Introduction
Sepsis is a life-threatening multiple organ failure
caused by an uncontrolled inflammatory response in response to
infection and has become a leading cause of mortality in intensive
care units (1,2). The lungs are the most vulnerable
organs in patients with sepsis, with >50% of these patients
developing acute lung injury (ALI) (3,4). ALI
often progresses into its severe form acute respiratory distress
(ARDS), which is associated with a high mortality rate globally
(5). Epidemiological data
demonstrate that >210,000 individuals in the US are diagnosed
with sepsis every year and the fatality rate of severe ARDS [the
ratio of the partial pressure of oxygen in the blood to the
fraction of inspired oxygen delivered (FIO2) <100]
approaches 40% in 50 countries in 2014 (6). Although current medical technologies
and pharmacological approaches including corticosteroids, omega
fatty acids, statins, have achieved notable improvements, effective
therapeutic drugs that shorten the duration of ventilation and
improve the mortality rate are limited (7). Therefore, it is necessary to fully
elucidate the molecular mechanism associated with initiation and
development of sepsis-induced ALI to identify effective
intervention targets.
Heat-shock proteins (HSPs) are a family of
structurally conserved proteins expressed at high levels in various
tissues, such as heart, muscle and brain, that protect the cell
from numerous stressors and stimuli (8). HSPs prevent and reduce apoptosis,
oxidative stress and human inflammatory diseases (9,10).
HSPB8, also known as HSP22, is a HSP expressed at high levels in
the myocardium, endothelium and motoneurons, and has been shown to
exert protective effects on sepsis-induced myocardial dysfunction,
diabetes-induced endothelial injury, and motoneuron diseases
(11–14). However, to the best of our
knowledge, the regulatory role of HSPB8 in lung disease has been
rarely investigated. In lung ischemia-reperfusion injury, HSPB8 was
demonstrated to inhibit cell apoptosis and lipid peroxidation,
indicating that increasing HSPB8 expression can protect lung cells
from external damage (15).
Notably, the levels of HSPB8 are elevated following sepsis, and the
upregulation of HSPB8 improves sepsis-induced myocardial
dysfunction and alleviates cognitive dysfunction in
sepsis-associated encephalopathy, suggesting a role of HSPB8 in
response to sepsis-associated diseases (13,16).
To the best of our knowledge, however, the specific roles of HSPB8
in sepsis-induced ALI and whether HSPB8 overexpression can
attenuate sepsis-induced ALI have not been reported.
In the present study, an in vitro
sepsis-induced ALI model was developed using lipopolysaccharide
(LPS)-induced A549 cells (17). To
the best of our knowledge, the present study was the first to
assess the regulatory role of HSPB8 in the LPS-induced inflammatory
response, oxidative stress and apoptosis in A549 cells, and its
mechanism of action. The present study aimed to provide a potential
novel approach for therapeutic intervention in sepsis-induced
ALI.
Materials and methods
Cell culture, treatment and
transfection
A549 human alveolar type II epithelial cells were
cultured in Ham's F-12K medium (both iCell Bioscience, Inc.)
supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.) and 1% penicillin/streptomycin in a 5/95%
CO2/air incubator at 37°C. A549 cells were treated with
1 µg/ml LPS (MilliporeSigma) for 24 h at 37°C to simulate
sepsis-induced ALI. To assess the molecular mechanism associated
with mitophagy, A549 cells were treated with 10 µM mitochondrial
division inhibitor-1 (Mdivi-1; Abcam) (18,19)
for 2 h at 37°C before LPS stimulation.
The full length of HSPB8 was cloned into the
pcDNA3.1 plasmid to construct the HSPB8 overexpression vector
(oe-HSPB8; Shanghai GenePharma Co., Ltd.). The empty pcDNA3.1
vector was used as a negative control (oe-NC; Shanghai GenePharma
Co., Ltd.). A549 cells were transfected with 15 nM oe-NC or
oe-HSPB8 using Lipofectamine® 3000 (Invitrogen; Thermo
Fisher Scientific, Inc.) at 37°C for 6 h according to the
manufacturer's instructions. At 48 h post-transfection, the
transfection efficacy was determined by western blotting.
Western blotting
Total protein was isolated from cells using RIPA
lysis buffer (Beyotime, Shanghai, China) containing protease
inhibitor cocktail (Roche Applied Science). Total protein was
quantified using a BCA Protein Assay kit (Pierce; Thermo Fisher
Scientific, Inc.), and 30 µg/lane protein was separated by sodium
dodecyl sulfate-polyacrylamide gel electrophoresis on a 12% gel and
transferred onto a polyvinylidene fluoride membrane
(MilliporeSigma). The membranes were blocked with 5% skimmed milk
at room temperature for 2 h, and probed with primary antibodies
against HSPB8 (1:2,000; cat. no. 15287-1-AP; Wuhan Sanying
Biotechnology), Bcl-2 (1:1,500; cat. no. 26593-1-AP; Wuhan Sanying
Biotechnology), Bax (1:8,000; cat. no. 50599-2-Ig; Wuhan Sanying
Biotechnology), caspase 3 (1:800; cat. no. 19677-1-AP; Wuhan
Sanying Biotechnology), Parkin (1:2,000; cat. no. 14060-1-AP; Wuhan
Sanying Biotechnology), cytochrome c oxidase (COX) IV (1:10,000;
cat. no. 11242-1-AP; Wuhan Sanying Biotechnology), PTEN-induced
kinase 1 (PINK1; 1:600; cat. no. 23274-1-AP; Wuhan Sanying
Biotechnology), LC3II/I (1:2,500; cat. no. 14600-1-AP; Wuhan
Sanying Biotechnology), Beclin-1 (1:1,000; cat. no. 11306-1-AP;
Wuhan Sanying Biotechnology), p62 (1:10,000; cat. no. 18420-1-AP;
Wuhan Sanying Biotechnology) and β-actin (1:5,000; cat. no.
20536-1-AP; Wuhan Sanying Biotechnology) at 4°C overnight.
Subsequently, the membranes were incubated with HRP-conjugated
secondary antibody (1:5,000; cat. no. SA00001-2; Wuhan Sanying
Biotechnology) for 2 h at room temperature. The bands were
visualized using an Amersham ECL Western Blotting Detection Kit
(Amersham; Cytiva) on a Bio-Rad ChemiDoc XRS+ System (Bio-Rad
Laboratories, Inc.) and quantified using ImageJ version 1.52
software (NIH, USA). COX IV was used to normalize protein
expression of Parkin and β-actin was used as the loading control
for other proteins.
ELISA
The levels of TNF-α, IL-1β and IL-6 in culture
medium were measured using Human TNF-alpha Quantikine ELISA Kit
(cat. no. STA00D), Human IL-1 beta/IL-1F2 Quantikine ELISA Kit
(cat. no. SLB50) and Human IL-6 Quantikine ELISA Kit (cat. no.
S6050B) according to the manufacturer's instructions (R&D
Systems, Inc.), respectively. The absorbance was measured at 450 nm
using a microplate reader (Bio-Rad Laboratories, Inc.).
Measurement of reactive oxygen species
(ROS), malondialdehyde (MDA), superoxide dismutase (SOD) and
catalase (CAT)
To assess ROS production, A549 cells were incubated
with 5 µM dichlorofluorescin-diacetate (MilliporeSigma) for 1 h at
37°C in the dark. Images were captured under a fluorescence
microscope (Olympus Corporation). Commercial kits from Nanjing
Jiancheng Bioengineering Institute were used to measure MDA content
(cat. no. A003-4-1), and the activities of SOD (cat. no. A001-3-2)
and CAT (cat. no. A007-1-1) in the culture medium according to the
manufacturer's instructions.
Flow cytometry
Cell apoptosis was examined using an Annexin V-FITC
cell apoptosis kit (Nanjing KeyGen Biotech Co., Ltd.) according to
the manufacturer's instructions. In brief, cells were washed with
pre-chilled PBS and resuspended with 500 µl 1X binding buffer.
Subsequently, 5 µl Annexin V-FITC and 10 µl PI were added to the
suspension. After mixing for 10 min at room temperature in the
dark, apoptotic cells (early + late apoptotic cells) were detected
using a FACS Canto™ II flow cytometer (Becton, Dickinson
and Company) and the data were analyzed using the Cell Quest
software (version 5.1; BD Biosciences).
Immunofluorescence assay
A549 cells (5×104 cells/ml) were seeded
on glass coverslips and stained with 100 µM MitoTracker Deep Red
(Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C for 10 min to
assess the mitochondrial morphology. A549 cells were washed with
PBS, fixed with 4% paraformaldehyde at room temperature for 10 min,
permeabilized with 0.2% Triton-X 100 for 5 min and blocked with 10%
normal goat serum (Solarbio Life Sciences, Beijing, China) at 37°C
for 1 h. Cells were incubated with primary antibodies against
Parkin (1:100; cat. no. 14060-1-AP; Wuhan Sanying Biotechnology) at
4°C overnight, followed by incubation with CoraLite488-conjugated
Goat Anti-Rabbit IgG secondary antibody (1:500; cat. no. SA00013-2;
Wuhan Sanying Biotechnology) at room temperature for 1 h in the
dark. The nuclei were counterstained with
4′,6-diamidino-2-phenylindole at room temperature for 5 min. Images
were captured under a fluorescence microscope (Olympus
Corporation).
Measurement of the mitochondrial
membrane potential
The mitochondrial membrane potential was detected by
JC-1 staining (Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. In brief, A549 cells were seeded into
6-well plates (2×105 cells/well) and treated with 1
µg/ml LPS at 37°C for 24 h, then 10 mg/ml JC-1 staining solution
was added to each well for 10 min at 3°C in the dark. Images were
captured under a fluorescence microscope (Olympus Corporation). Red
fluorescence indicated normal mitochondrial membrane potential and
green fluorescence indicated decreased mitochondrial membrane
potential.
Statistical analysis
All data are presented as the mean ± standard
deviation. All experiments were repeated at least three times. Data
were analyzed using Student's unpaired t-test or one-way analysis
of variance followed by Tukey's post hoc test. GraphPad Prism
(version 8.0; Dotmatics) was used for data analysis. P<0.05 was
considered to indicate a statistically significant difference.
Results
HSPB8 overexpression attenuates
LPS-induced inflammatory cytokine production and oxidative stress
in A549 cells
HSPB8 protein expression was significantly increased
in A549 cells following LPS stimulation compared with that in the
control group (Fig. 1A). To assess
the role of HSPB8 overexpression in attenuating LPS-induced ALI
in vitro, A549 cells were transfected with oe-HSPB8 to
overexpress HSPB8 before LPS stimulation. Successful transfection
was demonstrated by a significant increase in HSPB8 protein
expression in the oe-HSPB8 group compared with the oe-NC group
(Fig. 1B). The LPS group exhibited
a significant increase in HSPB8 protein expression compared with
the control group. HSPB8 protein expression in the LPS + oe-HSPB8
group was significantly increased compared with that in the LPS +
oe-NC group (Fig. 1C).
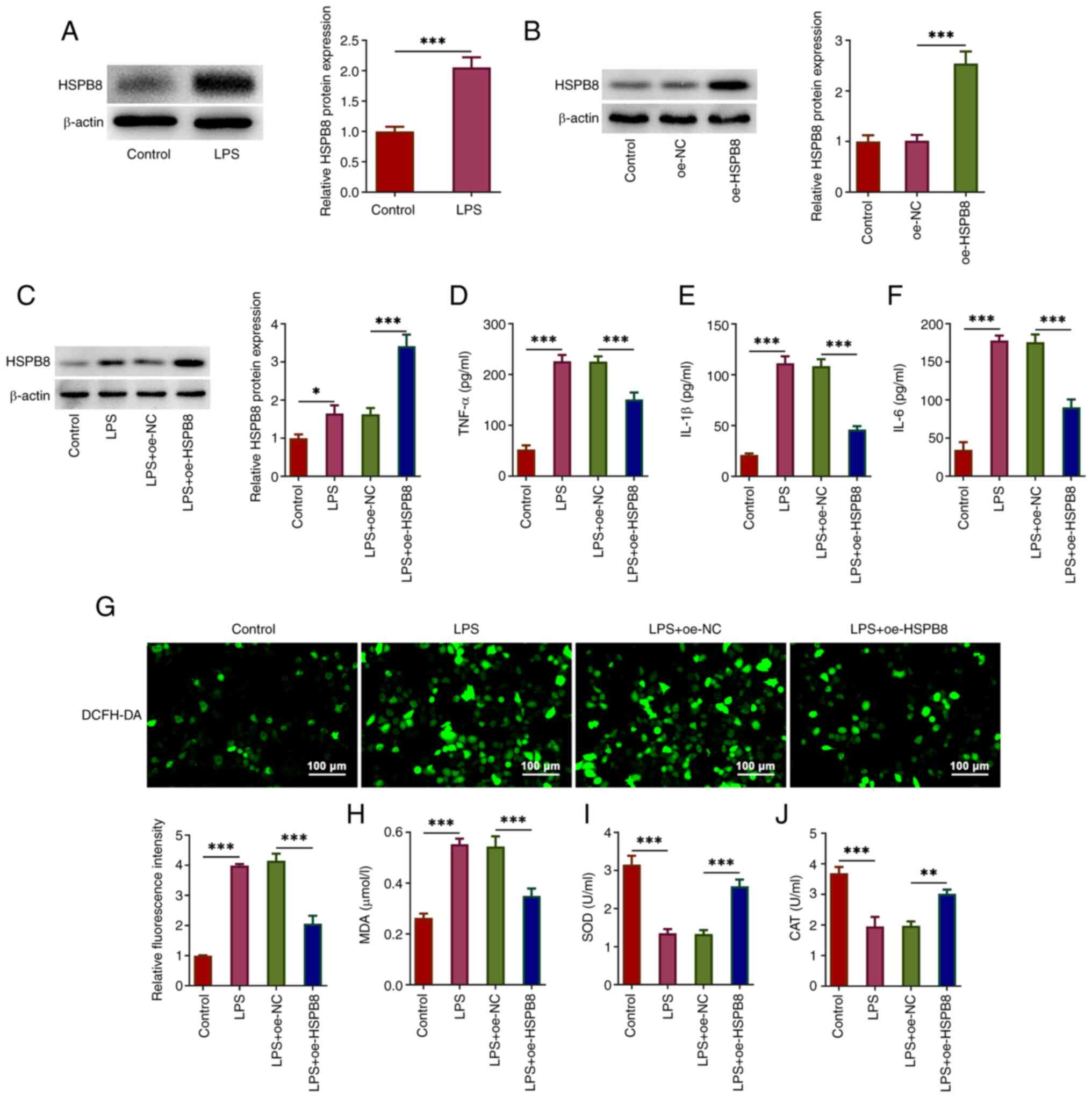 | Figure 1.HSPB8 overexpression attenuates
LPS-induced inflammatory cytokine levels and oxidative stress in
A549 cells. (A) A549 cells were stimulated by LPS to simulate
sepsis-induced acute lung injury. The protein expression levels of
HSPB8 were assessed by western blotting. (B) A549 cells were
transfected with oe-NC or oe-HSPB8, and the HSPB8 expression was
assessed. (C) A549 cells and HSPB8-overexpressing A549 cells were
treated with LPS. The protein expression levels of HSPB8 were
assessed using western blotting. The concentration of (D) TNF-α,
(E) IL-1β and (F) IL-6 in culture medium of A549 cells was detected
using ELISAs. (G) Production of intracellular reactive oxygen
species was detected by DCFH-DA staining. Scale bar, 100 µm. (H)
MDA content, and (I) SOD and (J) CAT activity were examined using
commercial kits. *P<0.05, **P<0.01 and ***P<0.001. CAT,
catalase; DCFH-DA, 2′-7′-dichlorodihydrofluorescein diacetate; HSP,
heat-shock protein; LPS, lipopolysaccharide; MDA malondialdehyde;
NC, negative control; oe, overexpression; SOD, superoxide
dismutase. |
ELISAs demonstrated that LPS treatment significantly
increased the production of TNF-α, IL-1β and IL-6 in A549 cells
compared with that in the control group. The LPS + oe-HSPB8 group
exhibited significantly decreased TNF-α, IL-1β and IL-6 levels
compared with the LPS + oe-NC group (Fig. 1D-F), suggesting that HSPB8 could
alleviate LPS-induced inflammatory cytokine production in A549
cells. Furthermore, increased ROS fluorescence intensity was
observed in the LPS group, with decreased fluorescence intensity in
the LPS + oe-HSPB8 group (Fig.
1G). MDA content was significantly increased, and SOD and CAT
activities were significantly decreased in the LPS group compared
with the control group, indicating the occurrence of oxidative
stress in LPS-induced A549 cells. However, this was mitigated by
oe-HSPB8 transfection, with significantly decreased MDA levels, and
significantly increased SOD and CAT activity compared with the LPS
+ oe-NC group (Fig. 1H-J). These
results suggested that HSPB8 could partly decrease LPS-induced
oxidative stress in A549 cells.
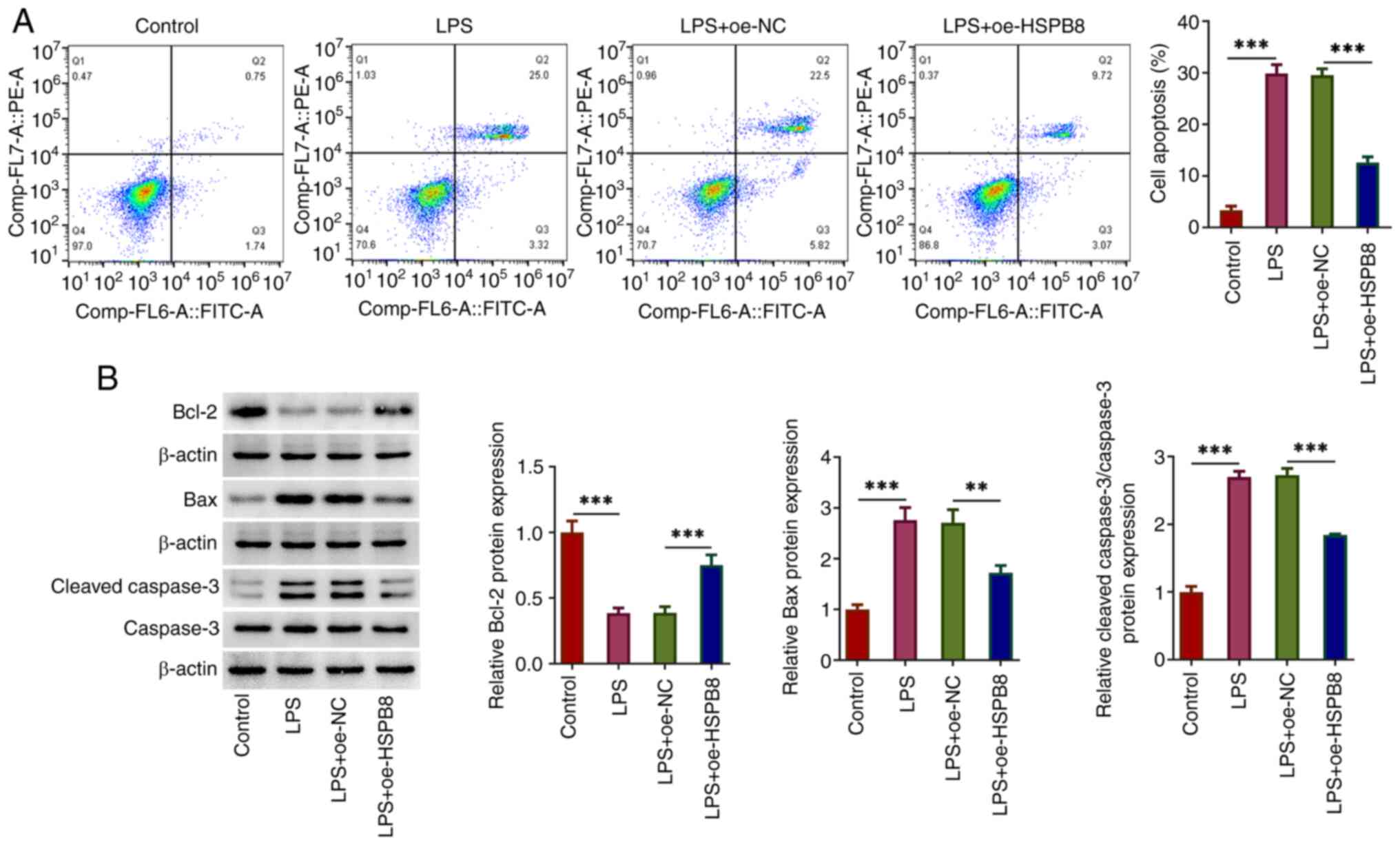
HSPB8 overexpression decreases
LPS-induced apoptosis in A549 cells
The impact of HSPB8 on LPS-induced apoptosis in A549
cells was assessed. According to the results of the flow cytometry
analysis, LPS significantly promoted cell apoptosis compared with
the control group, which was partly blocked by HSPB8 overexpression
compared with the LPS + oe-NC group (Fig. 2A). LPS significantly decreased the
expression levels of the anti-apoptotic protein Bcl-2, and
significantly increased the expression levels of the pro-apoptotic
proteins Bax and cleaved-caspase 3 compared with those in the
control group. These changes were significantly reversed by
oe-HSPB8 transfection (Fig. 2B).
Therefore, oe-HSPB8 transfection exerted anti-apoptotic activity in
A549 cells exposed to LPS.
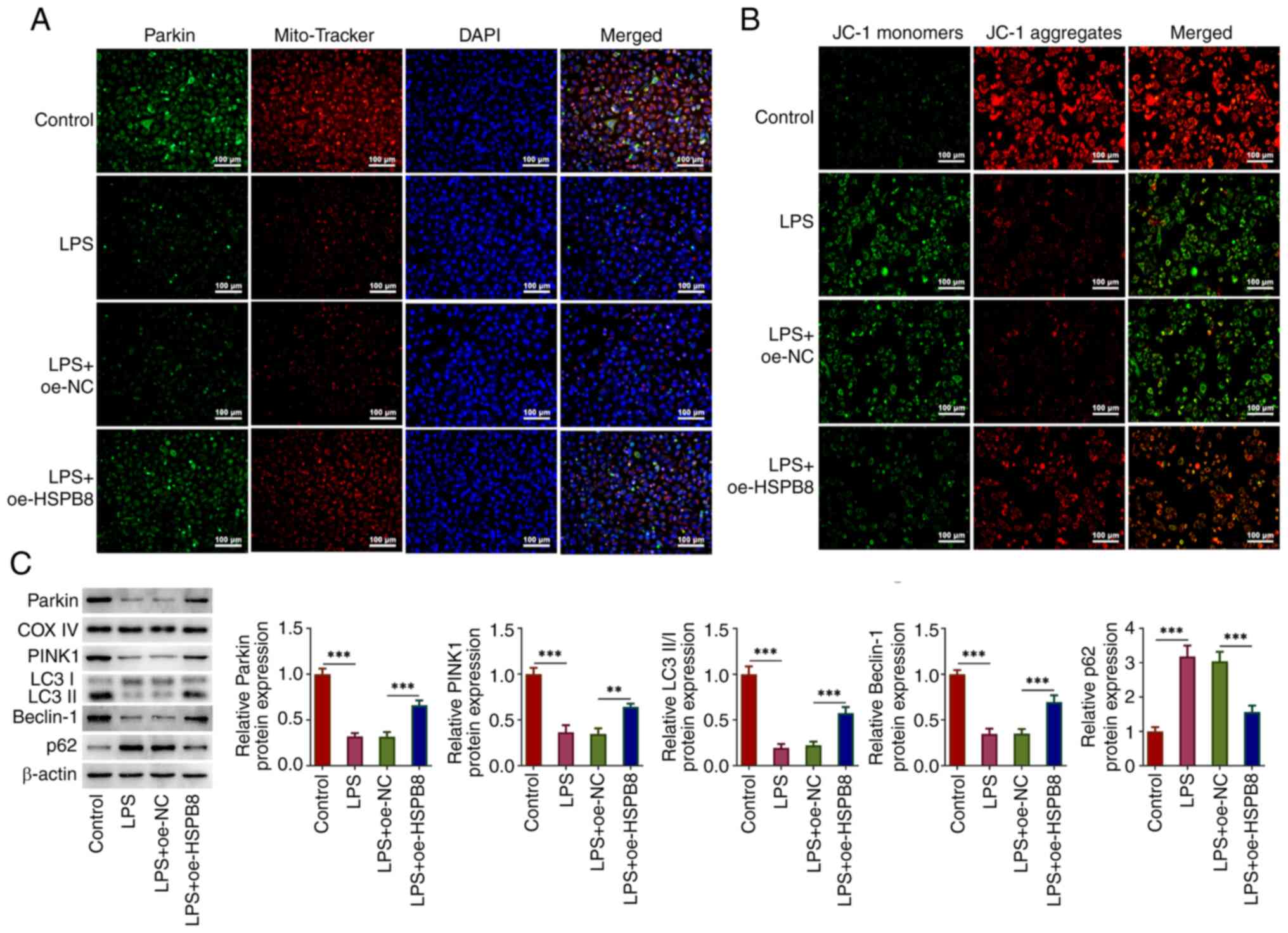
HSPB8 overexpression activates
mitophagy in LPS-exposed A549 cells
The potential regulatory mechanism of HSPB8 was
subsequently investigated. MitoTracker was used to indicate the
location of mitochondria. Parkin fluorescence signal was markedly
reduced following LPS treatment and partly restored by oe-HSPB8
transfection (Fig. 3A). Compared
to the control group, the low red immunofluorescent intensity in
LPS group revealed that LPS caused a reduction of the mitochondrial
membrane potential, which was partly restored by oe-HSPB8
transfection as the red immunofluorescent intensity was enhanced
compared with the LPS + oe-NC group (Fig. 3B). Furthermore, the protein levels
of Parkin, PINK1, LC3II/I and Beclin-1 were significantly
decreased, and the protein expression levels of p62 were
significantly increased in the LPS group compared with the control
group; however, this was mitigated by oe-HSPB8 transfection, with
significantly decreased p62 expression, and significantly increased
protein levels of Parkin, PINK1, LC3II/I and Beclin-1 compared with
the LPS + oe-NC group (Fig. 3C).
This suggested that mitophagy was inhibited in LPS-induced A549
cells, whereas oe-HSPB8 transfection activated mitophagy.
Mdivi-1 decreases the inhibitory
effects of HSPB8 on the inflammatory response, oxidative stress and
apoptosis in LPS-treated A549 cells
Finally, the role of mitophagy in the protective
mechanism of HSPB8 against LPS-mediated cell injury was confirmed
using the mitophagy inhibitor Mdivi-1. The inhibitory effects of
oe-HSPB8 transfection on TNF-α, IL-1β and IL-6 in LPS-treated A549
cells were significantly inhibited by additional treatment with
Mdivi-1, evidenced by elevated concentrations of TNF-α, IL-1β and
IL-6 in the Mdivi-1 + LPS + oe-HSPB8 group compared with the LPS +
oe-HSPB8 group (Fig. 4A-C).
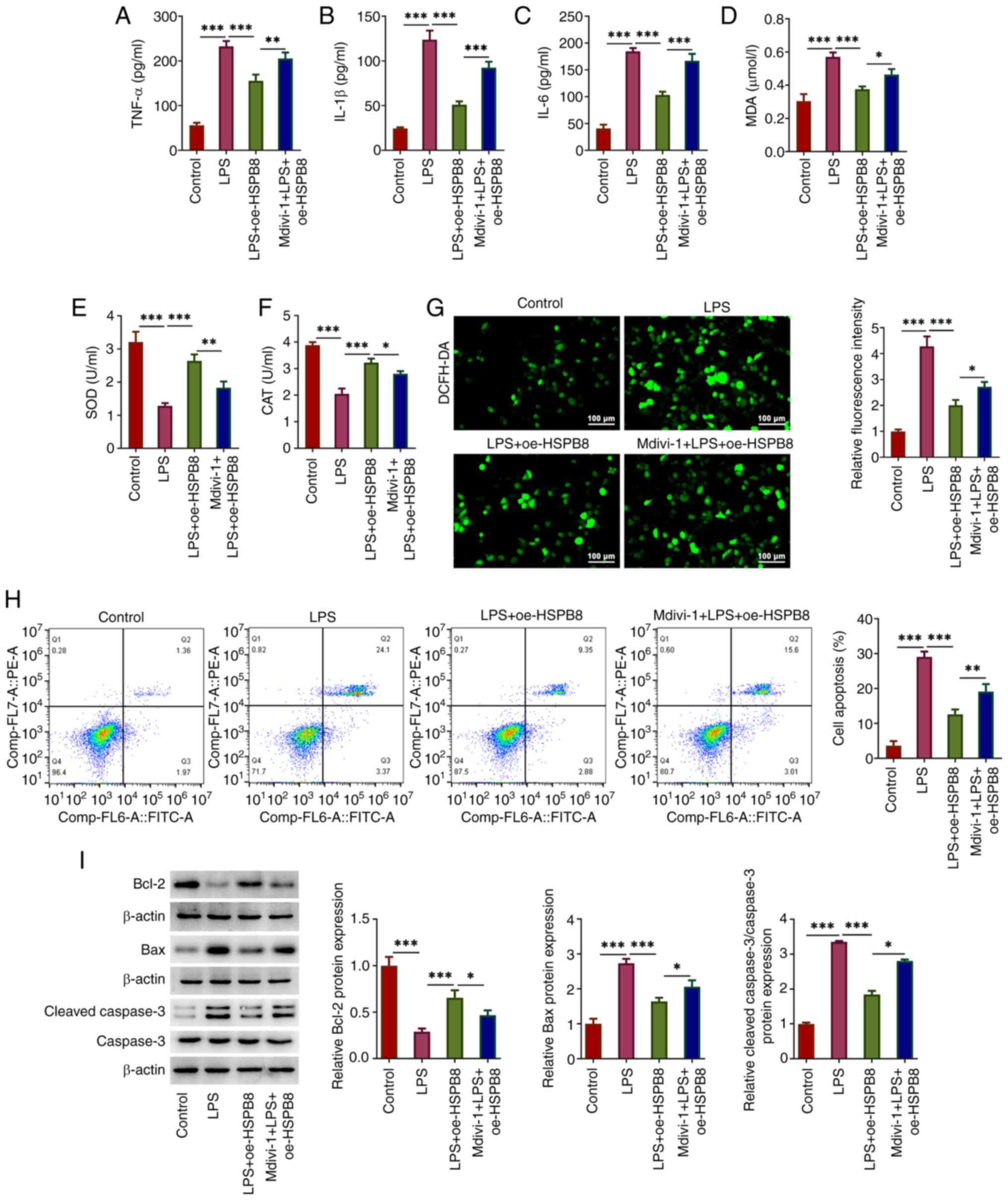 | Figure 4.Mdivi-1 decreases the inhibitory
effects of HSPB8 on the inflammatory response, oxidative stress and
apoptosis in LPS-exposed A549 cells. The concentration of (A)
TNF-α, (B) IL-1β and (C) IL-6 in the culture medium of A549 cells
was detected using ELISAs. (D) MDA content, and (E) SOD and (F) CAT
activities were measured using commercial kits. (G) Production of
intracellular ROS was detected by DCFH-DA staining. Scale bar, 100
µm. (H) Flow cytometry was performed to investigate the cell
apoptosis rate. (I) Expression levels of apoptosis-related proteins
were assessed using western blotting. *P<0.05, **P<0.01 and
***P<0.001. CAT, catalase; DCFH-DA,
2′-7′-dichlorodihydrofluorescein diacetate; HSP, heat-shock
protein; LPS, lipopolysaccharide; MDA malondialdehyde; Mdivi-1,
mitochondrial division inhibitor-1; oe, overexpression; ROS,
reactive oxygen species; SOD, superoxide dismutase. |
MDA levels were significantly increased, and SOD and
CAT activity was significantly decreased in the Mdivi-1 + LPS +
oe-HSPB8 group compared with the LPS + oe-HSPB8 group (Fig. 4D-F). An increase in ROS
fluorescence was observed in the Mdivi-1 + LPS + oe-HSPB8 group
compared with the LPS + oe-HSPB8 group (Fig. 4G), suggesting that Mdivi-1 partly
reversed the inhibitory effect of HSPB8 on oxidative stress in
LPS-exposed A549 cells. Furthermore, the cell apoptosis rate in the
Mdivi-1 + LPS + oe-HSPB8 group was significantly increased compared
with that in the LPS + oe-HSPB8 group (Fig. 4H), accompanied by significantly
increased Bax and cleaved-caspase 3 expression, and significantly
decreased Bcl-2 protein expression in the Mdivi-1 + LPS + oe-HSPB8
group compared with the LPS + oe-HSPB8 group (Fig. 4I), demonstrating that Mdivi-1
decreased the anti-apoptotic effect of oe-HSPB8 transfection.
Discussion
Sepsis is a life-threatening condition with a
complex pathological mechanism. The sepsis-mediated inflammatory
response causes alveolar epithelial cell damage, epithelial barrier
dysfunction and fluid extravasation into the alveolar space,
ultimately leading to alveolar epithelial cell death and ALI
progression (20). LPS is the
primary component of the outer membrane of Gram-negative bacteria.
As a primary pathogenic factors of sepsis, LPS could trigger the
inflammatory cascade, inducing necrosis and apoptosis of epithelial
cells (21). Accordingly,
LPS-induced A549 cells were used in the present study to establish
an in vitro cellular model that simulates sepsis-induced
ALI.
It is widely recognized that oxidative stress and
inflammatory responses serve roles in facilitating sepsis-induced
ALI, and decreasing excessive production of ROS and proinflammatory
cytokines can attenuate pathological injury of lung tissues and
improve the survival rate of mice receiving a lethal dose of LPS
(22–24). Furthermore, as a major form of cell
death, apoptosis is involved in the pathogenesis of sepsis-induced
ALI (25). Decreasing cell
apoptosis is an option to improve sepsis-associated pulmonary
epithelial barrier dysfunction (26). At present, numerous potential
targets have been demonstrated to exert protective effects against
sepsis-stimulated ALI due to anti-inflammatory, anti-oxidation and
anti-apoptotic properties. For example, topiroxostat has been
reported to inhibit oxidative stress, inflammation and apoptosis,
and decrease lung damage in a rat model of sepsis (27). Protocatechuic acid could
effectively counteract sepsis-mediated lung injury by reducing the
inflammatory response, oxidative stress and apoptotic events, with
the potential to alleviate sepsis-induced ALI (28). HSPB8 serves a role in
cytoprotection and resistance to oxidative stress and inflammation
(13,14). Yu et al (14) reported that HSPB8 attenuated
diabetes-induced endothelial injury by decreasing mitochondrial ROS
generation. Yu et al (13)
reported that HSPB8 alleviated LPS-induced myocardial injury by
inhibiting inflammation, oxidative stress and apoptosis in
cardiomyocytes. In agreement with the aforementioned studies, the
present study demonstrated that LPS treatment increased HSPB8
expression in A549 cells. oe-HSPB8 transfection mitigated the
LPS-induced inflammatory response, oxidative stress and apoptosis
in A549 cells, suggesting that HSBP8 may attenuate sepsis-induced
ALI. This is in line with a previous study, which has reported that
increased HSPB8 expression can protect lung cells from external
damage (15). A previous study
reported that HSPB8 deficiency increased mitochondrial oxidative
stress and mitochondrial damage, while HSPB8 overexpression
inhibited mitochondrial oxidative stress and impairment in A549
cells (29), further confirming
that HSPB8 exerts a cytoprotective function in A549 cells under
both normal and inflammatory conditions.
Mitophagy removes damaged mitochondria via autophagy
and serves a role in maintaining mitochondrial homeostasis and cell
survival (30). Mitophagy is
associated with sepsis-induced ALI (31–33).
The PINK1/Parkin signaling is essential to maintain mitochondrial
quality control through activating mitophagy. During this process,
PINK1 can promote the translocation of Parkin from the cytoplasm to
mitochondria for mitophagy activation (34). Hydrogen has been demonstrated to
relieve sepsis-induced ALI by promoting PINK1/Parkin-mediated
mitophagy (35). Kahweol (a
natural diterpene extracted from coffee beans) treatment can
alleviate oxidative stress and the inflammatory response in
sepsis-induced ALI by increasing mitophagy and improving
mitochondrial homeostasis (34).
Previous studies have reported that HSPB8 was directly involved in
mitophagy and attenuated myocardial ischemia-reperfusion injury
through mitophagy (36,37). Consistently, in the present study,
mitophagy was inhibited in LPS-treated A549 cells, demonstrated by
decreased expression levels of Parkin, PINK1, Beclin-1 and LC3II/I,
and increased p62 expression. oe-HSPB8 transfection increased
mitophagy through enhancing Parkin, PINK1, Beclin-1 and LC3II/I,
and reducing p62 expression and inhibited the inflammatory
response, oxidative stress and apoptosis in LPS-induced A549 cells,
suggesting that the protective role of HSPB8 against sepsis-induced
ALI impacts mitophagy. The present study demonstrated that the
inhibitory effects of oe-HSPB8 transfection on LPS-induced
oxidative stress, the inflammatory response and apoptosis in
LPS-induced A549 cells were mitigated by treatment with Mdivi-1,
suggesting that HSPB8 alleviated sepsis-induced ALI by activating
mitophagy.
The present study had a number of limitations.
Firstly, the present study only assessed the protective mechanism
of oe-HSPB8 transfection; in future, this should be confirmed by
HSPB8 knockdown to achieve comprehensive understanding of the
molecular function of HSPB8 in lung injury. Secondly, the present
study only assessed the effect of oe-HSPB8 transfection on A549
cells under inflammatory conditions; the effect of oe-HSPB8
transfection under normal conditions should also be evaluated.
Furthermore, in vivo studies are required to validate the
present findings and develop drugs targeting HSPB8 for clinical
treatment of sepsis-related ALI.
In summary, the present study reported the
regulatory role of HSPB8 in sepsis-induced ALI. oe-HSPB8
transfection could attenuate the LPS-mediated inflammatory
response, oxidative stress and apoptosis in A549 cells by promoting
mitophagy. HSPB8 may serve as a potential therapeutic target in
sepsis-induced ALI, and drugs targeting HSPB8 may be potential
candidates for the clinical treatment of sepsis-associated ALI.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
XZ contributed to study conception and design. Data
collection and analysis were performed by XZ, MW, MS and NY. The
manuscript was drafted by MW and revised by XZ. XZ and MW confirm
the authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rudd KE, Johnson SC, Agesa KM, Shackelford
KA, Tsoi D, Kievlan DR, Colombara DV, Ikuta KS, Kissoon N, Finfer
S, et al: Global, regional, and national sepsis incidence and
mortality, 1990-2017: Analysis for the global burden of disease
study. Lancet. 395:200–211. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Markwart R, Saito H, Harder T, Tomczyk S,
Cassini A, Fleischmann-Struzek C, Reichert F, Eckmanns T and
Allegranzi B: Epidemiology and burden of sepsis acquired in
hospitals and intensive care units: A systematic review and
meta-analysis. Intensive Care Med. 46:1536–1551. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jiang W, Ma C, Bai J and Du X: Macrophage
SAMSN1 protects against sepsis-induced acute lung injury in mice.
Redox Biol. 56:1024322022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Singer M, Deutschman CS, Seymour CW,
Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche
JD, Coopersmith CM, et al: The third international consensus
definitions for sepsis and septic shock (sepsis-3). JAMA.
315:801–810. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pham T and Rubenfeld GD: Fifty years of
research in ARDS. The epidemiology of acute respiratory distress
syndrome. A 50th birthday review. Am J Respir Crit Care Med.
195:860–870. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Englert JA, Bobba C and Baron RM:
Integrating molecular pathogenesis and clinical translation in
sepsis-induced acute respiratory distress syndrome. JCI Insight.
4:e1240612019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
He YQ, Deng JL, Zhou CC, Jiang SG, Zhang
F, Tao X and Chen WS: Ursodeoxycholic acid alleviates
sepsis-induced lung injury by blocking PANoptosis via STING
pathway. Int Immunopharmacol. 125:1111612023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
de Thonel A, Le Mouël A and Mezger V:
Transcriptional regulation of small HSP-HSF1 and beyond. Int J
Biochem Cell Biol. 44:1593–1612. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kalmar B and Greensmith L: Induction of
heat shock proteins for protection against oxidative stress. Adv
Drug Deliv Rev. 61:310–318. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ikwegbue PC, Masamba P, Oyinloye BE and
Kappo AP: Roles of heat shock proteins in apoptosis, oxidative
stress, human inflammatory diseases, and cancer. Pharmaceuticals
(Basel). 11:22017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li F, Xiao H, Hu Z, Zhou F and Yang B:
Exploring the multifaceted roles of heat shock protein B8 (HSPB8)
in diseases. Eur J Cell Biol. 97:216–229. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rusmini P, Cristofani R, Galbiati M,
Cicardi ME, Meroni M, Ferrari V, Vezzoli G, Tedesco B, Messi E,
Piccolella M, et al: The role of the heat shock protein B8 (HSPB8)
in motoneuron diseases. Front Mol Neurosci. 10:1762017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yu Y, Hu LL, Liu L, Yu LL, Li JP, Rao JA,
Zhu LJ, Bao HH and Cheng XS: Hsp22 ameliorates
lipopolysaccharide-induced myocardial injury by inhibiting
inflammation, oxidative stress, and apoptosis. Bioengineered.
12:12544–12554. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yu L, Liang Q, Zhang W, Liao M, Wen M,
Zhan B, Bao H and Cheng X: HSP22 suppresses diabetes-induced
endothelial injury by inhibiting mitochondrial reactive oxygen
species formation. Redox Biol. 21:1010952019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang S, Tian J, Zhang F, Liu A, Xie B and
Chen Q: The protective effects of heat shock protein 22 in lung
ischemia-reperfusion injury mice. Biochem Biophys Res Commun.
512:698–704. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ling J, Yu S, Xiong F and Li S: HSPB8
up-regulation alleviates cognitive dysfunction in a mouse model of
sepsis-associated encephalopathy. Int Immunopharmacol.
122:1104482023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nan CC, Zhang N, Cheung KCP, Zhang HD, Li
W, Hong CY, Chen HS, Liu XY, Li N and Cheng L: Knockdown of lncRNA
MALAT1 alleviates LPS-induced acute lung injury via inhibiting
apoptosis through the miR-194-5p/FOXP2 axis. Front Cell Dev Biol.
8:5868692020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Givvimani S, Munjal C, Tyagi N, Sen U,
Metreveli N and Tyagi SC: Mitochondrial division/mitophagy
inhibitor (Mdivi) ameliorates pressure overload induced heart
failure. PLoS One. 7:e323882012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ji Y, Leng Y, Lei S, Qiu Z, Ming H, Zhang
Y, Zhang A, Wu Y and Xia Z: The mitochondria-targeted antioxidant
MitoQ ameliorates myocardial ischemia-reperfusion injury by
enhancing PINK1/Parkin-mediated mitophagy in type 2 diabetic rats.
Cell Stress Chaperones. 27:353–367. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Park I, Kim M, Choe K, Song E, Seo H,
Hwang Y, Ahn J, Lee SH, Lee JH, Jo YH, et al: Neutrophils disturb
pulmonary microcirculation in sepsis-induced acute lung injury. Eur
Respir J. 53:18007862019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li K, He Z, Wang X, Pineda M, Chen R, Liu
H, Ma K, Shen H, Wu C, Huang N, et al: Apigenin C-glycosides of
Microcos paniculata protects lipopolysaccharide induced apoptosis
and inflammation in acute lung injury through TLR4 signaling
pathway. Free Radic Biol Med. 124:163–175. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen R, Cao C, Liu H, Jiang W, Pan R, He
H, Ding K and Meng Q: Macrophage Sprouty4 deficiency diminishes
sepsis-induced acute lung injury in mice. Redox Biol.
58:1025132022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang HH, Duan JX, Liu SK, Xiong JB, Guan
XX, Zhong WJ, Sun CC, Zhang CY, Luo XQ, Zhang YF, et al: A
COX-2/sEH dual inhibitor PTUPB alleviates
lipopolysaccharide-induced acute lung injury in mice by inhibiting
NLRP3 inflammasome activation. Theranostics. 10:4749–4761. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jiang L, Yang D, Zhang Z, Xu L, Jiang Q,
Tong Y and Zheng L: Elucidating the role of Rhodiola rosea L. In
sepsis-induced acute lung injury via network pharmacology: Emphasis
on inflammatory response, oxidative stress, and the PI3K-AKT
pathway. Pharm Biol. 62:272–284. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jiang WY, Ren J, Zhang XH, Lu ZL, Feng HJ,
Yao XL, Li DH, Xiong R, Fan T and Geng Q: CircC3P1 attenuated
pro-inflammatory cytokine production and cell apoptosis in acute
lung injury induced by sepsis through modulating miR-21. J Cell Mol
Med. 24:11221–11229. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li X, Jamal M, Guo P, Jin Z, Zheng F, Song
X, Zhan J and Wu H: Irisin alleviates pulmonary epithelial barrier
dysfunction in sepsis-induced acute lung injury via activation of
AMPK/SIRT1 pathways. Biomed Pharmacother. 118:1093632019.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fu H, Zhang J and Huang M: Topiroxostat
ameliorates oxidative stress and inflammation in sepsis-induced
lung injury. Z Naturforsch C J Biosci. 75:425–431. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Alsharif KF, Almalki AA, Alsanie WF,
Alzahrani KJ, Kabrah SM, Elshopakey GE, Alghamdi AAA, Lokman MS,
Sberi HA, Bauomy AA, et al: Protocatechuic acid attenuates
lipopolysaccharide-induced septic lung injury in mice: The possible
role through suppressing oxidative stress, inflammation and
apoptosis. J Food Biochem. 45:e139152021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yu LL, Wang Y, Xiao ZK and Chen SS: Heat
shock protein B8 promotes proliferation and migration in lung
adenocarcinoma A549 cells by maintaining mitochondrial function.
Mol Cell Biochem. 476:187–197. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shefa U, Jeong NY, Song IO, Chung HJ, Kim
D, Jung J and Huh Y: Mitophagy links oxidative stress conditions
and neurodegenerative diseases. Neural Regen Res. 14:749–756. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mannam P, Shinn AS, Srivastava A, Neamu
RF, Walker WE, Bohanon M, Merkel J, Kang MJ, Dela Cruz CS, Ahasic
AM, et al: MKK3 regulates mitochondrial biogenesis and mitophagy in
sepsis-induced lung injury. Am J Physiol Lung Cell Mol Physiol.
306:L604–L619. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mohsin M, Tabassum G, Ahmad S, Ali S and
Ali Syed M: The role of mitophagy in pulmonary sepsis.
Mitochondrion. 59:63–75. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhu QJ, Wang J, Li Y, Bai ZJ, Guo XB and
Pan T: PRKCA promotes mitophagy through the miR-15a-5p/PDK4 axis to
relieve sepsis-induced acute lung injury. Infect Immun.
91:e00465222023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li G, Fu T, Wang W, Xiong R, Liu B, He R,
Xu C, Wang W, Li N and Geng Q: Pretreatment with kahweol attenuates
sepsis-induced acute lung injury via improving mitochondrial
homeostasis in a CaMKKII/AMPK-dependent pathway. Mol Nutr Food Res.
67:e23000832023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen H, Lin H, Dong B, Wang Y, Yu Y and
Xie K: Hydrogen alleviates cell damage and acute lung injury in
sepsis via PINK1/Parkin-mediated mitophagy. Inflamm Res.
70:915–930. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li F, Tan J, Zhou F, Hu Z and Yang B: Heat
shock protein B8 (HSPB8) reduces oxygen-glucose
deprivation/reperfusion injury via the induction of mitophagy. Cell
Physiol Biochem. 48:1492–1504. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cheng J, Ji M, Jing H and Lin H: DUSP12
ameliorates myocardial ischemia-reperfusion injury through
HSPB8-induced mitophagy. J Biochem Mol Toxicol. 37:e233102023.
View Article : Google Scholar : PubMed/NCBI
|