by incomplete or absent tooth eruption despite
the presence of an unobstructed pathway for eruption due to
dysfunction in the eruption mechanism (54). The association between PFE and
PTH1R was initially identified by Decker et al (55). Despite the incomplete understanding
of PFE pathogenesis, further investigation of PTH1R has
increasingly implicated an aberrant PTHrP-PTH1R signaling pathway
in DFCs as being a contributing factor to the development of PFE
(29,35). Tooth eruption depends on an
unobstructed pathway and sufficient force (56,57).
A characteristic of PFE is its unimpeded pathway, thus suggesting
that a lack of adequate force may be the underlying cause. The
eruption force is driven by the coordinated actions of the DF,
alveolar bone formation at the tooth root, and periodontal tissue
(29). DFCs serve a vital role in
this intricate process, particularly through their involvement in
the PTHrP-PTH1R pathway, which directly governs DFPC proliferation,
and subsequent DFPC differentiation into cementoblasts, alveolar
osteoblasts and periodontal ligament cells (35,58).
PTH1R is abundant in DFCs and is particularly enriched in
PTHrP+ DFPC (48,51).
These findings highlight the importance of the PTHrP-PTH1R
signaling pathway in guiding PTHrP+ DFPC differentiation
during tooth eruption (51). To
confirm that PTH1R deletion leads to PFE, a previous study
specifically utilized PTHrP-CreER to delete the receptor in
PTHrP+ DFC (59). Due
to the fact that the PFE of the first molar in mice shows a similar
phenotype to human PFE in adulthood (specifically that of open
occlusion) (52,60), mice were selected to establish a
model to determine the role of PTH1R in tooth eruption. The results
showed that, compared with in the control mice, the PTH1R-deficient
mice exhibited a phenotype characteristic of PFE. Subsequent
examination demonstrated that PTH1R deficiency in PTHrP+
DFPCs resulted in the abnormal formation of cementoblasts, thus
leading to premature cellular cementum formation on the root
surface and subsequent loss of periodontal attachment (59). Although previous studies have
traditionally considered tooth eruption to be a distinct process
from root formation (61),
previous studies have established an interrelationship between them
(48,59). These findings provide information
on the involvement of DFCs in the mechanisms underlying PFE;
however, further exploration is necessary to elucidate additional
underlying mechanisms (Fig.
2).
Abnormal Wnt/β-catenin signaling
pathway activity in DFCs
The Wnt signaling pathway comprises two distinct
pathways: The classical β-catenin-dependent pathway and the
nonclassical pathway (62).
Wnt/β-catenin signaling serves a crucial role in tooth development
and eruption, with active expression of Wnt/β-catenin signaling
observed in MSCs, including DFCs (63). Wnt signaling is crucial in multiple
stages of tooth development and it guides tooth development during
fetal formation (64). Aberrant
Wnt signaling can impede tooth development, while overactivation
can lead to misplaced tooth eruption. After birth, normal tooth
root and periodontal tissue formation depend on Wnt signaling.
Inactivation of Wnt/β-catenin signaling causes tooth root loss or
short roots with increased periodontal space. Proper bone
resorption and formation are essential for normal tooth eruption,
and Wnt/β-catenin signaling is crucial (63,65).
In MSCs, classical Wnt signaling promotes the differentiation of
DFPCs into osteoblasts rather than chondrocytes and adipocytes
(63). Studies have highlighted
the dual role of the Wnt signaling pathway in osteoclast formation;
β-catenin activation promotes the proliferation of osteoclast
progenitor cells at an early stage, after which β-catenin is
inactivated to promote osteoclast differentiation (66,67).
This process ensures that osteoclasts perform normal functions and
supports smooth bone resorption.
Studies have demonstrated that excessive
Wnt/β-catenin signaling in MSCs can result in tooth eruption
disorders (68,69). Activation of β-catenin in DFPCs and
osteoblasts, under the influence of Wnt/β-catenin signal
transduction, leads to upregulation of OPG expression (70), and OPG inhibits the RANK-RANKL
pathway, thereby suppressing osteoclast differentiation and
maturation, and ultimately contributing to tooth eruption disorder.
Conversely, the constitutive activation of β-catenin (Ocn-cre;
CtnnbLOX (EX3)/+, Col1a1-cre; CtnnbLOX
(EX3)/+) or the homozygous deletion of Axin2, which is a
negative regulator of Wnt signaling, can promote DFC
differentiation, and increase cementoblasts and cellular
cementogenesis. Eventually, excessive cementum and tooth stiffness
can occur (63,68,71,72).
Therefore, the upregulation of Wnt/β-catenin signaling in DFCs can
impair tooth eruption by disrupting osteoclast function and
promoting the excessive formation of cementoblasts. The latter
effect can be reflected in the distortion of periodontal tissue. As
the DF is crucial for periodontal tissue formation, Wnt/β-catenin
signaling within the DF is indispensable for periodontal tissue
homeostasis. Previous studies have shown that mice with continuous
Wnt/β-catenin signaling upregulation in dental tissues fail to
exhibit tooth eruption (68,71).
Upon excluding cases not attributed to disrupted osteoclast
activity, it was observed that these mice experienced calcification
of the periodontal ligament and functional periodontal ligament,
which obliterated the distinction between alveolar bone and
cellular cementum, thus leading to tooth rigidity and subsequent
failure of tooth eruption (72).
Additionally, aberrant osteoblast differentiation can result in
tooth eruption failure. Osteoblasts are derived from DFPCs, and
their differentiation is also regulated by the Wnt pathway
(73). Overexpression of Wnt10b in
the Ocn promoter in mice was shown to enhance postnatal bone mass
by promoting osteoblast differentiation, which consequently impairs
tooth eruption (74).
Abnormal TGF-β signaling pathway
activity in DFCs
The interaction between the epithelium and
mesenchyme is crucial for tooth morphogenesis and eruption
(75,76). This process involves multiple
signaling pathways, including the TGF-β signaling pathway, with
members of the TGF-β family playing crucial roles in normal and
pathological tooth development. Among these members, TGF-β type 2
receptor (Tgfbr2), which is one of the receptors for TGF-β, is
expressed in both epithelial and neural crest-derived mesenchyme. A
mouse model with conditional deletion of Tgfbr2 in mesenchymal
cells using Osterix (Osx) promoter-driven Cre recombinase exhibited
delayed tooth eruption. Simultaneously, aberrant differentiation of
osteoblasts and dentinal cells was observed, along with a
significant decrease in the number of osteoclasts (77). The expression of Osx is primarily
localized in the dental mesenchyme, specifically in the apex of the
dental papilla and DFC (48).
Therefore, it may be hypothesized that Osx is localized in DFCs,
thus the Osx-driven Cre recombinase can result in the deletion of
Tgfbr2 and the inhibition of TGF-β signaling in the aforementioned
models. This leads to the abnormal differentiation of DFCs into
osteoblasts and dentinogenic cells, as well as the abnormal
formation of osteoclasts, which ultimately causes tooth eruption
disorders. Additionally, aberrant expression of Smad4 can disrupt
tooth development through the TGF-β signaling pathway, as Smad acts
as an intracellular mediator within the TGF-β signaling pathway
(78). The conditional deletion of
Smad4 in mesenchymal cells derived from the neural crest has been
shown to halt tooth development (79). In addition, in a previous study,
the deletion of Smad4 in the dental mesenchyme using Ocn-Cre led to
abnormal root formation and delayed odontoblast differentiation
(80). Due to the fact that MSCs,
such as DFPCs, are critical for tooth root development, Smad4
deficiency in DFCs is likely to lead to abnormal tooth development
with impaired eruption. Moreover, due to the fact that Smad4
impacts the bone morphogenetic protein (BMP) signaling cascade, it
has been implicated in the regulation of the BMP signaling
pathway.
Abnormal BMP signaling pathways in
DFCs
The BMP signaling pathway is an essential component
of osteoblast differentiation and bone development, and it exhibits
extensive interplay with TGF-β signaling (81,82).
Research has demonstrated that selective knockout of BMP2 in DFPC
leads to impaired formation of tooth tissue and periodontal tissue,
thus suggesting that the BMP signaling pathway in DFPCs serves an
important role in maintaining the normal physiological function of
DFPCs (83). In addition, mouse
models with deletion of the BMP1 and TLL1 genes have shown impaired
tooth eruption (84). BMP1 and
TLL1 are encoded by distinct genes, but share similar structures
and functions, and belong to a small family of extracellular
metalloproteinases (85).
Following gene knockout of BMP1 and TLL1, mice with impaired tooth
eruption displayed reduced osteoclasts, which was potentially due
to impaired osteoclast induction. Mesenchymal cells exhibit high
levels of BMP7, and the removal of BMP7 from these cells can lead
to impaired tooth eruption and abnormal mineralization (86). One possibility for this effect is
that the timing of tooth eruption is directly related to
mineralization onset. Another plausible explanation is that BMP7
function in dental pulp and DFPCs affects tooth eruption; however,
the specific mechanism involved remains unclear. Additionally,
muscle segment homeobox like 2 (Msx2), which is a target of BMP
signaling, has been reported to be expressed in mesenchymal cells
(87,88), and Msx2-null mice also exhibited
tooth eruption failure (89).
Experiments have suggested the alteration of the RANK osteoclast
differentiation pathway in Msx2-null mice, thus implying the effect
of Msx2 on the potential regulation of this pathway, which impacts
osteoclast function and causes tooth eruption failure. These
findings suggest that dysregulation of the TGF-β and BMP signaling
pathways within DFCs often results in abnormal tooth eruption.
Abnormal Hedehog (Hh) signaling
pathways in DFCs and other cells
Hh signaling has a crucial role in the development
of various organs, including teeth, by mediating interactions
between epithelial and mesenchymal cells. Hertwig's epithelial root
sheath is surrounded by dental papilla and DFPCs that express
receptor patched 1 (Ptch1) for Hh. During tooth development,
Hh-expressing cells are strictly localized in the dental
epithelium, whereas Ptch-positive cells are found in dental
mesenchymal cells without Sonic hedgehog protein (Shh) expression
(90). Analysis of mice with
mesoblastic dysplasia revealed abnormalities in the C-terminus of
the Ptch1 protein. In these mutants, the proliferation of
mesenchymal cells around the teeth was inhibited. Additionally,
they exhibited disrupted molar eruption and shorter roots. These
findings indicate that abnormal transmission of the Shh signal
between the epithelium and DF mesenchyme may impact tooth root
development and eruption (91).
The involvement and functions of DFCs in tooth eruption exhibit
temporal and spatial variations. Temporal variations divide tooth
eruption into intraosseous and extraskeletal stages, with DFCs
playing distinct roles (92).
Intraosseous eruption mainly results from alveolar bone resorption
and remodeling. During this period, different regions of the DF are
thought to play different roles, and DFCs around the crown induce
different types of osteoclast differentiation by upregulating the
expression of factors such as CSF-1, VEGF and RANKL (24), which leads to an increase in the
number of osteoclasts, thus facilitating crown bone resorption to
establish unobstructed eruption pathways. By contrast, the DF
tissue near the developing root apex serves a key role in alveolar
bone formation, thus providing upward force for tooth eruption.
This process is intricately linked to the differentiation of DFCs
into osteoblasts. Spatial effects on DFs may be the result of
regional differences in gene expression. In a previous study, DFCs
were isolated from both the crown and basal regions of rat teeth,
and RNA was extracted from each region for analysis. The expression
of RANKL in the crown region was greater than that in the basal
region, whereas the expression of BMP-2 in the basal region was
greater than that in the crown region (24). Thus, the spatial localization of
gene expression in the DF may modulate osteoclast generation and
osteoblast differentiation. The coordinated activity of different
regions of the tooth, which is mediated by DFCs, facilitates tooth
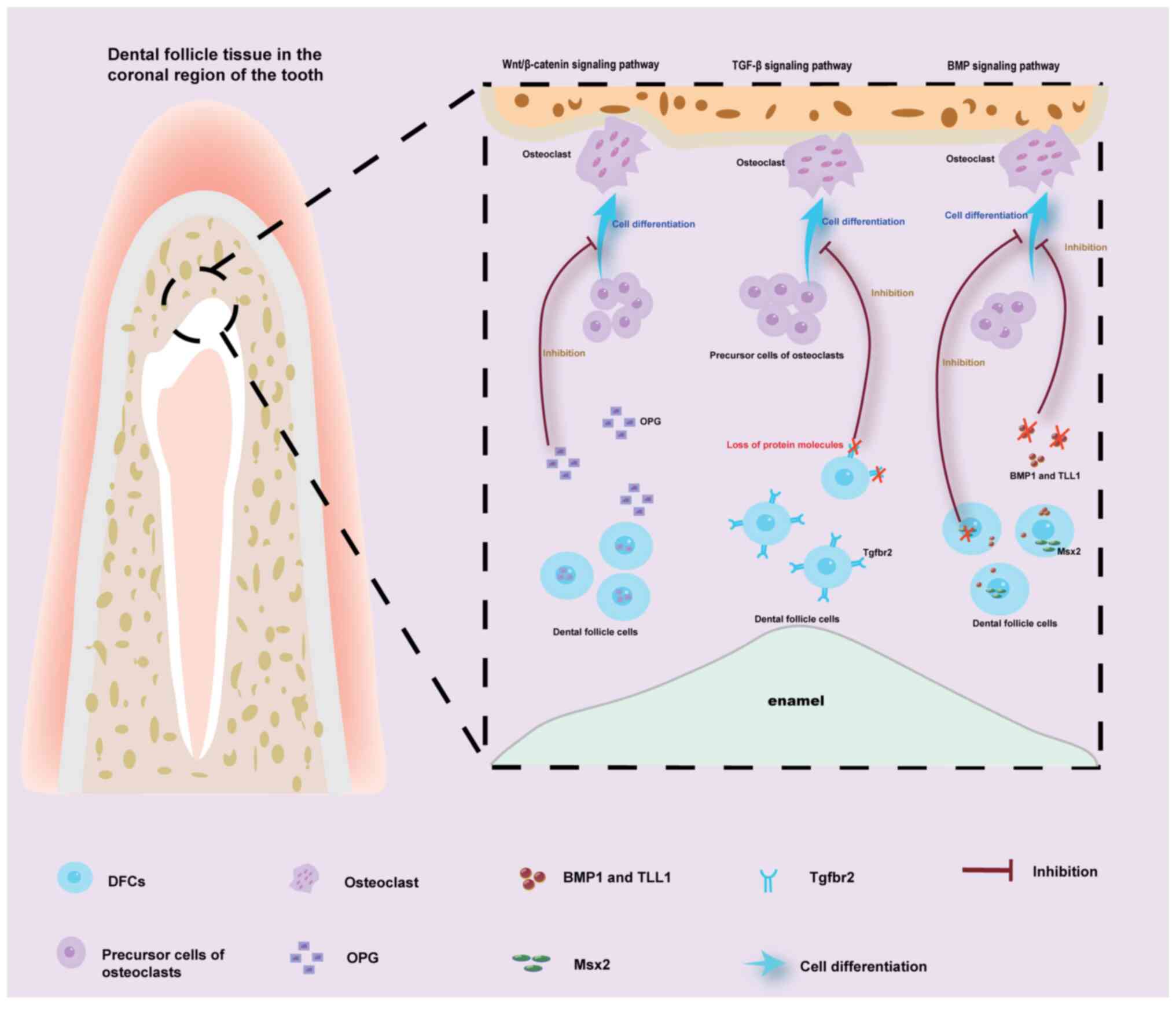
movement toward the oral cavity. However, the removal of the crown
or root region of DF can impede successful eruption; specifically,
crown removal disrupts pathway formation, whereas root removal
inhibits bone formation (16). The
DFs in the two regions exert influences on tooth eruption through
distinct signaling pathways. In the crown region, the main pathways
involved are the Wnt/β-catenin pathway, TGF-β signaling pathway and
BMP signaling pathway. In the root region, in addition to the
aforementioned pathways, the Hh signaling pathway also serves a
significant role (Figs. 3 and
4). When the tooth is exposed to
the oral cavity, it enters the stage of extraosseous eruption. In
this stage, the force of tooth upward eruption is mainly derived
from the periodontal ligament (93). Furthermore, periodontal tissue
derived from DFs and normal follicle development crucially support
the tooth eruption progression outside the alveolar bone.
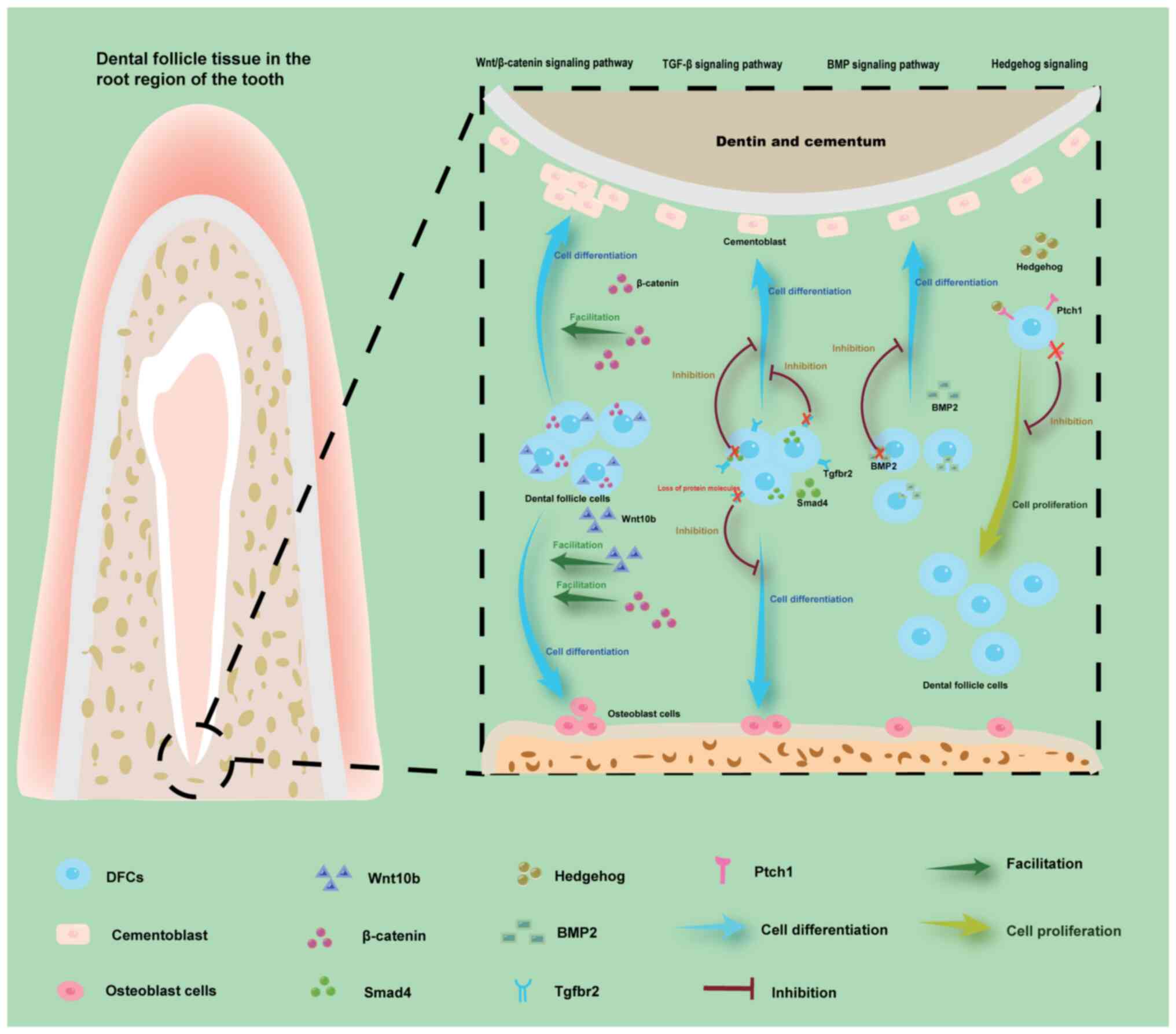 | Figure 4.Depicted regions primarily reside
within the dental follicle tissue surrounding the root apex.
Overexpression of β-catenin in the Wnt/β-catenin signaling pathway
promotes differentiation of DFCs into cementoblasts, thereby
enhancing cementum formation. Conversely, loss of Tgfbr2 and Smad4
in the TGF-β signaling pathway, as well as BMP2 deficiency in the
BMP signaling pathway inhibits the formation of cementoblasts,
Eventually, an excess or deficiency of cementum can result in
abnormal root development. The excessive upregulation of Wnt10b and
β-catenin in the Wnt/β-catenin signaling pathway leads to a
significant increase in bone mass by promoting osteoblast
differentiation. The loss of the Tgfbr2 in the TGF-β signaling
pathway inhibits the differentiation of DFC into osteoblasts,
leading to the block of alveolar bone formation. Loss of Ptch1
receptors inhibits the proliferation of DFCs, thereby affecting
tooth eruption. BMP, bone morphogenetic protein; DFC, dental
follicle cell; Ptch1, patched 1; Tgfbr2, TGF-β type 2 receptor. |
Syndromes and genetic disorders
Abnormal tooth eruption can be classified into two
main categories, failed and delayed tooth eruption, and is
associated with numerous systemic syndromes and genetic diseases,
the majority of which are caused by pathogenic gene mutations.
Table I presents a comprehensive
list of syndromes and genetic disorders associated with aberrant
tooth eruption. According to the data, 48 diseases are known to be
linked to abnormal tooth eruption (23,25,28,29).
There is convincing evidence for a strong relationship between
abnormal tooth eruption and the presence of DFs in seven of these
diseases. Furthermore, five of these disorders have been attributed
to mutations in specific genes: Cleidocranial dysplasia (CCD),
osteopetrosis, mucopolysaccharidosis VI, enamel renal syndrome and
dentin dysplasia (DD). Moreover, regional odontodysplasia (RO) and
multiple calcifying hyperplastic DFs are two distinct types of
tooth eruption disorders that are not associated with genetic
mutations.
 | Table I.Syndromes and genetic disorders
associated with abnormal tooth eruption. |
Table I.
Syndromes and genetic disorders
associated with abnormal tooth eruption.
| Disease name | OMIM
number(s)a | Orphanet
numberb | Association with
the DF | Risk
factorsb | Tooth eruption
status | (Refs.) |
|---|
| Cleidocranial
dysplasia | 119600; 620099 | 1452 | Yes | RUNX2 gene
mutation | Delayed
eruption | (23,25,27–29) |
| Albers-Schönberg
osteopetrosis | 166600 | 53 | Yes | CLCN7 gene
heterozygous mutations | Failure of
eruption | (25,28) |
|
Mucopolysac-charidosis VI | 253200 | 583 | Yes | ARSB gene
mutation | Delayed
eruption | (23) |
| Enamel renal
syndrome | 204690 | 1031 | Yes | FAM20A gene
mutation | Failure of
eruption | (27) |
| Dentin
dysplasia | 125400; 125420 | 1653 | Yes | VPS4B gene
mutation | Failure of
eruption | (27,29) |
| Regional
odontodysplasia | / | 83450 | Yes | Local circulatory
disorders, viral infections, local trauma, pharmacotherapy during
pregnancy, facial asymmetry or a combination of these factors | Failure or delay in
eruption | (28,29) |
| Multiple calcifying
hyperplastic dental follicles | / | / | Yes | Unknown | Failure of
eruption | (25) |
| Gorlin
syndrome | 109400 | 377 | Unknown | Ptch1 gene
mutation | Failure of
eruption | (27,29) |
| Oculodental
syndrome, Rutherfurd type | 180900 | 2709 | Unknown | Unknown | Failure of
eruption | (28) |
| Cherubism | 118400 | 184 | Unknown | SH3BP2 gene
mutation in ~80% of cases | Failure of
eruption | (27–29) |
| Albright hereditary
osteodystrophy | 612462 | 79444 | Unknown | GNAS gene
mutation | Delayed
eruption | (25) |
| Gardner
syndrome | 175100 | 79665 | Unknown | APC gene
mutation | Failure of
eruption | (23,27,29) |
| Osteoglophonic
dysplasia | 166250 | 2645 | Unknown | FGFR1 gene
mutation | Failure of
eruption | (28,29) |
| Nance-Horan
syndrome | 302350 | 627 | Unknown | NHS gene
mutation | Failure of
eruption | (28) |
| McCune-Albright
syndrome | 174800 | 562 | Unknown | Somatic mutations
of the GNAS gene | Failure of
eruption | (28) |
|
Hypodontia-dysplasia of nails
syndrome | 189500 | 2228 | Unknown | MSX1 gene
mutation | Failure of
eruption | (28) |
| GAPO syndrome | 230740 | 2067 | Unknown | Homozygous nonsense
or splicing mutations in the ANTXR1 gene | Failure of
eruption | (23,25,27–29) |
| Osteopathia striata
with cranial sclerosis | 300373 | 2780 | Unknown | Mutations in the
Wilms tumor gene on the X chromosome | Failure of
eruption | (25) |
| Singleton-Merten
syndrome | 182250; 616298 | 85191 | Unknown | Unknown | Delayed
eruption | (25,27,29) |
| Aarskog
syndrome | 100050; 305400 | 915 | Unknown | FGD1 gene
mutation | Delayed
eruption | (25,27,29) |
| Acrodysostosis | 101800; 614613 | 950 | Unknown | Heterozygous
mutations in either the PRKAR1A or PDE4D genes | Delayed
eruption | (25) |
| Apert syndrome | 101200 | 87 | Unknown | FGFR2 gene
mutation | Delayed
eruption | (25,27,29) |
| Chondroectodermal
dysplasia | 225500; 617088;
618123 | 289 | Unknown | EVC and EVC2 gene
mutations | Delayed
eruption | (25) |
| Cockayne
syndrome | 133540; 214150;
216400; 216411; 278780; 610756; 610758; 616570 | 191 | Unknown | ERCC6 and ERCC8
gene mutations | Delayed
eruption | (25) |
| Dubowitz
syndrome | 223370 | 235 | Unknown | Unknown | Delayed
eruption | (25) |
| Frontometa physeal
dysplasia | 305620; 617137 | 1826 | Unknown | Unknown | Delayed
eruption | (25) |
| Goltz syndrome | 305600 | 2092 | Unknown | PORCN gene
mutation | Delayed
eruption | (25) |
| Hunter's
syndrome | 309900 | 580 | Unknown |
Iduronate-2-sulfatase deficiency | Delayed
eruption | (25) |
| Incontinentia
pigmenti | 308300 | 464 | Unknown | IKBKG gene
mutation | Delayed
eruption | (25,29) |
| Levy-Hollister
syndrome | 149730; 620192;
620193 | 2363 | Unknown | Unknown | Delayed
eruption | (25) |
| Osteogenesis
imperfecta | 166200; 166210;
166220; 166230; 259420; 259440; 610682; 610915; 610967; 610968;
613848; 613849; 613982; 614856; 615066; 615220; 616229; 616507;
619131; 619795 | 666 | Unknown | COL1A1 and COL1A2
gene mutations | Delayed
eruption | (25,27,29) |
| Hutchinson-Gilford
syndrome | 176670 | 740 | Unknown | Unknown | Delayed
eruption | (25) |
|
Pyknodysostosis | 265800 | 763 | Unknown | Encoding cathepsin
K gene mutations | Delayed
eruption | (25) |
| Carpenter
syndrome | 201000; 614976 | 65759 | Unknown | RAB23 and MEGF8
gene mutations | Failure of
eruption | (27,29) |
| Down syndrome | 190685 | 870 | Unknown | Additional
independent chromosome 21 (47,+21) | Failure of
eruption | (29) |
| Hypertrichosis
lanuginosa congenita | 145700; 145701;
307150 | 2222 | Unknown | Unknown | Failure of
eruption | (29) |
| Costello
syndrome | 218040 | 3071 | Unknown | HRAS gene
mutation | Failure of
eruption | (27,29) |
| Junctional
epidermolysis bullosa | / | 305 | Unknown | mutations in
various genes, including COL17A1, ITGA6, ITGB4, LAMA3, LAMB3, LAMC2
and ITGA3 | Failure of
eruption | (27,29) |
| Gaucher
disease | 230800; 230900;
231000; 231005; 608013; 610539 | 355 | Unknown | GBA gene
mutation | Failure of
eruption | (29) |
| Hereditary gingival
fibromatosis | 135300; 605544;
609955; 611010; 617626 | 2024 | Unknown | Unknown | Failure of
eruption | (27,29) |
| Hallermann-Streiff
syndrome | 234100 | 2108 | Unknown | Unknown | Failure of
eruption | (27,29) |
|
Hyperimmuno-globulinemia | 252500 | 576 | Unknown | GNPTAB gene
mutation | Failure of
eruption | (29) |
| Menkes disease | 309400 | 565 | Unknown | ATP7A gene
mutation | Failure of
eruption | (29) |
| Neurofibro-matosis
type 1 | 162200; 162210;
613675 | 636 | Unknown | NF1 gene
mutation | Failure of
eruption | (29) |
| Parry-Romberg
syndrome | 141300 | 1214 | Unknown | Unknown | Failure of
eruption | (29) |
| Sclerosteosis | 269500; 614305 | 3152 | Unknown | Unknown | Failure of
eruption | (29) |
| SHORT syndrome | 269880 | 3163 | Unknown | PIK3R1 gene
mutation | Failure of
eruption | (27,29) |
| Infantile spasms
syndrome (West Syndrome) | 300672; 308350;
613477; 613722; 615006; 616139; 616341; 617065; 617929; 618298 | 3451 | Unknown | Gene mutation of
STXBP1, TSC1, TSC2 and trisomy 21 | Failure of
eruption | (29) |
CCD
CCD, which was identified by Marie and Sainton in
1897, is an autosomal dominant disorder characterized by hypoplasia
of the clavicle and skull, widening of the suture and fontanelle,
and short stature (49). In
addition to skeletal abnormalities, patients with CCD often have
dental issues, such as supernumerary teeth accompanied by severe
malocclusion and crossbite, retention of primary dentition,
impacted teeth and failed tooth eruption (94,95).
In a recent study, 50 patients with CCD were examined, 41 of whom
had symptoms of tooth eruption failure. These patients presented
with a total of 665 teeth displaying abnormal eruption patterns.
The most commonly affected teeth were canines (79.5%), followed by
permanent premolars (71.0 and 62.5%, first and second permanent
premolars, respectively), and superdeciduous teeth and/or retained
primary teeth were often observed in this area. Conversely, the
first and second molars were less affected (6.0 and 24.0%,
respectively) (27).
Genetic studies have shown that mutations in a
single allele of RUNX2 cause CCD. These mutations commonly arise
from deletions, missense mutations and substitutions occurring
within the DNA binding region of RUNX2. The RUNX2 gene, also known
as core binding factor a1 (Cbfa1), is located on chromosome 6p21
(96). RUNX2 acts as a crucial
transcriptional regulator of osteoblast differentiation during bone
formation (97). In addition,
heterozygous Runx2-knockout mice were found to exhibit the majority
of bone abnormalities observed in human patients with CCD. It has
been reported that Runx2 is expressed in preosteogenic mesenchyme
and active osteogenesis sites in mice (98–100). Mice with complete deficiency of
Runx2 [Runx2 (−/-)] have been shown to exhibit severe osteogenesis
imperfecta and often succumb to respiratory distress at birth due
to defects in the ribs. Heterozygous mutant mice [Runx2 (+/-)] can
survive but exhibit skeletal abnormalities, including an open
fontanelle and clavicular defects. This phenotype suggests that a
mutation in one allele of Runx2 in mice is sufficient to produce an
osteogenic malformation (101).
These mice recapitulate the bone abnormalities that are commonly
observed in most cases of CCD. To investigate whether these mice
can also replicate tooth eruption abnormalities similar to those
found in CCD, a heterozygous Runx2-knockout mouse model was
generated to observe tooth eruption (101). Compared with wild-type mice,
mutant mice exhibited a significant delay in tooth eruption.
Further investigations into the impact of Runx2 on skeletal and
dental anomalies, and its primary cellular targets, have
demonstrated that Runx2 is expressed in osteoblasts and DFs but not
in osteoclasts (102,103). Therefore, the abnormal tooth
eruption in Runx2 (+/-) young adult mice may be attributed to two
factors: i) Due to the inhibition of DF-mediated osteoclast
signaling during tooth eruption in Runx2 (+/-) mice; and ii) due to
the impaired osteogenic differentiation of DFCs leading to
defective bone deposition in osteoblasts and consequently resulting
in abnormal eruption.
Further elucidation of the molecular basis of the
abnormal eruption observed in Runx2 (+/-) mice is required to test
these two possibilities. First, impaired osteoclast recruitment is
a possible cellular mechanism for delayed tooth eruption in
patients with CCD. Previous research has demonstrated active
resorption of alveolar bone and an increase in osteoclasts during
eruption in both wild-type mice and Runx2 (+/-) mutant mice;
however, this increase was significantly inhibited in the mutant
mice. Additionally, this previous study indicated that Runx2 may
serve a role in osteoclastogenesis by activating the expression of
RANKL and receptor activators of RANK-RANKL signaling (101). It may be hypothesized that the
two alleles of Runx2 promote RANK-RANKL signaling, which is
essential for active osteoclast recruitment in the tooth
germination pathway, and that DFCs play a crucial role in
osteoclast recruitment and express Runx2. This finding suggested
that Runx2 mutations in DFCs may hinder active alveolar bone
resorption by affecting osteoclast numbers, thus contributing to
abnormal tooth eruption. Additionally, the effect of Runx2
mutations on osteoblasts was investigated by examining its effect
on the osteogenic differentiation of DFCs. The findings
demonstrated that a Runx2 mutation decreased the mineralization
capacity of DFCs and downregulated the expression of genes
associated with osteoblast function, such as ALP, Osx, OCN, ColIα1
and OPN. Furthermore, it disrupted bone formation during tooth
eruption, consequently diminishing the osteogenic potential of
DFCs. These effects may contribute to abnormal tooth eruption in
patients with CCD (104).
Osteopetrosis
Osteopetrosis is a disease caused by disruption of
the bone remodeling process with osteoclastic bone resorption
defects, and can be divided into intermediate autosomal recessive
osteopetrosis (global incidence, 1/250,000) and autosomal dominant
osteopetrosis (global incidence, 1/20,000) (105) depending on how it occurs. The
clinical manifestations of osteopetrosis commonly include
fractures, scoliosis, osteoarthritis, bone marrow insufficiency,
developmental delays, tooth eruption disorders and a range of
neurological symptoms. Additionally, heightened bone density can
lead to compression of cranial nerves and subsequent abnormal
innervation (106,107). The eruption of teeth may be
delayed or completely absent due to decreased bone resorption and
abnormal opening of tooth eruption pathways. Additionally, dental
deformities, enamel hypoplasia, dentin abnormalities, inadequate
mineralization of enamel and dentin, and defects in the periodontal
membrane have been observed (108). A statistical analysis of patients
with osteopetrosis demonstrated that the maxillary second molars
(66.7%) and mandibular second molars (58.3%) exhibited the highest
incidence of tooth eruption failure, whereas anterior teeth and
first premolars were rarely affected (27).
Osteopetrosis arises from gene mutations that cause
abnormalities in the rough marginal region and dysfunction of
osteoclasts, which fail to mediate extracellular acidification in
this area, thus resulting in obstructed osteolysis (23). The genes involved in the formation
and function of the rough marginal region of osteoclasts include
CLCN7, TCIRG1, OSTM1, SNX10 and PLEKHM1. Mutations in these genes
impair the transport of endosomal and lysosomal vesicles, thereby
disrupting rough marginal regions, as well as osteoclast formation
and function (109). Osteoclasts
serve a crucial role in tooth eruption, and abnormal tooth eruption
in patients with osteopetrosis may be attributed to dysfunctional
osteoclasts. Among the aforementioned mutated genes, CLCN7
mutations are the most common cause of osteopetrosis (110), and their impact on osteoclasts is
closely related to DFCs. A CLCN7-deficient mouse model was
established via injection of chitosan-CLCN7-small interfering RNA
nanoparticles, and the mice exhibited abnormal tooth eruption.
Coincidentally, these dental changes have also been observed in
patients with CLCN7 mutations (111,112). Subsequent experiments have
demonstrated that CLCN7 regulates tooth eruption through the
DFC-mediated osteoclast pathway by decreasing CLCN7 expression in
the DFC, thus leading to reduced numbers of osteoclasts and bone
resorption pits (111).
Therefore, the lack of CLCN7 in DFCs may inhibit osteoclast
formation. This relationship may be mediated through the RANKL-OPG
pathway. The RANK-RANKL-OPG signaling axis and downstream
transcription factors are important pathways through which DFCs
regulate osteoclast generation. OPG secreted by DFCs may inhibit
osteoclast generation (45,113),
whereas RANKL secreted by DFCs is an important positive regulator
of osteoclast differentiation (114,115). RANKL and OPG have been reported
to be expressed in DFCs, and CLCN7-deficient mice exhibited
downregulated RANKL expression and upregulated OPG expression,
which inhibited osteoclast generation (111). Thus, mutations in CLCN7 may
result in diminished osteoclasts and aberrant tooth eruption
through the RANK-RANKL-OPG signaling pathway mediated by DFCs.
Furthermore, in vitro investigations of DFCs
have demonstrated that defects in CLCN7 can impede DFC
differentiation. Previous research has indicated that DFCs can
differentiate into various cell types, including osteoblasts
(116). Normally, induced DFCs
exhibit upregulation of osteoblast-related genes, such as ALP, BSP,
OPN and TGFB1, thus confirming their potential for osteoblastic
differentiation. However, the expression levels of these proteins
have been shown to be reduced in a CLCN7-deficient cell group
(111). Thus, CLCN7 mutations may
be involved in regulating the osteogenic differentiation of DFCs to
influence tooth eruption.
Mucopolysaccharidosis VI
Mucopolysaccharidosis represents a cluster of
hereditary disorders characterized by impaired degradation of
mucopolysaccharides [also known as glycosaminoglycans (GAGs)] due
to deficiency of specific enzymes, thus resulting in increased
accumulation of mucopolysaccharides across diverse tissues
(117). Mucopolysaccharidosis
types I–VII are classified based on clinical and biochemical
characteristics, and exhibit a high degree of variability. Clinical
manifestations include developmental delay, growth retardation and
skeletal abnormalities (118).
Initially, the accumulation of mucopolysaccharides in various
organs leads to progressive intellectual disability and
neurodevelopmental deficiency. The most severe consequences occur
when excessive GAG accumulation affects the heart, thus resulting
in severe cardiovascular disease and even death. Additionally, an
excessive buildup of GAG in the DF can impede tooth eruption
(23).
Maroteaux-Lamy syndrome (mucopolysaccharidosis type
VI), which was initially reported in 1965 (119), is an uncommon autosomal recessive
disorder, with a global incidence ranging from 0.0132:100,000 to
20:100,000 (120,121). This disorder arises due to a
deficiency of arylsulfatase B (ARSB), which is a crucial gene
involved in the degradation of dermatan sulfate. Mutations in this
gene lead to the accumulation of undegraded or partially degraded
mucopolysaccharides that disrupt cellular function and give rise to
various symptoms. Patients with mucopolysaccharidosis VI exhibit
physical characteristics resembling those of other types of
mucopolysaccharidosis, including short stature, joint stiffness,
corneal opacity, and cardiac and respiratory dysfunction (122). However, in contrast to patients
with other subtypes, patients with this condition exhibit normal
cognitive abilities, metachromatic inclusions in white blood cells
and deficiencies in ARSB (123).
Furthermore, dental abnormalities are significant manifestations of
Maroteaux-Lamy syndrome. These abnormalities are commonly described
as dysplastic and widely spaced permanent molars with abnormal root
eruption and calcification. Such aberrant teeth often coincide with
DF irregularities, wherein excessive deposition of dermatan sulfate
impairs the normal morphology and function of the DF. Consequently,
the DF becomes tougher and thicker due to the dense fibrous
connective tissue observed upon histopathological examination. This
abnormal DF increases resistance to tooth eruption, thus ultimately
leading to failed tooth eruption (23,118,123).
Enamel renal syndrome
Enamel renal syndrome is an uncommon genetic
disorder inherited in an autosomal recessive pattern due to
biallelic mutations in the FAM20A gene (124). It is characterized by
amelogenesis imperfecta (AI), delayed tooth eruption,
intramedullary calcification, gingival enlargement, gingival
fibromatosis and nephrocalcinosis (125). Among them, AI and
nephrocalcinosis are the most common characteristics of these
patients. AI refers to a group of genetic disorders ranging in
incidence from 1:700 to 1:14,000 in the United States that affects
both the quality and quantity of enamel. Symptoms can be observed
in some or all teeth, with AI uniformly affecting enamel across
individuals, thus resulting in either hypoplastic or
undermineralized enamel. The affected teeth may exhibit
discoloration, sensitivity, or increased susceptibility to
disintegration prior to or after eruption (126). Nephrocalcinosis is a disease
characterized by calcium salt deposition in the kidney, which may
be predominantly cortical or medullary in nature; it is often
associated with primary hyperparathyroidism, distal renal tubular
acidosis and other diseases (127). In addition to enamel and kidney
lesions, abnormal tooth eruption is also a prevalent clinical
manifestation. Patients with enamel renal syndrome exhibit an
aberrant eruption pathway for their posterior teeth (125). Although the root is fully formed,
the eruption of the tooth stops halfway, and pericoronal
radiolucency manifests around the impacted teeth. Previous case
studies have demonstrated that the DF associated with mandibular
posterior molars exhibits an atypical structure that is
characterized by dense connective tissue and mineralized tissue
(128–130). Therefore, delayed tooth eruption
can be attributed to the pathological condition of the DF. One
possibility for this effect is that DFs may exhibit impaired
synthesis of essential molecular components required for proper
tooth eruption. Previous studies have demonstrated that FAM20A is
localized in the DF above the cusp, and its deficiency has been
linked to unsuccessful tooth eruption, thus suggesting a potential
role for FAM20A-catalyzed phosphorylation in regulating the pathway
involved in shaping the pathway of tooth eruption. Additionally,
the presence of pericoronal radiolucency around the impacted teeth
can be associated with mutations in FAM20A within the DF (130–132). Another factor is that tooth
eruption may be hindered by the DF due to mechanical retention
caused by cystic or fibrous transformation. Additionally, the
presence of calcification in the DF of patients could contribute to
abnormal eruption (133). FAM20A
mutations are responsible for enamel renal syndrome and are also
associated with calcification in the DF. The gene normally
suppresses mineralization; however, in patients with homozygous
FAM20A mutations, increased promoter activity and reduced
inhibition of oxalate crystal growth cause mineralization of the
DF, thus impairing its ability to support normal tooth eruption
(127).
DD
Genetic dentin disorders have been well documented
and include two primary types: Dentinogenesis imperfecta (DI) and
DD (134). Based on the clinical
classification, DI can be further categorized into three subgroups
(types I–III), whereas DD can be divided into two subgroups
(135). The present review
specifically focused on DD, which was previously referred to as a
‘rootless tooth’; however, with advancements in understanding of
this disorder, this condition has become known as DD. This disorder
is classified into type I (DD1) and type II (DD2) (136). DD1 is a rare autosomal dominant
nonsyndromic disorder in human dentinal diseases, with an estimated
incidence of 1/100,000 (137). In
DD1, the patient's crown exhibits a normal shape, morphology and
coloration; however, the patient presents with premature tooth
loss, tooth loosening and abnormal tooth eruption (138,139). Imaging demonstrates structural
abnormalities, including bulbous crowns, occlusion of the
endodontic compartment, shortened roots and periapical
radiolucency. Pulp remnants in permanent teeth may show
crescent-shaped radiolucence, whereas deciduous teeth show complete
pulp occlusion (139). The
clinical appearance of teeth in patients with DD2 is also normal;
however, the primary teeth may appear to be amber and translucent
(140). In DD2, the roots exhibit
a normal shape and morphological features. Therefore, delayed tooth
eruption is rarely reported as being a feature of DD2, but it is
often observed in patients with DD1 (139). This is due to the fact that root
development has a certain impact on tooth eruption, thus
necessitating further investigations into the potential causes of
delayed eruption in DD1.
To date, mutations in the VPS4B, SMOC2 and SSUH2
genes have been identified via genetic screening to be associated
with the pathogenesis of DD1 (141–143). Among them, VPS4B has been shown
to be closely related to the formation of alveolar bone and
cementum, and the normal differentiation of DFCs is also an
essential component of cementogenesis and the development and
formation of surrounding alveolar bone (144). Therefore, VPS4B mutations may
lead to abnormal osteogenesis by affecting the normal
differentiation and proliferation of DFCs, and eventually leading
to abnormal tooth eruption. Ultimately, a comparative analysis of
the proliferation and osteogenic induction capacity of DFCs derived
from patients with VPS4B-mutant DD1 and healthy controls was
conducted (145). The growth
rates of DFCs were found to be significantly greater in patients
with DD1 than in controls; however, compared with those from
control individuals, DFCs from patients with DD1 exhibited lower
expression levels of osteogenic genes, such as ALP, OCN, BSP and
RUNX2, as well as fewer calcium nodules, as observed via Alizarin
red S and ALP staining. These findings suggest that VPS4B may have
a crucial role in regulating the osteogenic differentiation of DFCs
and that mutations in VPS4B could lead to reduced osteogenic
capacity in patients with DD1. Consequently, impaired root
formation and bone remodeling during development may ultimately
contribute to tooth eruption disorders.
RO
RO is a rare developmental anomaly that was first
described by Zegarelli et al in 1963 (146). The etiology of RO remains
incompletely elucidated, although it is not believed to have a
hereditary basis (147). Various
potential pathogenic factors have been postulated in the
literature, including local trauma, radiation exposure, high fever
episodes, vascular disorders, prenatal drug administration,
localized or systemic viral infections, reactivation of latent
viruses, impaired migration or differentiation of neural crest
cells, nutritional or metabolic deficiencies, ischemia events and
Rhesus disease (148,149). The clinical manifestations of RO
include discoloration of teeth (yellow or brown), impaired tooth
eruption, atypical tooth morphology, tooth mobility, and the
presence of swelling or abscess formation (150). The main radiographic
characteristics include an enlarged pulp cavity, open root apices,
indistinct borders and a ghost-like appearance of the affected
tooth (151). Histologically,
enamel and dentin show hypoplasia and insufficient calcification,
the pulp is larger than normal, and the DF appears to be calcified
(152,153). In general, the disease affects
both primary and permanent dentition. The mandible is generally
more susceptible than the maxilla. Among the clinical
manifestations, tooth eruption failure commonly occurs (149). The failure of tooth eruption may
be attributed to dental deformities hindering the process, abnormal
calcification and swelling of the DF causing mechanical
obstruction, or dysregulation of signaling pathways in DFPCs during
the eruption induction pathway resulting from calcification of the
DF (154,155). According to the literature,
imaging studies have demonstrated abnormal hyperplasia and fibrous
tissue swelling in the vicinity of nonerupted teeth (154–156), which is associated with aberrant
calcification of the DF tissue. In addition, histological studies
have demonstrated various types of calcification within the DF of
patients with RO, including fibrous or nonfibrous osteoid chains,
as well as fused calcified spheres attached to larger calcified
masses or osteoid chains. These calcifications are predominantly
located in areas typically occupied by enamel formation, some of
which are formed independently of collagen involvement, whereas
others result from collagen fiber mineralization (154,157). The accumulation of calcified
tissue in the DF is closely associated with both the enlargement of
the DF and an increase in periodontal fibrous tissue. These
abnormal DF tissues may eventually lead to tooth eruption
disorder.
Multiple calcifying hyperplastic DFs
(MCHDFs)
MCHDFs are rare, and their etiology is still
unclear (158); they are
clinically defined by multiple unerupted teeth and large DFs
(159). Radiographically, these
follicles are observed as radiolucency surrounding the crown of the
unerupted tooth and may also exhibit radiopaque lesions within the
inner part of the DF (160,161). The histological features of this
condition include extensive cemento-like calcification and the
presence of residual odontogenic epithelium within the fibrous
connective tissue matrix (162).
The process of calcification is usually performed in DFs because
DFPCs in DFs can differentiate into cementoblasts or osteoblasts
(163). Impacted teeth may result
from incomplete digestion of fibrous tissue (164) and abnormal structure or
enlargement of the DF, which obstructs tooth eruption.
Additionally, calcified tissue within the DF could disrupt
DFPC-related signal transduction pathways that are crucial for
proper tooth eruption (162). The
reported data have suggested that the incidence of type I
calcification is greater in patients with MCHDF than in patients
with type II calcification. However, type I calcification may also
occur in DFs with hypoplasia and regional odontodysplasia, thus
suggesting similar etiologies for tooth eruption disorders in these
conditions (165). Following the
excision of abnormal DFs, successful eruption of impacted teeth in
patients with MCHDF further underscores the pivotal role of
diseased DFs in eruption failure (165).
Conclusion
The DF serves a crucial role in tooth eruption, and
regulates the formation and resorption of alveolar bone.
Abnormalities in DFCs are closely associated with abnormal eruption
patterns, and disturbances in signaling pathways within the DF
represent an important factor contributing to tooth eruption
disorders. PTHrP signaling can modulate osteoclast differentiation
by influencing the CSF-1-RANK-RANKL-OPG pathway. Moreover, PTH1R is
abundantly expressed in DFCs, and interacts with both PTHrP and
PTH. Notably, mutations in PTH1R are associated with PFE.
Wnt/β-catenin, TGF-β and BMP signaling pathways are essential for
tooth development and eruption, with disruptions in these pathways
impairing osteoclast and osteoblast functions, and leading to
eruption disorders. Furthermore, disrupted Shh signaling
transmission between the epithelium and DF mesenchyme may also
impact tooth root development and eruption. Moreover, DF
abnormalities are clearly associated with various clinical
syndromes exhibiting tooth eruption disorder symptoms. These
include skull dysplasia, osteopetrosis, mucopolysaccharidosis VI,
enamel renal syndrome and DD, which are caused by mutations in
related genes. Moreover, conditions such as regional tooth
dysplasia, MCHDFs and some odontogenic cysts are not attributed to
genetic mutations or have an unknown etiology; instead, they mostly
arise from structural anomalies within the DF that mechanically
impede normal tooth eruption.
Future perspectives
A deeper understanding of the mechanisms involving
DFCs in tooth eruption is crucial. This present review may improve
knowledge and aid in resolving clinical issues related to the
regulation of tooth eruption. The application of single-cell
epigenomic technology may facilitate a more comprehensive
understanding of the epigenetic regulation governing DFPCs and
determine the patterns of epigenetic modifications that are
potentially implicated in tooth eruption disorders. Additionally,
the DF organoid model has emerged as an experimental paradigm for
investigating tooth development and regeneration in recent years
(166,167). The application of the this model
is expected to gradually expand. Currently, DF organoid models
mainly target tooth development issues in children and adolescents;
however, with technological advancements and the increase in
clinical practice, this model may also serve a significant role in
investigating tooth eruption. By integrating these techniques, we
aim to identify the potential molecular mechanisms of DFPCs in
tooth eruption disorders, and provide crucial theoretical support
and a scientific basis for future developments in tooth
regeneration treatments.
Acknowledgements
Not applicable.
Funding
This work was supported by the Key R&D Program of Zhejiang
(grant no. 2023C03072), the National Natural Science Foundation of
China (grant no. 81400511), the Zhejiang Provincial Natural Science
Foundation of China (grant no. LY18H140001), and the R&D
Program of the Stomatology Hospital of Zhejiang University School
of Medicine (grant no. RD2022JCEL04). XPC is sponsored by the
Zhejiang Provincial Program for the Cultivation of High-level
Innovative Health Talents.
Availability of data and materials
Not applicable.
Authors' contributions
WZ, XC and JC conceptualized the study. YY
validated the reliability of the topic selection. JC, YY, JL, HL,
ZS and YZ performed the literature review and wrote the manuscript.
WZ, XC, YoW and YaW completed the review and editing of the
manuscript. JC and YZ completed the supervision of the work. HL and
ZS participated in generating the figures. WZ conduct the project
administration. XC and WZ provided funding. Data authentication is
not applicable. All authors have read and approved the final
version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Use of artificial intelligence tools
During the preparation of this work, AI tools were
used to improve the readability and language of the manuscript, and
subsequently, the authors revised and edited the content produced
by the AI tools as necessary, taking full responsibility for the
ultimate content of the present manuscript.
References
|
1
|
Chai Y, Jiang X, Ito Y, Bringas P Jr, Han
J, Rowitch DH, Soriano P, McMahon AP and Sucov HM: Fate of the
mammalian cranial neural crest during tooth and mandibular
morphogenesis. Development. 127:1671–1679. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen G, Sun Q, Xie L, Jiang Z, Feng L, Yu
M, Guo W and Tian W: Comparison of the odontogenic differentiation
potential of dental follicle, dental papilla, and cranial neural
crest cells. J Endod. 41:1091–1099. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bastos VC, Gomez RS and Gomes CC:
Revisiting the human dental follicle: From tooth development to its
association with unerupted or impacted teeth and pathological
changes. Dev Dyn. 251:408–423. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wise GE and Yao S: Regional differences of
expression of bone morphogenetic protein-2 and RANKL in the rat
dental follicle. Eur J Oral Sci. 114:512–516. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhou T, Pan J, Wu P, Huang R, Du W, Zhou
Y, Wan M, Fan Y, Xu X, Zhou X, et al: Dental follicle cells: roles
in development and beyond. Stem Cells Int. 2019:91596052019.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Morsczeck C, Götz W, Schierholz J,
Zeilhofer F, Kühn U, Möhl C, Sippel C and Hoffmann KH: Isolation of
precursor cells (PCs) from human dental follicle of wisdom teeth.
Matrix Biol. 24:155–165. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bi R, Lyu P, Song Y, Li P, Song D, Cui C
and Fan Y: Function of dental follicle progenitor/stem cells and
their potential in regenerative medicine: From mechanisms to
applications. Biomolecules. 11:9972021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yao S, Pan F, Prpic V and Wise GE:
Differentiation of stem cells in the dental follicle. J Dent Res.
87:767–771. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu J, Yu F, Sun Y, Jiang B, Zhang W, Yang
J, Xu GT, Liang A and Liu S: Concise reviews: Characteristics and
potential applications of human dental tissue-derived mesenchymal
stem cells. Stem Cells. 33:627–638. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Morsczeck C, Völlner F, Saugspier M,
Brandl C, Reichert TE, Driemel O and Schmalz G: Comparison of human
dental follicle cells (DFCs) and stem cells from human exfoliated
deciduous teeth (SHED) after neural differentiation in vitro. Clin
Oral Investig. 14:433–440. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Richman JM: Shedding new light on the
mysteries of tooth eruption. Proc Natl Acad Sci USA. 116:353–355.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zeng L, He H, Sun M, Gong X, Zhou M, Hong
Y, Wu Y, Chen X and Chen Q: Runx2 and Nell-1 in dental follicle
progenitor cells regulate bone remodeling and tooth eruption. Stem
Cell Res Ther. 13:4862022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yu Y, Cui C, Guan SY, Xu RS, Zheng LW,
Zhou XD and Fan Y: Function of orofacial stem cells in tooth
eruption: An evolving perspective. Chin J Dent Res. 24:143–152.
2021.PubMed/NCBI
|
|
14
|
Suri L, Gagari E and Vastardis H: Delayed
tooth eruption: Pathogenesis, diagnosis, and treatment. A
literature review. Am J Orthod Dentofacial Orthop. 126:432–445.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Marks SC Jr and Cahill DR: Regional
control by the dental follicle of alterations in alveolar bone
metabolism during tooth eruption. J Oral Pathol. 16:164–169. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cahill DR and Marks SC Jr: Tooth eruption:
Evidence for the central role of the dental follicle. J Oral
Pathol. 9:189–200. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Roulias P, Kalantzis N, Doukaki D, Pachiou
A, Karamesinis K, Damanakis G, Gizani S and Tsolakis AI: Teeth
eruption disorders: A critical review. Children (Basel).
9:7712022.PubMed/NCBI
|
|
18
|
Rasmussen P and Kotsaki A: Inherited
retarded eruption in the permanent dentition. J Clin Pediatr Dent.
21:205–211. 1997.PubMed/NCBI
|
|
19
|
Raghoebar GM, Boering G, Vissink A and
Stegenga B: Eruption disturbances of permanent molars: A review. J
Oral Pathol Med. 20:159–166. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Raghoebar GM, Boering G and Vissink A:
Clinical, radiographic and histological characteristics of
secondary retention of permanent molars. J Dent. 19:164–170. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jain S, Raza M, Sharma P and Kumar P:
Unraveling impacted maxillary incisors: The why, when, and how. Int
J Clin Pediatr Dent. 14:149–157. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Morsczeck C, De Pellegrin M, Reck A and
Reichert TE: Evaluation of current studies to elucidate processes
in dental follicle cells driving osteogenic differentiation.
Biomedicines. 11:27872023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Oosterkamp BC, Ockeloen CW, Carels CE and
Kuijpers-Jagtman AM: Tooth eruption disturbances and syndromes. Ned
Tijdschr Tandheelkd. 121:233–238. 2014.(In Dutch). View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wise GE: Cellular and molecular basis of
tooth eruption. Orthod Craniofac Res. 12:67–73. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wise GE, Frazier-Bowers S and D'Souza RN:
Cellular, molecular, and genetic determinants of tooth eruption.
Crit Rev Oral Biol Med. 13:323–334. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li XX, Wang MT, Wu ZF, Sun Q, Ono N,
Nagata M, Zang XL and Ono W: Etiological mechanisms and
genetic/biological modulation related to PTH1R in primary failure
of tooth eruption. Calcif Tissue Int. Jun 4–2024.(Epub ahead of
print). View Article : Google Scholar
|
|
27
|
Guo X and Duan X: Genotype-phenotype
analysis of selective failure of tooth eruption-A systematic
review. Clin Genet. 104:287–297. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hanisch M, Hanisch L, Kleinheinz J and
Jung S: Primary failure of eruption (PFE): A systematic review.
Head Face Med. 14:52018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yamaguchi T, Hosomichi K, Shirota T,
Miyamoto Y, Ono W and Ono N: Primary failure of tooth eruption:
Etiology and management. Jpn Dent Sci Rev. 58:258–267. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Librizzi M, Naselli F, Abruscato G,
Luparello C and Caradonna F: Parathyroid hormone related protein
(PTHrP)-associated molecular signatures in tissue differentiation
and non-tumoral diseases. Biology (Basel). 12:9502023.PubMed/NCBI
|
|
31
|
Wysolmerski JJ, Broadus AE, Zhou J, Fuchs
E, Milstone LM and Philbrick WM: Overexpression of parathyroid
hormone-related protein in the skin of transgenic mice interferes
with hair follicle development. Proc Natl Acad Sci USA.
91:1133–1137. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wysolmerski JJ, McCaughern-Carucci JF,
Daifotis AG, Broadus AE and Philbrick WM: Overexpression of
parathyroid hormone-related protein or parathyroid hormone in
transgenic mice impairs branching morphogenesis during mammary
gland development. Development. 121:3539–3547. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vasavada RC, Cavaliere C, D'Ercole AJ,
Dann P, Burtis WJ, Madlener AL, Zawalich K, Zawalich W, Philbrick W
and Stewart AF: Overexpression of parathyroid hormone-related
protein in the pancreatic islets of transgenic mice causes islet
hyperplasia, hyperinsulinemia, and hypoglycemia. J Biol Chem.
271:1200–1208. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Foley J, Longely BJ, Wysolmerski JJ,
Dreyer BE, Broadus AE and Philbrick WM: PTHrP regulates epidermal
differentiation in adult mice. J Invest Dermatol. 111:1122–1128.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nagata M, Ono N and Ono W: Mesenchymal
progenitor regulation of tooth eruption: A view from PTHrP. J Dent
Res. 99:133–142. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang J, Liao L, Li Y, Xu Y, Guo W, Tian W
and Zou S: Parathyroid hormone-related peptide (1–34) promotes
tooth eruption and inhibits osteogenesis of dental follicle cells
during tooth development. J Cell Physiol. 234:11900–11911. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Obara N, Suzuki Y and Takeda M: Gene
expression of beta-catenin is up-regulated in inner dental
epithelium and enamel knots during molar tooth morphogenesis in the
mouse. Cell Tissue Res. 325:197–201. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
MacDonald BT, Tamai K and He X:
Wnt/beta-catenin signaling: Components, mechanisms, and diseases.
Dev Cell. 17:9–26. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wodarz A and Nusse R: Mechanisms of Wnt
signaling in development. Annu Rev Cell Dev Biol. 14:59–88. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ouyang H, McCauley LK, Berry JE, Saygin
NE, Tokiyasu Y and Somerman MJ: Parathyroid hormone-related protein
regulates extracellular matrix gene expression in cementoblasts and
inhibits cementoblast-mediated mineralization in vitro. J Bone
Miner Res. 15:2140–2153. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Philbrick WM, Dreyer BE, Nakchbandi IA and
Karaplis AC: Parathyroid hormone-related protein is required for
tooth eruption. Proc Natl Acad Sci USA. 95:11846–11851. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Heinrich J, Bsoul S, Barnes J, Woodruff K
and Abboud S: CSF-1, RANKL and OPG regulate osteoclastogenesis
during murine tooth eruption. Arch Oral Biol. 50:897–908. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ibáñez L, Nácher-Juan J, Terencio MC,
Ferrándiz ML and Alcaraz MJ: Osteostatin inhibits
M-CSF+RANKL-induced human osteoclast differentiation by modulating
NFATc1. Int J Mol Sci. 23:85512022. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shiyan H, Nanquan R, Shuhao X and Xiaobing
L: Research progress on the cellular and molecular mechanisms of
tooth eruption. Hua Xi Kou Qiang Yi Xue Za Zhi. 34:317–321.
2016.(In Chinese). PubMed/NCBI
|
|
45
|
Udagawa N, Koide M, Nakamura M, Nakamichi
Y, Yamashita T, Uehara S, Kobayashi Y, Furuya Y, Yasuda H, Fukuda C
and Tsuda E: Osteoclast differentiation by RANKL and OPG signaling
pathways. J Bone Miner Metab. 39:19–26. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Huang H, Wang J, Zhang Y, Zhu G, Li YP,
Ping J and Chen W: Bone resorption deficiency affects tooth root
development in RANKL mutant mice due to attenuated IGF-1 signaling
in radicular odontoblasts. Bone. 114:161–171. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Cui W, Cuartas E, Ke J, Zhang Q, Einarsson
HB, Sedgwick JD, Li J and Vignery A: CD200 and its receptor,
CD200R, modulate bone mass via the differentiation of osteoclasts.
Proc Natl Acad Sci USA. 104:14436–14441. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ono W, Sakagami N, Nishimori S, Ono N and
Kronenberg HM: Parathyroid hormone receptor signalling in
osterix-expressing mesenchymal progenitors is essential for tooth
root formation. Nat Commun. 7:112772016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Dean T, Vilardaga JP, Potts JT Jr and
Gardella TJ: Altered selectivity of parathyroid hormone (PTH) and
PTH-related protein (PTHrP) for distinct conformations of the
PTH/PTHrP receptor. Mol Endocrinol. 22:156–166. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Martin TJ, Sims NA and Seeman E:
Physiological and pharmacological roles of PTH and PTHrP in bone
using their shared receptor, PTH1R. Endocr Rev. 42:383–406. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Aziz S, Hermann NV, Dunø M, Risom L,
Daugaard-Jensen J and Kreiborg S: Primary failure of eruption of
teeth in two siblings with a novel mutation in the PTH1R gene. Eur
Arch Paediatr Dent. 20:295–300. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kanno CM, de Oliveira JA, Garcia JF, Roth
H and Weber BH: Twenty-year follow-up of a familial case of
PTH1R-associated primary failure of tooth eruption. Am J Orthod
Dentofacial Orthop. 151:598–606. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Frazier-Bowers SA, Simmons D, Wright JT,
Proffit WR and Ackerman JL: Primary failure of eruption and PTH1R:
The importance of a genetic diagnosis for orthodontic treatment
planning. Am J Orthod Dentofacial Orthop. 137:160–161. e1–e7. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Stutz C, Wagner D, Gros CI, Sayeh A,
Gegout H, Kuchler-Bopp S and Strub M: Primary failure of eruption
and tooth resorption. Orthod Fr. 93:283–288. 2022.(In French).
PubMed/NCBI
|
|
55
|
Decker E, Stellzig-Eisenhauer A, Fiebig
BS, Rau C, Kress W, Saar K, Rüschendorf F, Hubner N, Grimm T and
Weber BH: PTHR1 loss-of-function mutations in familial,
nonsyndromic primary failure of tooth eruption. Am J Hum Genet.
83:781–786. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wise GE and King GJ: Mechanisms of tooth
eruption and orthodontic tooth movement. J Dent Res. 87:414–434.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wise GE, Yao S and Henk WG: Bone formation
as a potential motive force of tooth eruption in the rat molar.
Clin Anat. 20:632–639. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Li J, Parada C and Chai Y: Cellular and
molecular mechanisms of tooth root development. Development.
144:374–384. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Takahashi A, Nagata M, Gupta A, Matsushita
Y, Yamaguchi T, Mizuhashi K, Maki K, Ruellas AC, Cevidanes LS,
Kronenberg HM, et al: Autocrine regulation of mesenchymal
progenitor cell fates orchestrates tooth eruption. Proc Natl Acad
Sci USA. 116:575–580. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Tokavanich N, Gupta A, Nagata M, Takahashi
A, Matsushita Y, Yatabe M, Ruellas A, Cevidanes L, Maki K,
Yamaguchi T, et al: A three-dimensional analysis of primary failure
of eruption in humans and mice. Oral Dis. 26:391–400. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wang XP: Tooth eruption without roots. J
Dent Res. 92:212–214. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Vuong LT and Mlodzik M: Different
strategies by distinct Wnt-signaling pathways in activating a
nuclear transcriptional response. Curr Top Dev Biol. 149:59–89.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Tokavanich N, Wein MN, English JD, Ono N
and Ono W: The role of Wnt signaling in postnatal tooth root
development. Front Dent Med. 2:7691342021. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Liu F, Chu EY, Watt B, Zhang Y, Gallant
NM, Andl T, Yang SH, Lu MM, Piccolo S, Schmidt-Ullrich R, et al:
Wnt/beta-catenin signaling directs multiple stages of tooth
morphogenesis. Dev Biol. 313:210–224. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Zhang R, Yang G, Wu X, Xie J, Yang X and
Li T: Disruption of Wnt/β-catenin signaling in odontoblasts and
cementoblasts arrests tooth root development in postnatal mouse
teeth. Int J Biol Sci. 9:228–236. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Weivoda MM, Ruan M, Hachfeld CM, Pederson
L, Howe A, Davey RA, Zajac JD, Kobayashi Y, Williams BO, Westendorf
JJ, et al: Wnt signaling inhibits osteoclast differentiation by
activating canonical and noncanonical cAMP/PKA pathways. J Bone
Miner Res. 31:65–75. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Wei W, Zeve D, Suh JM, Wang X, Du Y,
Zerwekh JE, Dechow PC, Graff JM and Wan Y: Biphasic and
dosage-dependent regulation of osteoclastogenesis by β-catenin. Mol
Cell Biol. 31:4706–4719. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Kim TH, Bae CH, Jang EH, Yoon CY, Bae Y,
Ko SO, Taketo MM and Cho ES: Col1a1-cre mediated activation of
β-catenin leads to aberrant dento-alveolar complex formation. Anat
Cell Biol. 45:193–202. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Glass DA II, Bialek P, Ahn JD, Starbuck M,
Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA and
Karsenty G: Canonical Wnt signaling in differentiated osteoblasts
controls osteoclast differentiation. Dev Cell. 8:751–764. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Nie B, Zhang SY, Guan SM, Zhou SQ and Fang
X: Role of Wnt/β-catenin pathway in the arterial medial
calcification and its effect on the OPG/RANKL system. Curr Med Sci.
39:28–36. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Kim TH, Lee JY, Baek JA, Lee JC, Yang X,
Taketo MM, Jiang R and Cho ES: Constitutive stabilization of
ß-catenin in the dental mesenchyme leads to excessive dentin and
cementum formation. Biochem Biophys Res Commun. 412:549–555. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Wu Y, Yuan X, Perez KC, Hyman S, Wang L,
Pellegrini G, Salmon B, Bellido T and Helms JA: Aberrantly elevated
Wnt signaling is responsible for cementum overgrowth and dental
ankylosis. Bone. 122:176–183. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Bennett CN, Longo KA, Wright WS, Suva LJ,
Lane TF, Hankenson KD and MacDougald OA: Regulation of
osteoblastogenesis and bone mass by Wnt10b. Proc Natl Acad Sci USA.
102:3324–3329. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Bennett CN, Ouyang H, Ma YL, Zeng Q, Gerin
I, Sousa KM, Lane TF, Krishnan V, Hankenson KD and MacDougald OA:
Wnt10b increases postnatal bone formation by enhancing osteoblast
differentiation. J Bone Miner Res. 22:1924–1932. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Thesleff I and Nieminen P: Tooth
morphogenesis and cell differentiation. Curr Opin Cell Biol.
8:844–850. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Sui BD, Zheng CX, Zhao WM, Xuan K, Li B
and Jin Y: Mesenchymal condensation in tooth development and
regeneration: A focus on translational aspects of organogenesis.
Physiol Rev. 103:1899–1964. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Wang Y, Cox MK, Coricor G, MacDougall M
and Serra R: Inactivation of Tgfbr2 in Osterix-Cre expressing
dental mesenchyme disrupts molar root formation. Dev Biol.
382:27–37. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Massagué J: TGF-beta signal transduction.
Annu Rev Biochem. 67:753–791. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Ko SO, Chung IH, Xu X, Oka S, Zhao H, Cho
ES, Deng C and Chai Y: Smad4 is required to regulate the fate of
cranial neural crest cells. Dev Biol. 312:435–447. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Gao Y, Yang G, Weng T, Du J, Wang X, Zhou
J, Wang S and Yang X: Disruption of Smad4 in odontoblasts causes
multiple keratocystic odontogenic tumors and tooth malformation in
mice. Mol Cell Biol. 29:5941–5951. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Beederman M, Lamplot JD, Nan G, Wang J,
Liu X, Yin L, Li R, Shui W, Zhang H, Kim SH, et al: BMP signaling
in mesenchymal stem cell differentiation and bone formation. J
Biomed Sci Eng. 6:32–52. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Fabregat I, Herrera B and Sánchez A:
Editorial special issue TGF-beta/BMP signaling pathway. Cells.
9:23632020. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Rakian A, Yang WC, Gluhak-Heinrich J, Cui
Y, Harris MA, Villarreal D, Feng JQ, Macdougall M and Harris SE:
Bone morphogenetic protein-2 gene controls tooth root development
in coordination with formation of the periodontium. Int J Oral Sci.
5:75–84. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Wang J, Muir AM, Ren Y, Massoudi D,
Greenspan DS and Feng JQ: Essential roles of bone morphogenetic
protein-1 and mammalian tolloid-like 1 in postnatal root dentin
formation. J Endod. 43:109–115. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Ge G and Greenspan DS: Developmental roles
of the BMP1/TLD metalloproteinases. Birth Defects Res C Embryo
Today. 78:47–68. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Malik Z, Roth DM, Eaton F, Theodor JM and
Graf D: Mesenchymal Bmp7 controls onset of tooth mineralization: A
novel way to regulate molar cusp shape. Front Physiol. 11:6982020.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Semba I, Nonaka K, Takahashi I, Takahashi
K, Dashner R, Shum L, Nuckolls GH and Slavkin HC:
Positionally-dependent chondrogenesis induced by BMP4 is
co-regulated by Sox9 and Msx2. Dev Dyn. 217:401–414. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Cai C, Wang J, Huo N, Wen L, Xue P and
Huang Y: Msx2 plays an important role in BMP6-induced osteogenic
differentiation of two mesenchymal cell lines: C3H10T1/2 and C2C12.
Regen Ther. 14:245–251. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Aïoub M, Lézot F, Molla M, Castaneda B,
Robert B, Goubin G, Néfussi JR and Berdal A: Msx2 -/- transgenic
mice develop compound amelogenesis imperfecta, dentinogenesis
imperfecta and periodental osteopetrosis. Bone. 41:851–859. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Hosoya A, Shalehin N, Takebe H, Shimo T
and Irie K: Sonic hedgehog signaling and tooth development. Int J
Mol Sci. 21:15872020. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Nakatomi M, Morita I, Eto K and Ota MS:
Sonic hedgehog signaling is important in tooth root development. J
Dent Res. 85:427–431. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Jain P and Rathee M: Anatomy, Head and
Neck, Tooth Eruption. StatPearls [Internet]. StatPearls Publishing;
Treasure Island, FL: 2024, https://www.ncbi.nlm.nih.gov/books/NBK549878/
|
|
93
|
Kasugai S, Suzuki S, Shibata S, Yasui S,
Amano H and Ogura H: Measurements of the isometric contractile
forces generated by dog periodontal ligament fibroblasts in vitro.
Arch Oral Biol. 35:597–601. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Kalliala E and Taskinen PJ: Cleidocranial
dysostosis. Report of six typical cases and one atypical case. Oral
Surg Oral Med Oral Pathol. 15:808–822. 1962. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Shih-Wei Cheng E, Tsuji M, Suzuki S and
Moriyama K: An overview of the intraoral features and craniofacial
morphology of growing and adult Japanese cleidocranial dysplasia
subjects. Eur J Orthod. 44:711–722. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Jaruga A, Hordyjewska E, Kandzierski G and
Tylzanowski P: Cleidocranial dysplasia and RUNX2-clinical
phenotype-genotype correlation. Clin Genet. 90:393–402. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Komori T: Regulation of proliferation,
differentiation and functions of osteoblasts by Runx2. Int J Mol
Sci. 20:16942019. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Harada H, Tagashira S, Fujiwara M, Ogawa
S, Katsumata T, Yamaguchi A, Komori T and Nakatsuka M: Cbfa1
isoforms exert functional differences in osteoblast
differentiation. J Biol Chem. 274:6972–6978. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Komori T, Yagi H, Nomura S, Yamaguchi A,
Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, et al:
Targeted disruption of Cbfa1 results in a complete lack of bone
formation owing to maturational arrest of osteoblasts. Cell.
89:755–764. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Otto F, Thornell AP, Crompton T, Denzel A,
Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen
BR, et al: Cbfa1, a candidate gene for cleidocranial dysplasia
syndrome, is essential for osteoblast differentiation and bone
development. Cell. 89:765–771. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Yoda S, Suda N, Kitahara Y, Komori T and
Ohyama K: Delayed tooth eruption and suppressed osteoclast number
in the eruption pathway of heterozygous Runx2/Cbfa1 knockout mice.
Arch Oral Biol. 49:435–442. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
D'Souza RN, Aberg T, Gaikwad J, Cavender
A, Owen M, Karsenty G and Thesleff I: Cbfa1 is required for
epithelial-mesenchymal interactions regulating tooth development in
mice. Development. 126:2911–2920. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Bronckers AL, Engelse MA, Cavender A,
Gaikwad J and D'Souza RN: Cell-specific patterns of Cbfa1 mRNA and
protein expression in postnatal murine dental tissues. Mech Dev.
101:255–258. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Liu Y, Sun X, Zhang X, Wang X, Zhang C and
Zheng S: RUNX2 mutation impairs osteogenic differentiation of
dental follicle cells. Arch Oral Biol. 97:156–164. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Nadyrshina DD and Khusainova RI: Clinical,
genetic aspects and molecular pathogenesis of osteopetrosis.
Vavilovskii Zhurnal Genet Selektsii. 27:383–392. 2023.PubMed/NCBI
|
|
106
|
Aker M, Rouvinski A, Hashavia S, Ta-Shma
A, Shaag A, Zenvirt S, Israel S, Weintraub M, Taraboulos A,
Bar-Shavit Z and Elpeleg O: An SNX10 mutation causes malignant
osteopetrosis of infancy. J Med Genet. 49:221–226. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Keng LT and Liang SK: Albers-Schönberg
disease. Korean J Intern Med. 34:1167–1168. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Luzzi V, Consoli G, Daryanani V, Santoro
G, Sfasciotti GL and Polimeni A: Malignant infantile osteopetrosis:
Dental effects in paediatric patients. Case reports. Eur J Paediatr
Dent. 7:39–44. 2006.PubMed/NCBI
|
|
109
|
Sobacchi C, Schulz A, Coxon FP, Villa A
and Helfrich MH: Osteopetrosis: Genetics, treatment and new
insights into osteoclast function. Nat Rev Endocrinol. 9:522–536.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Polgreen LE, Imel EA and Econs MJ:
Autosomal dominant osteopetrosis. Bone. 170:1167232023. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Wang H, Pan M, Ni J, Zhang Y, Zhang Y, Gao
S, Liu J, Wang Z, Zhang R, He H, et al: ClC-7 deficiency impairs
tooth development and eruption. Sci Rep. 6:199712016. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Xue Y, Wang W, Mao T and Duan X: Report of
two Chinese patients suffering from CLCN7-related osteopetrosis and
root dysplasia. J Craniomaxillofac Surg. 40:416–420. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Wise GE, Lumpkin SJ, Huang H and Zhang Q:
Osteoprotegerin and osteoclast differentiation factor in tooth
eruption. J Dent Res. 79:1937–1942. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Suzuki T, Suda N and Ohyama K:
Osteoclastogenesis during mouse tooth germ development is mediated
by receptor activator of NFKappa-B ligand (RANKL). J Bone Miner
Metab. 22:185–191. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Yasuda H: Discovery of the RANKL/RANK/OPG
system. J Bone Miner Metab. 39:2–11. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Morsczeck C, Moehl C, Götz W, Heredia A,
Schäffer TE, Eckstein N, Sippel C and Hoffmann KH: In vitro
differentiation of human dental follicle cells with dexamethasone
and insulin. Cell Biol Int. 29:567–575. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Nagpal R, Goyal RB, Priyadarshini K,
Kashyap S, Sharma M, Sinha R and Sharma N: Mucopolysaccharidosis: A
broad review. Indian J Ophthalmol. 70:2249–2261. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Smith KS, Hallett KB, Hall RK, Wardrop RW
and Firth N: Mucopolysaccharidosis: MPS VI and associated delayed
tooth eruption. Int J Oral Maxillofac Surg. 24:176–180. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Andersson HC: 50 Years ago in the journal
of pediatrics: Hurler's disease, Morquio's disease and related
mucopolysaccharidoses. J Pediatr. 167:3372015. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Costa-Motta FM, Bender F, Acosta A,
Abé-Sandes K, Machado T, Bomfim T, Boa Sorte T, da Silva D, Bittles
A, Giugliani R and Leistner-Segal S: A community-based study of
mucopolysaccharidosis type VI in Brazil: The influence of founder
effect, endogamy and consanguinity. Hum Hered. 77:189–196. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Vairo F, Federhen A, Baldo G, Riegel M,
Burin M, Leistner-Segal S and Giugliani R: Diagnostic and treatment
strategies in mucopolysaccharidosis VI. Appl Clin Genet. 8:245–255.
2015.PubMed/NCBI
|
|
122
|
Tomanin R, Karageorgos L, Zanetti A,
Al-Sayed M, Bailey M, Miller N, Sakuraba H and Hopwood JJ:
Mucopolysaccharidosis type VI (MPS VI) and molecular analysis:
Review and classification of published variants in the ARSB gene.
Hum Mutat. 39:1788–1802. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Alpöz AR, Coker M, Celen E, Ersin NK,
Gökçen D, van Diggelenc OP and Huijmansc JG: The oral
manifestations of Maroteaux-Lamy syndrome (mucopolysaccharidosis
VI): A case report. Oral Surg Oral Med Oral Pathol Oral Radiol
Endod. 101:632–637. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Simancas Escorcia V, Guillou C, Abbad L,
Derrien L, Rodrigues Rezende Costa C, Cannaya V, Benassarou M,
Chatziantoniou C, Berdal A, Acevedo AC, et al: Pathogenesis of
enamel-renal syndrome associated gingival fibromatosis: A proteomic
approach. Front Endocrinol (Lausanne). 12:7525682021. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Roomaney IA, Kabbashi S and Chetty M:
Enamel renal syndrome: Protocol for a scoping review. JMIR Res
Protoc. 10:e297022021. View
Article : Google Scholar : PubMed/NCBI
|
|
126
|
Crawford PJ, Aldred M and Bloch-Zupan A:
Amelogenesis imperfecta. Orphanet J Rare Dis. 2:172007. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Farias MLM, Ornela GO, de Andrade RS,
Martelli DRB, Dias VO and Júnior HM: Enamel renal syndrome: A
systematic review. Indian J Nephrol. 31:1–8. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Khalifa R, Kammoun R, Mansour L, Ben Alaya
T and Ghoul S: Enamel renal syndrome: A case report with
calcifications in pulp, gingivae, dental follicle and kidneys. Spec
Care Dentist. 44:722–728. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
de la Dure-Molla M, Quentric M, Yamaguti
PM, Acevedo AC, Mighell AJ, Vikkula M, Huckert M, Berdal A and
Bloch-Zupan A: Pathognomonic oral profile of enamel renal syndrome
(ERS) caused by recessive FAM20A mutations. Orphanet J Rare Dis.
9:842014. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Wang SK, Aref P, Hu Y, Milkovich RN,
Simmer JP, El-Khateeb M, Daggag H, Baqain ZH and Hu JC: FAM20A
mutations can cause enamel-renal syndrome (ERS). PLoS Genet.
9:e10033022013. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Wang SK, Reid BM, Dugan SL, Roggenbuck JA,
Read L, Aref P, Taheri AP, Yeganeh MZ, Simmer JP and Hu JC: FAM20A
mutations associated with enamel renal syndrome. J Dent Res.
93:42–48. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Nitayavardhana I, Theerapanon T,
Srichomthong C, Piwluang S, Wichadakul D, Porntaveetus T and
Shotelersuk V: Four novel mutations of FAM20A in amelogenesis
imperfecta type IG and review of literature for its genotype and
phenotype spectra. Mol Genet Genomics. 295:923–931. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Normand de la Tranchade I, Bonarek H,
Marteau JM, Boileau MJ and Nancy J: Amelogenesis imperfecta and
nephrocalcinosis: A new case of this rare syndrome. J Clin Pediatr
Dent. 27:171–175. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Alhilou A, Beddis HP, Mighell AJ and Durey
K: Dentin dysplasia: Diagnostic challenges. BMJ Case Rep.
2018:bcr20172239422018. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Shields ED, Bixler D and el-Kafrawy AM: A
proposed classification for heritable human dentine defects with a
description of a new entity. Arch Oral Biol. 18:543–553. 1973.
View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Akhil Jose EJ, Palathingal P, Baby D and
Thachil JM: Dentin dysplasia type I: A rare case report. J Oral
Maxillofac Pathol. 23:3092019. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Barron MJ, McDonnell ST, Mackie I and
Dixon MJ: Hereditary dentine disorders: Dentinogenesis imperfecta
and dentine dysplasia. Orphanet J Rare Dis. 3:312008. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Chen D, Li X, Lu F, Wang Y, Xiong F and Li
Q: Dentin dysplasia type I-a dental disease with genetic
heterogeneity. Oral Dis. 25:439–446. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Kalk WW, Batenburg RH and Vissink A:
Dentin dysplasia type I: Five cases within one family. Oral Surg
Oral Med Oral Pathol Oral Radiol Endod. 86:175–178. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Song YL and Bian Z: Recognition on dentin
dysplasia type II. Zhonghua Kou Qiang Yi Xue Za Zhi. 58:766–771.
2023.(In Chinese). PubMed/NCBI
|
|
141
|
Yang Q, Chen D, Xiong F, Chen D, Liu C,
Liu Y, Yu Q, Xiong J, Liu J, Li K, et al: A splicing mutation in
VPS4B causes dentin dysplasia I. J Med Genet. 53:624–633. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Bloch-Zupan A, Jamet X, Etard C, Laugel V,
Muller J, Geoffroy V, Strauss JP, Pelletier V, Marion V, Poch O, et
al: Homozygosity mapping and candidate prioritization identify
mutations, missed by whole-exome sequencing, in SMOC2, causing
major dental developmental defects. Am J Hum Genet. 89:773–781.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Xiong F, Ji Z, Liu Y, Zhang Y, Hu L, Yang
Q, Qiu Q, Zhao L, Chen D, Tian Z, et al: Mutation in SSUH2 causes
autosomal-dominant dentin dysplasia type I. Hum Mutat. 38:95–104.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Handa K, Saito M, Yamauchi M, Kiyono T,
Sato S, Teranaka T and Sampath Narayanan A: Cementum matrix
formation in vivo by cultured dental follicle cells. Bone.
31:606–611. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Li Q, Lu F, Chen T, Zhang K, Lu Y, Li X,
Wang Y, Liu L, Tian Q, Xiong F and Chen D: VPS4B mutation impairs
the osteogenic differentiation of dental follicle cells derived
from a patient with dentin dysplasia type I. Int J Oral Sci.
12:222020. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Zegarelli EV, Kutscher AH, Applebaum E and
Archard HO: Odontodysplasia. Oral Surg Oral Med Oral Pathol.
16:187–193. 1963. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Crawford PJ and Aldred MJ: Regional
odontodysplasia: A bibliography. J Oral Pathol Med. 18:251–263.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Nijakowski K, Woś P and Surdacka A:
Regional odontodysplasia: A systematic review of case reports. Int
J Environ Res Public Health. 19:16832022. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Alotaibi O, Alotaibi G and Alfawaz N:
Regional odontodysplasia: An analysis of 161 cases from 1953 to
2017. Saudi Dent J. 31:306–310. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Marques AC, Castro WH and do Carmo MA:
Regional odontodysplasia: An unusual case with a conservative
approach. Br Dent J. 186:522–524. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Rushton MA: Odontodysplasia: ‘Ghost
teeth’. Br Dent J. 119:109–113. 1965.PubMed/NCBI
|
|
152
|
Carlos R, Contreras-Vidaurre E, Almeida
OP, Silva KR, Abrahão PG, Miranda AM and Pires FR: Regional
odontodysplasia: morphological, ultrastructural, and
immunohistochemical features of the affected teeth, connective
tissue, and odontogenic remnants. J Dent Child (Chic). 75:144–150.
2008.PubMed/NCBI
|
|
153
|
Kerebel B, Kerebel LM, Heron D and Le
Cabellec MT: Regional odontodysplasia: New histopathological data.
J Biol Buccale. 17:121–128. 1989.PubMed/NCBI
|
|
154
|
Kerebel LM and Kerebel B: Soft-tissue
calcifications of the dental follicle in regional odontodysplasia:
A structural and ultrastructural study. Oral Surg Oral Med Oral
Pathol. 56:396–404. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Barbería E, Sanz Coarasa A, Hernández A
and Cardoso-Silva C: Regional odontodysplasia. A literature review
and three case reports. Eur J Paediatr Dent. 13:161–166.
2012.PubMed/NCBI
|
|
156
|
Mathew A, Dauravu LM, Reddy SN, Kumar KR
and Venkataramana V: Ghost teeth: Regional odontodysplasia of
maxillary first molar associated with eruption disorders in a
10-year-old girl. J Pharm Bioallied Sci. 7 (Suppl 2):S800–S803.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Sapp JP and Gardner DG: Regional
odontodysplasia: An ultrastructural and histochemical study of the
soft-tissue calcifications. Oral Surg Oral Med Oral Pathol.
36:383–392. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Gomez RS, Silva EC, Silva-Filho EC and
Castro WH: Multiple calcifying hyperplastic dental follicles. J
Oral Pathol Med. 27:333–334. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Gardner DG and Radden B: Multiple
calcifying hyperplastic dental follicles. Oral Surg Oral Med Oral
Pathol Oral Radiol Endod. 79:603–606. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Jamshidi S, Zargaran M and Mohtasham N:
Multiple calcifying hyperplastic dental follicle (MCHDF): A case
report. J Dent Res Dent Clin Dent Prospects. 7:174–176.
2013.PubMed/NCBI
|
|
161
|
Rodrigues LG, da Silva VB, Carmelo JC,
Khouri MS, Mendes PA and Manzi FR: An imaging perspective to
multiple calcifying hyperplastic dental follicles-a report of three
cases. Ann Maxillofac Surg. 12:227–230. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Ulutürk H, Yücel E, Akinci HO, Calisan EB,
Yildirim B and Gizli A: Multiple calcifying hyperplastic dental
follicles. J Stomatol Oral Maxillofac Surg. 120:77–79. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Davari D, Arzhang E and Soltani P:
Multiple calcifying hyperplastic dental follicles: A case report. J
Oral Maxillofac Surg. 77:757–761. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Fukuta Y, Totsuka M, Takeda Y and Yamamoto
H: Pathological study of the hyperplastic dental follicle. J Nihon
Univ Sch Dent. 33:166–173. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Cho YA, Yoon HJ, Hong SP, Lee JI and Hong
SD: Multiple calcifying hyperplastic dental follicles: Comparison
with hyperplastic dental follicles. J Oral Pathol Med. 40:243–249.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Hemeryck L, Hermans F, Chappell J,
Kobayashi H, Lambrechts D, Lambrichts I, Bronckaers A and
Vankelecom H: Organoids from human tooth showing epithelial
stemness phenotype and differentiation potential. Cell Mol Life
Sci. 79:1532022. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Hemeryck L, Lambrichts I, Bronckaers A and
Vankelecom H: Establishing organoids from human tooth as a powerful
tool toward mechanistic research and regenerative therapy. J Vis
Exp. 182:e636712022.PubMed/NCBI
|