Heart failure leads to increased morbidity and
mortality and cardiac hypertrophy is an independent risk factor for
the development of heart failure (1,2).
Cardiac hypertrophy results from long-term pressure overload and is
characterized by the enlargement of myocardial cells and enhanced
contractility, which enable the heart to maintain normal blood
pumping ability (1,3,4).
Chronic concentric hypertrophy can lead to changes in the
expression of hypertrophy-related genes such as Nppa, Nppb
and myosin heavy chain 7, systolic dysfunction and extracellular
remodeling (4,5). Reactivation of Nppa and
Nppb can promote sodium excretion, lower blood pressure and
ameliorate the burden on the heart, resulting in antihypertrophic
effects. Long-term cardiac hypertrophy eventually leads to heart
failure (2,6). Studies on the molecular mechanisms of
cardiac hypertrophy are beneficial for the prevention and treatment
of heart failure.
Previous studies have shown that small GTPases, a
known type of molecular switch in cells, play a critical role in
cardiac hypertrophy (7,8). They have a molecular weight of only
20–30 kDa and GTPase activity (9).
These proteins can be divided into five families based on their
amino acid sequence: Ras, Rho, Rab, Arf and Ran (9,10).
Since the discovery of the Ras protein in the early 1980s, more
than 100 types of small GTPases have been identified (10,11).
Small GTPases are in GDP-bound (inactive) and GTP-bound (active)
states, which links upstream signaling molecules with downstream
effectors to mediate cell proliferation, differentiation,
transportation and cytoskeleton regulation (12). Ras mainly regulates cell signaling
pathways, including the Ras/mitogen-activated protein kinase kinase
kinase (MEKK)/c-Jun N-terminal kinase (JNK) and p38
mitogen-activated protein kinase (MAPK) pathways and plays
important roles in cell proliferation, differentiation, morphology
and apoptosis (13–17). Rho is not only involved in
regulating actin, cell polarity, cell migration, vesicle transport
and cytokinesis, but is also involved in hematopoiesis, especially
in the classical and noncanonical Wnt signaling pathways (18–21).
Rab is involved in vesicle and endocytic membrane trafficking and
can regulate plasma membrane delivery, organelle biogenesis and
degradation pathways, lysosomes and autophagy (22). In addition to its basic role in
membrane transport, Arf also regulates mitosis, plasma membrane
signaling, ciliary transport and lipid droplet functions (23). Ran mainly controls the entry and
exit of cargoes between the cytoplasm and nucleus via the nuclear
pore complex (24).
Since the discovery of the role of Ras in cardiac
hypertrophy in 1993, small GTPases, mainly Ras, Rho and Rab, have
been confirmed to be involved in cardiac hypertrophy. Ras can
mediate cardiac hypertrophy through multiple pathways, including
the MAPK and Ca2+/calcineurin/nuclear factor of
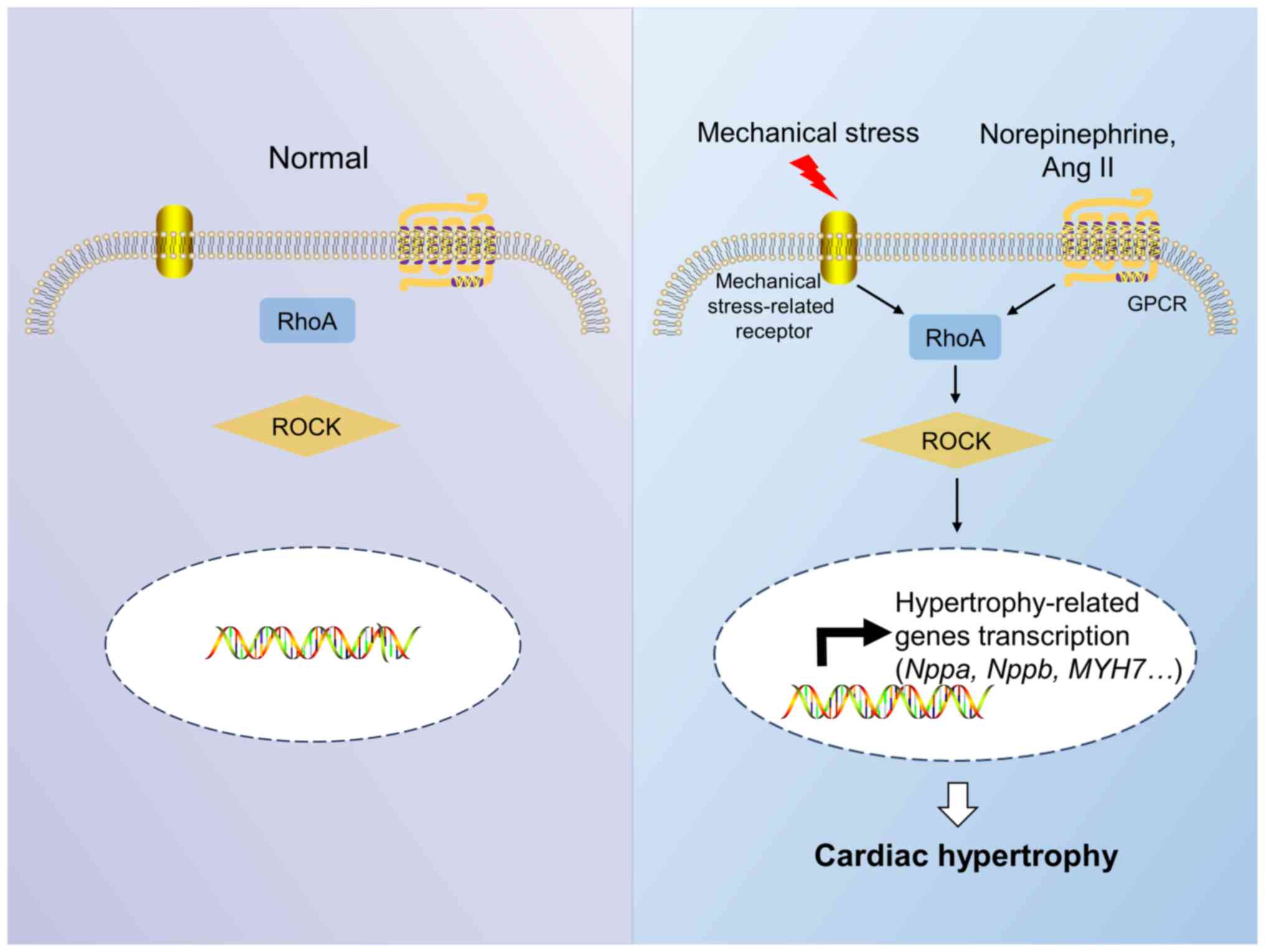
activated T cells (NFAT) pathways (14). The Rho A/Rho-associated protein
kinase (ROCK) pathway is the main pathway involved in Rho
A-mediated cardiac hypertrophy (25). Rac1, a member of the Rho family,
can cause cardiac hypertrophy by regulating the MAPK signaling
pathway and nicotinamide adenine dinucleotide phosphate (NADPH)
oxidase activity (26–30). By contrast, Cdc42 can antagonize
cardiac hypertrophy by activating JNK and inhibiting
calcineurin-NFAT activity (31).
At present, Rab1a, Rab4 and Rab4a from the Rab family have been
found to lead to myocardial hypertrophy (32). However, the relationships between
the other two small GTPases (Arf and Ran) and cardiac hypertrophy
remain unreported. The present study reviewed recent research
progress on the roles of small GTPases in cardiac hypertrophy,
which is helpful for the prevention and treatment of cardiac
hypertrophy and heart failure.
The Ras family contains 36 members that can be
classified into six types: Ras, Ral, Rit, Rap, Rheb and Rad (Ras
associated with diabetes) (14).
Ras is involved in regulating cell division, cell migration,
adhesion, differentiation and apoptosis (Table I) (33). Ral can regulate neuronal
plasticity, the immune response and glucose and lipid homeostasis
(34). Rit is involved in cell
transformation, cell survival, neuromorphogenesis and the
regulation of dopamine transporters and plays important roles in
autism and schizophrenia (35).
Rap plays a role in the normal tissue system, cancer, immune system
and hematopoietic system (36).
Rheb is highly expressed in the brain, can activate mammalian
target of rapamycin complex 1 in response to a variety of growth
factor stimuli and is associated with cell growth, protein
synthesis and regeneration (37,38).
Rad acts as a calcium channel regulator in the normal heart by
interacting with cardiac L-type calcium (Table I) (39). Ras and Rad play important roles in
cardiac hypertrophy (14,40). However, the relationships between
the other four members of the Ras subfamily and cardiac hypertrophy
are still unknown.
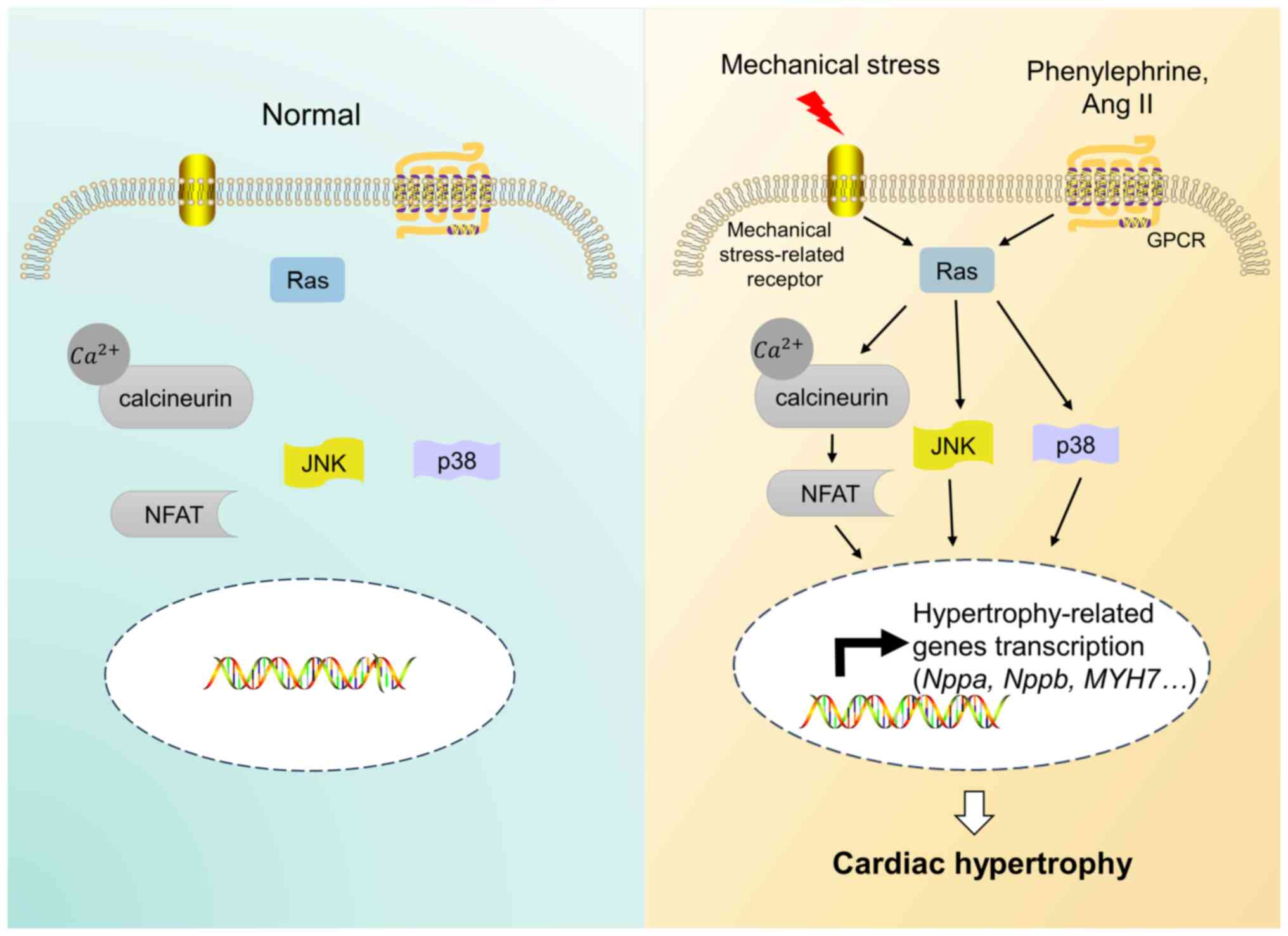
Ras can induce cardiac hypertrophy mainly through
activating the JNK, p38 MAPK and Ca2+/calcineurin/NFAT
signaling pathways both in vitro and in vivo
(Fig. 1) (15,17,43,44).
The activation of the Ras/MEKK/JNK pathway can mediate ANF gene
expression, which causes cardiac hypertrophy both in vitro
and in vivo (15). MEKK is
a serine triphosphate kinase that can be activated by active Ras
in vitro and can regulate stress-activated protein kinases
and mitogen-activated protein kinases (45,46).
The activation of JNK by MEKK can promote c-Jun transcriptional
activity and upregulate ANF gene expression in
phenylephrine-treated cardiomyocytes in vitro (15). In activated Ras transgenic mice,
JNK activity is upregulated in the left ventricle, which can result
in increased ventricular ANF and cardiac hypertrophy in vivo
(15). Mechanical stress-activated
integrins can trigger autophosphorylation of focal adhesion kinase
(FAK) at Tyr397, which can promote the interaction between FAK and
c-Src in vitro (17).
Tyr925 of FAK is then phosphorylated by c-Src (17). It has been confirmed that the
phosphorylation of Tyr397 and Tyr925 in FAK is required for p38
MAPK activation (17). In
addition, p38 MAPK activation induced by integrins also requires
activated Ras (17). Therefore, it
has been suggested that mechanical stress can activate p38 MAPK
through the integrin/FAK/Src/Ras pathway to induce cardiomyocyte
hypertrophy in vitro (17).
In addition, the downregulation of Ras can inhibit the calcineurin
activity stimulated by phenylephrine and its upregulation can lead
to an increase in the activity of the cytosolic transcription
factor NFAT in vitro, which indicates that Ras can activate
the Ca2+/calcineurin/NFAT signaling pathway in
cardiomyocytes (44). Rad was
originally identified in the skeletal muscle of patients with type
2 diabetes (47). Rad is
significantly downregulated in pressure overload and
phenylephrine-induced cardiac hypertrophy and decreasing Rad can
lead to significant enhancement of the phosphorylation and activity
of CaMKII, which induces cardiac hypertrophy both in vitro
and in vivo (40).
Therefore, Rad can antagonize cardiac hypertrophy by inhibiting the
activity of CaMKII (40).
Statins or farnesyltransferase inhibitors can
suppress Ras by intervening with its farnesylation or
geranylgeranylation at its carboxy terminus both in vitro
and in vivo (48–50). Previous studies have shown that
simvastatin can inhibit the progression of left ventricular
hypertrophy and that the farnesyltransferase inhibitor FTI-276 can
ameliorate cardiac remodeling in experimental animal models
(48–50). However, it has been proven that
statins may cause muscle problems, liver dysfunction, renal
insufficiency, poor uptake and short persistence in clinical
application (51). A study on
farnesyltransferase inhibitors is still at the experimental stage
and they have not undergone clinical tests (49). At present, there are fewer
effective Ras-targeting drugs with few side effects. Therefore, it
is worth developing more Ras-targeting drugs to prevent cardiac
hypertrophy.
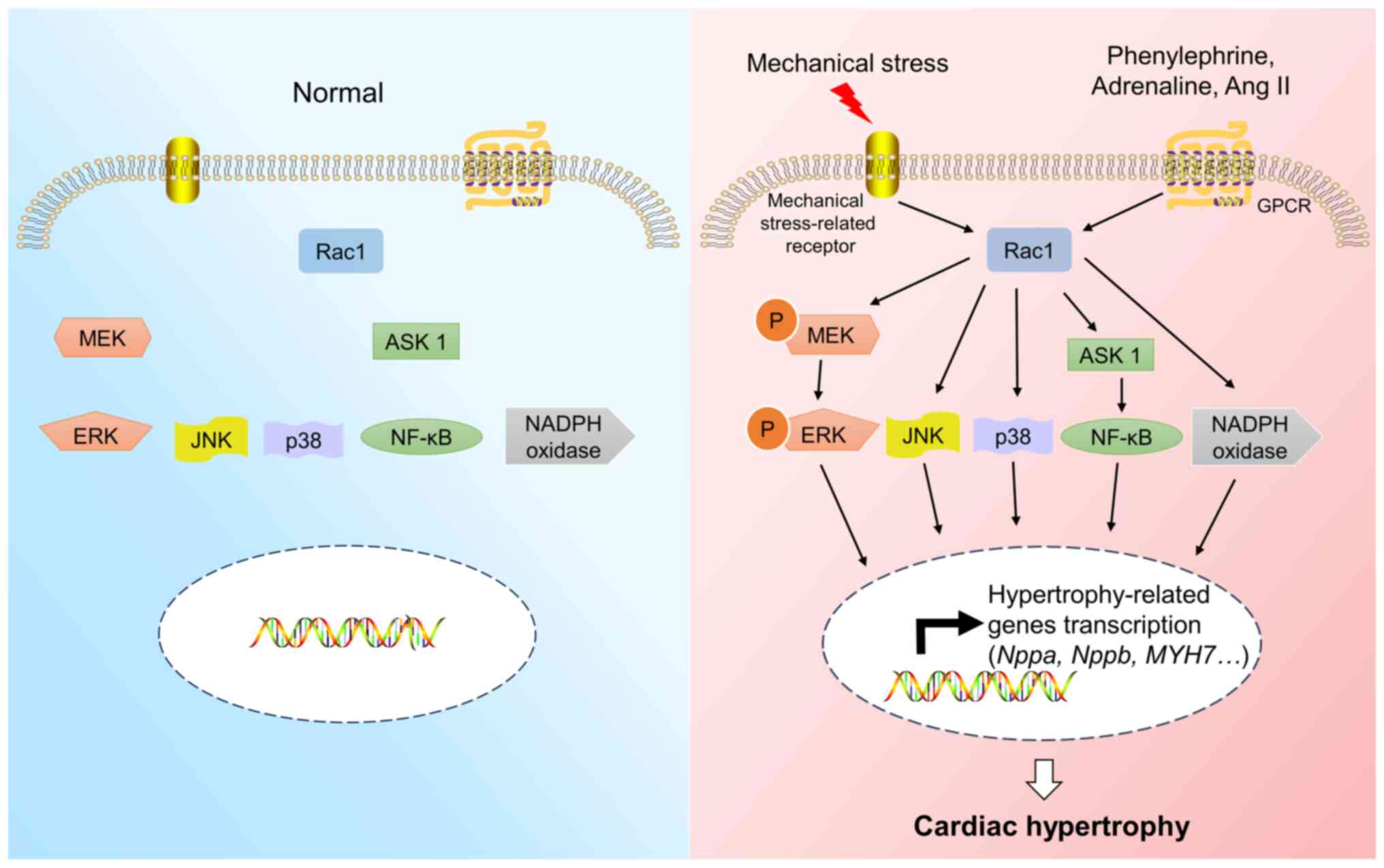
Rac1 can induce cardiac hypertrophy mainly through
the MAPK pathway, the apoptosis signal-regulated kinase 1
(ASK1)/NF-κB pathway and NADPH oxidase both in vitro and
in vivo (26–30,71,72).
To reveal the role of Rac1 in cardiac hypertrophy, an activating
mutant (V12 Rac1) and a negative mutant (N17 Rac1) were used in a
previous in vitro study (71). The overexpression of V12 Rac1 can
cause cardiac hypertrophy, while that of N17 Rac1 can inhibit the
hypertrophic responses induced by phenylephrine stimulation
(71). Previous studies have shown
that Rac1 can mediate cardiac hypertrophy by phosphorylating
mitogen-activated protein kinase kinase 1/2 (MEK1/2) and
extracellular signal-regulated kinase (ERK)1/2 (Fig. 3) both in vitro and in
vivo (26,73). The inhibition of the Rac1/JNK
pathway can reduce the reactivation of ANF and ameliorate
cardiomyocyte hypertrophy in vitro (27,74).
Mechanical stress can induce cardiac hypertrophy through the
Rac1/reactive oxygen species (ROS)/p38 MAPK pathway in vitro
(30). In addition, the
overexpression of Rac1 can induce the activation of ASK1 and NF-κB,
which leads to cardiomyocyte hypertrophy in vitro (28). ASK1 and NF-κB play important roles
in mediating the hypertrophic response of cardiomyocytes (28). NADPH oxidase activity is also
closely linked to cardiac hypertrophy induced by Rac1 both in
vitro and in vivo (29,75).
Rac1 can regulate NADPH oxidase activity through interaction with
its components, including gp91phox and
p67phox and the upregulation of NADPH oxidase activity
can lead to the development of cardiac hypertrophy (29).
The use of Rho-targeting medicines for treating
cardiac hypertrophy has been reported. Statins are powerful cardiac
protectants that have been found to inhibit cardiac hypertrophy by
blocking Rho isopentenylation as inhibitors of
3-hydroxy-3-methylglutaryl-CoA reductase both in vitro and
in vivo (76,77). 1,25-Dihydroxyvitamin D3 (VitD)
exerts its anti-cardiac hypertrophy effect by downregulating Rac1
in vivo (78). Alendronate,
a farnesyl pyrophosphate synthase inhibitor, can inhibit
angiotensin (Ang) II-induced neonatal cardiomyocyte hypertrophy by
inactivating RhoA both in vitro and in vivo (79). In addition, the RhoA inhibitor
Clostridium botulinum C3 exozyme and the ROCK inhibitor
Y-27632 exhibit anti-cardiac hypertrophy effects in vitro
(80). Glucagon like peptide 1 may
reverse cardiac hypertrophy by inhibiting the RhoA/ROCK2 signaling
pathway both in vitro and in vivo (81). The selective ROCK inhibitor fasudil
can treat various cardiovascular diseases, such as cerebral
ischemia and pulmonary hypertension, but its effect on cardiac
hypertrophy remains unknown (82,83).
The Rac1 inhibitor NSC23766 can antagonize cardiac hypertrophy
induced by active Rac1 both in vitro and in vivo
(26). There is no Cdc42-targeting
medicine for preventing cardiac hypertrophy. The aforementioned
Rho-targeting drugs and inhibitors do not undergo clinical tests
for treating cardiac hypertrophy and further studies are needed to
reveal their detailed roles in ameliorating cardiac
hypertrophy.
The Rab gene is widely distributed and encodes at
least 60 different members of the Rab family in humans (84,85).
The Rab family is mainly divided into 10 subfamilies: Rab1, Rab3,
Rab4, Rab5, Rab6, Rab8, Rab11, Rab22, Rab27 and Rab40 (85,86).
Rab1 is involved in vesicular trafficking from the endoplasmic
reticulum to the Golgi apparatus and its dysregulation is
associated with the development of cardiomyopathy (87). Rab3 can promote large dense-core
vesicle fusion (88). Rab4 can
mediate protein transport from the endosome to the plasma membrane
(Table I) (89). Rab5 plays a critical role in the
early steps of endocytosis (90).
Rab6 can regulate not only anterograde transport pathways from the
Golgi apparatus but also retrograde trafficking pathways to the
Golgi apparatus (91). Rab8 can
mediate anterograde membrane trafficking and plays an important
role in cell morphogenesis and cell migration (92). Rab11 can regulate the recycling of
endosomal cargo proteins to the plasma membrane and is involved in
cell migration (93,94). Rab22 plays a critical role in early
endosomal recycling (95). Rab27
can promote exocytosis (96).
Rab40 can mediate actin dynamics during cell migration (97). In addition, there are some Rab
proteins. The roles of Rab1a, Rab4 and Rab4a in cardiac hypertrophy
have been reported (89,98,99).
However, the relationship between other Rab proteins and cardiac
hypertrophy remains unclear.
In transgenic mice overexpressing Rab1a, the
endoplasmic reticulum and Golgi apparatus stack significantly
enlarged and secretory cardiac natriuretic peptide granules
increased, indicating that overexpression of Rab1a can lead to
cardiac hypertrophy both in vitro and in vivo
(98). Rab4 participates in the
transport of proteins from early endosomes to the plasma membrane
and promotes the cycle and activation of β-adrenergic receptor
(β-AR) (89,100). Cardiac-specific overexpression of
Rab4 can enhance β-AR signaling and lead to cardiac hypertrophy
both in vitro and in vivo (89). Rab4a participates in glucose
transport and induces β-AR recycling to the plasma membrane
(99). Increased myocardial Rab4a
expression activates β-adrenergic hypersensitivity and leads to
cardiac hypertrophy both in vitro and in vivo
(99). Other proteins in the Rab
family also play different roles in the heart, but their
relationship with cardiac hypertrophy remains to be further
studied.
Cardiac hypertrophy is an irreversible myocardial
injury accompanied by myocardial dysfunction and fibrosis due to
long-term pathological overload, eventually leading to heart
failure (4). As a molecular switch
in cell, small GTPases play an important role in the development of
cardiac hypertrophy (7,8). The roles of Ras, Rad, RhoA, Rac1,
Cdc42, Rab1a, Rab4 and Rab4a in cardiac hypertrophy have been
reported (14,31,40,65,71,89,98,99).
The roles of Ras, Rad, RhoA, Rac1 and Cdc42 in cardiac hypertrophy
are relatively clear, but further studies are needed (14,25,26,31,40).
By comparison, the molecular mechanisms of Rab1a, Rab4 and Rab4a in
cardiac hypertrophy remain elusive (89,98,99).
However, the associations between other small GTPases, including
Arf and Ran and cardiac hypertrophy remain unclear. Further studies
are needed.
Previous studies have shown that regulators of
G-protein signaling can suppress small GTPases by inactivating the
heterotrimeric G-proteins of G protein-coupled receptors (GPCRs),
which act as GTPase-activating proteins (101–103). In addition, it has been reported
that RGS2 and RGS4 can ameliorate cardiac hypertrophy by
suppressing hypertrophy-related signaling initiated by GPCRs in
response to upstream agonists such as angiotensin II and
phenylephrine in myocardial cells (102,104–106). However, direct evidence showing
that RGS can exert protective effects on cardiac hypertrophy
through mediating small GTPases is lacking. This topic is worthy of
further study.
Currently, statins are used in clinical treatment as
powerful cardioprotectants because they inhibit Ras and RhoA
(48). It has been reported that
Rac1-targeting VitD, RhoA-targeting alendronate and inhibitors of
Ras, RhoA and Rac1 can exhibit antihypertrophic effects in the
experimental stage, but these agents have not been tested
clinically for treating cardiac hypertrophy (26,49,78,79,83).
There are no reports on the use of Rad, Cdc42 and Rab-targeting
medicines for treating cardiac hypertrophy. Further studies on the
roles of small GTPases in cardiac hypertrophy and the development
of small GTPase-targeting medicines for treating cardiac
hypertrophy are needed. The present study is beneficial for further
studies on the molecular mechanisms of cardiac hypertrophy and
provided more references for the development of prospective
therapeutic targets for cardiac hypertrophy and heart failure.
Not applicable.
The present study was supported by the Natural Science
Foundation of Hubei Province (grant no. 2022CFB843), the Special
Project on Diabetes and Angiopathy (grant no. 2024TNB04) and Hubei
University of Science and Technology School-level Fund (grant no.
BK202220).
Not applicable.
XW, XN and HW edited the manuscript. ZR conceived,
edited and finalized the manuscript. Data authentication is not
applicable. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Shimizu I and Minamino T: Physiological
and pathological cardiac hypertrophy. J Mol Cell Cardiol.
97:245–262. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tham YK, Bernardo BC, Ooi JY, Weeks KL and
McMullen JR: Pathophysiology of cardiac hypertrophy and heart
failure: Signaling pathways and novel therapeutic targets. Arch
Toxicol. 89:1401–1438. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nakamura M and Sadoshima J: Mechanisms of
physiological and pathological cardiac hypertrophy. Nat Rev
Cardiol. 15:387–407. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Samak M, Fatullayev J, Sabashnikov A,
Zeriouh M, Schmack B, Farag M, Popov AF, Dohmen PM, Choi YH,
Wahlers T and Weymann A: Cardiac Hypertrophy: An introduction to
molecular and cellular basis. Med Sci Monit Basic Res. 22:75–79.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gallo S, Vitacolonna A, Bonzano A,
Comoglio P and Crepaldi T: ERK: A key player in the pathophysiology
of cardiac hypertrophy. Int J Mol Sci. 20:21642019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Oka T, Akazawa H, Naito AT and Komuro I:
Angiogenesis and cardiac hypertrophy: Maintenance of cardiac
function and causative roles in heart failure. Circ Res.
114:565–571. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lezoualc'h F, Métrich M, Hmitou I,
Duquesnes N and Morel E: Small GTP-binding proteins and their
regulators in cardiac hypertrophy. J Mol Cell Cardiol. 44:623–632.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Clerk A and Sugden PH: Small guanine
nucleotide-binding proteins and myocardial hypertrophy. Circ Res.
86:1019–1023. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Matozaki T, Nakanishi H and Takai Y: Small
G-protein networks: Their crosstalk and signal cascades. Cell
Signal. 12:515–524. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lundquist EA: Small GTPases. Greenwald I:
WormBook; pp. 1–18. 2006, PubMed/NCBI
|
|
11
|
Wennerberg K, Rossman KL and Der CJ: The
Ras superfamily at a glance. J Cell Sci. 118:843–846. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Reiner DJ and Lundquist EA: Small GTPases.
WormBook. 2018:1–65. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Karnoub AE and Weinberg RA: Ras oncogenes:
Split personalities. Nat Rev Mol Cell Biol. 9:517–531. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ramos-Kuri M, Meka SH, Salamanca-Buentello
F, Hajjar RJ, Lipskaia L and Chemaly ER: Molecules linked to Ras
signaling as therapeutic targets in cardiac pathologies. Biol Res.
54:232021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ramirez MT, Sah VP, Zhao XL, Hunter JJ,
Chien KR and Brown JH: The MEKK-JNK pathway is stimulated by
alpha1-adrenergic receptor and ras activation and is associated
with in vitro and in vivo cardiac hypertrophy. J Biol Chem.
272:14057–14061. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Matsuda T, Jeong JI, Ikeda S, Yamamoto T,
Gao S, Babu GJ, Zhai P and Del Re DP: H-Ras isoform mediates
protection against pressure overload-induced cardiac dysfunction in
part through activation of AKT. Circ Heart Fail. 10:e0036582017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Aikawa R, Nagai T, Kudoh S, Zou Y, Tanaka
M, Tamura M, Akazawa H, Takano H, Nagai R and Komuro I: Integrins
play a critical role in mechanical stress-induced p38 MAPK
activation. Hypertension. 39:233–238. 2022. View Article : Google Scholar
|
|
18
|
Heasman SJ and Ridley AJ: Mammalian Rho
GTPases: New insights into their functions from in vivo studies.
Nat Rev Mol Cell Biol. 9:690–701. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schlessinger K, Hall A and Tolwinski N:
Wnt signaling pathways meet Rho GTPases. Genes Dev. 23:265–277.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mosaddeghzadeh N and Ahmadian MR: The RHO
Family GTPases: Mechanisms of regulation and signaling. Cells.
10:18312021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mackay DJ and Hall A: Rho GTPases. J Biol
Chem. 273:20685–20688. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schwartz SL, Cao C, Pylypenko O, Rak A and
Wandinger-Ness A: Rab GTPases at a glance. J Cell Sci.
120:3905–3910. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jackson CL and Bouvet S: Arfs at a glance.
J Cell Sci. 127:4103–4109. 2014.PubMed/NCBI
|
|
24
|
Yoneda Y, Hieda M, Nagoshi E and Miyamoto
Y: Nucleocytoplasmic protein transport and recycling of Ran. Cell
Struct Funct. 24:425–433. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Na W, Peng G, Jianping Z, Yanzhong C,
Shengjiang G and Li C: RhoA/ROCK may involve in cardiac hypertrophy
induced by experimental hyperthyroidism. Toxicol Ind Health.
28:831–839. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sun Y, Xu C, Jiang Z and Jiang X:
DEF6(differentially exprehomolog) exacerbates pathological cardiac
hypertrophy via RAC1. Cell Death Dis. 14:4832023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lin KH, Kumar VB, Shanmugam T, Shibu MA,
Chen RJ, Kuo CH, Ho TJ, Padma VV, Yeh YL and Huang CY: miR-145-5p
targets paxillin to attenuate angiotensin II-induced pathological
cardiac hypertrophy via downregulation of Rac 1, pJNK, p-c-Jun,
NFATc3, ANP and by Sirt-1 upregulation. Mol Cell Biochem.
476:3253–3260. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Higuchi Y, Otsu K, Nishida K, Hirotani S,
Nakayama H, Yamaguchi O, Hikoso S, Kashiwase K, Takeda T, Watanabe
T, et al: The small GTP-binding protein Rac1 induces cardiac
myocyte hypertrophy through the activation of apoptosis
signal-regulating kinase 1 and nuclear factor-kappa B. J Biol Chem.
278:20770–20777. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Satoh M, Ogita H, Takeshita K, Mukai Y,
Kwiatkowski DJ and Liao JK: Requirement of Rac1 in the development
of cardiac hypertrophy. Proc Natl Acad Sci USA. 103:7432–7437.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Aikawa R, Nagai T, Tanaka M, Zou Y,
Ishihara T, Takano H, Hasegawa H, Akazawa H, Mizukami M, Nagai R
and Komuro I: Reactive oxygen species in mechanical stress-induced
cardiac hypertrophy. Biochem Biophys Res Commun. 289:901–907. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Maillet M, Lynch JM, Sanna B, York AJ,
Zheng Y and Molkentin JD: Cdc42 is an antihypertrophic molecular
switch in the mouse heart. J Clin Invest. 119:3079–3088. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu J, Zheng X and Wu X: The Rab GTPase in
the heart: Pivotal roles in development and disease. Life Sci.
306:1208062022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tomazini A and Shifman JM: Targeting Ras
with protein engineering. Oncotarget. 14:672–687. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Apken LH and Oeckinghaus A: The RAL
signaling network: Cancer and beyond. Int Rev Cell Mol Biol.
361:21–105. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shi GX, Cai W and Andres DA: Rit subfamily
small GTPases: Regulators in neuronal differentiation and survival.
Cell Signal. 25:2060–2068. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Minato N: Rap G protein signal in normal
and disordered lymphohematopoiesis. Exp Cell Res. 319:2323–2328.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhong Y, Zhou X, Guan KL and Zhang J: Rheb
regulates nuclear mTORC1 activity independent of farnesylation.
Cell Chem Biol. 29:1037–1045.e4. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang W, Pang D, Chen M, Du C, Jia L, Wang
L, He Y, Jiang W, Luo L, Yu Z, et al: Rheb mediates
neuronal-activity-induced mitochondrial energetics through
mTORC1-independent PDH activation. Dev Cell. 56:811–825.e6. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li Y, Chang Y, Li X, Li X, Gao J, Zhou Y,
Wu F, Bai R, Dong T, Ma S, et al: RAD-Deficient human
cardiomyocytes develop hypertrophic cardiomyopathy phenotypes due
to calcium dysregulation. Front Cell Dev Biol. 8:5858792020.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chang L, Zhang J, Tseng YH, Xie CQ, Ilany
J, Brüning JC, Sun Z, Zhu X, Cui T, Youker KA, et al: Rad GTPase
deficiency leads to cardiac hypertrophy. Circulation.
116:2976–2983. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Thorburn A, Thorburn J, Chen SY, Powers S,
Shubeita HE, Feramisco JR and Chien KR: HRas-dependent pathways can
activate morphological and genetic markers of cardiac muscle cell
hypertrophy. J Biol Chem. 268:2244–2249. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ramos-Kuri M, Rapti K, Mehel H, Zhang S,
Dhandapany PS, Liang L, García-Carrancá A, Bobe R, Fischmeister R,
Adnot S, et al: Dominant negative Ras attenuates pathological
ventricular remodeling in pressure overload cardiac hypertrophy.
Biochim Biophys Acta. 1853:2870–2884. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Petrich BG and Wang Y: Stress-activated
MAP kinases in cardiac remodeling and heart failure; new insights
from transgenic studies. Trends Cardiovasc Med. 14:50–55. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ichida M and Finkel T: Ras regulates NFAT3
activity in cardiac myocytes. J Biol Chem. 276:3524–3530. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lange-Carter CA and Johnson GL:
Ras-dependent growth factor regulation of MEK kinase in PC12 cells.
Science. 265:1458–1461. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Russell M, Lange-Carter CA and Johnson GL:
Direct interaction between Ras and the kinase domain of
mitogen-activated protein kinase kinase kinase (MEKK1). J Biol
Chem. 270:11757–11760. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Reynet C and Kahn CR: Rad: A member of the
Ras family overexpressed in muscle of type II diabetic humans.
Science. 262:1441–1444. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cho KJ, Hill MM, Chigurupati S, Du G,
Parton RG and Hancock JF: Therapeutic levels of the
hydroxmethylglutaryl-coenzyme A reductase inhibitor lovastatin
activate ras signaling via phospholipase D2. Mol Cell Biol.
31:1110–1120. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ding J, Chen YX, Chen Y, Mou Y, Sun XT,
Dai DP, Zhao CZ, Yang J, Hu SJ and Guo X: Overexpression of FNTB
and the activation of Ras induce hypertrophy and promote apoptosis
and autophagic cell death in cardiomyocytes. J Cell Mol Med.
24:8998–9011. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Li X, Han J, Li L, Wang KJ and Hu SJ:
Effect of farnesyltransferase inhibition on cardiac remodeling in
spontaneously hypertensive rats. Int J Cardiol. 168:3340–3347.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cai T, Abel L, Langford O, Monaghan G,
Aronson JK, Stevens RJ, Lay-Flurrie S, Koshiaris C, McManus RJ,
Hobbs FDR and Sheppard JP: Associations between statins and adverse
events in primary prevention of cardiovascular disease: Systematic
review with pairwise, network and dose-response meta-analyses. BMJ.
374:n15372021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Jaiswal M, Dvorsky R and Ahmadian MR:
Deciphering the molecular and functional basis of Dbl family
proteins: A novel systematic approach toward classification of
selective activation of the Rho family proteins. J Biol Chem.
288:4486–4500. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Strassheim D, Gerasimovskaya E, Irwin D,
Dempsey EC, Stenmark K and Karoor V: RhoGTPase in vascular disease.
Cells. 8:5512019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lee CF, Carley RE, Butler CA and Morrison
AR: Rac GTPase signaling in immune-mediated mechanisms of
atherosclerosis. Cells. 10:28082021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Nguyen DT, Gao L, Wong A and Chen CS:
Cdc42 regulates branching in angiogenic sprouting in vitro.
Microcirculation. 24:10.1111/micc.12372. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Lv J, Zeng J, Guo F, Li Y, Xu M, Cheng Y,
Zhang L, Cai S, Chen Y, Zheng Y and Hu G: Endothelial Cdc42
deficiency impairs endothelial regeneration and vascular repair
after inflammatory vascular injury. Respir Res. 19:272018.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Basbous S, Azzarelli R, Pacary E and
Moreau V: Pathophysiological functions of Rnd proteins. Small
GTPases. 12:336–357. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Blom M, Reis K and Aspenström P: RhoD
localization and function is dependent on its GTP/GDP-bound state
and unique N-terminal motif. Eur J Cell Biol. 97:393–401. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ahmad Mokhtar AM, Hashim IF, Mohd Zaini
Makhtar M, Salikin NH and Amin-Nordin S: The Role of RhoH in TCR
signalling and its involvement in diseases. Cells. 10:9502021.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kilian LS, Voran J, Frank D and Rangrez
AY: RhoA: A dubious molecule in cardiac pathophysiology. J Biomed
Sci. 28:332021. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Miyamoto S, Del Re DP, Xiang SY, Zhao X,
Florholmen G and Brown JH: Revisited and revised: is RhoA always a
villain in cardiac pathophysiology? J Cardiovasc Transl Res.
3:330–343. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zhou Q, Wei SS, Wang H, Wang Q, Li W, Li
G, Hou JW, Chen XM, Chen J, Xu WP, et al: Crucial Role of
ROCK2-Mediated phosphorylation and upregulation of FHOD3 in the
pathogenesis of angiotensin II-induced cardiac hypertrophy.
Hypertension. 69:1070–1083. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Sakaguchi T, Takefuji M, Wettschureck N,
Hamaguchi T, Amano M, Kato K, Tsuda T, Eguchi S, Ishihama S, Mori
Y, et al: Protein Kinase N promotes stress-induced cardiac
dysfunction through phosphorylation of myocardin-related
transcription factor A and disruption of its interaction with
actin. Circulation. 140:1737–1752. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Huang J, Qu Q, Dai Y, Ren D, Qian J and Ge
J: Detrimental Role of PDZ-RhoGEF in pathological cardiac
hypertrophy. Hypertension. 80:403–415. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Aoki H, Izumo S and Sadoshima J:
Angiotensin II activates RhoA in cardiac myocytes: A critical role
of RhoA in angiotensin II-induced premyofibril formation. Circ Res.
82:666–676. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Nakagawa O, Fujisawa K, Ishizaki T, Saito
Y, Nakao K and Narumiya S: ROCK-I and ROCK-II, two isoforms of
Rho-associated coiled-coil forming protein serine/threonine kinase
in mice. FEBS Lett. 392:189–193. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Zhang YM, Bo J, Taffet GE, Chang J, Shi J,
Reddy AK, Michael LH, Schneider MD, Entman ML, Schwartz RJ and Wei
L: Targeted deletion of ROCK1 protects the heart against pressure
overload by inhibiting reactive fibrosis. FASEB J. 20:916–925.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Okamoto R, Li Y, Noma K, Hiroi Y, Liu PY,
Taniguchi M, Ito M and Liao JK: FHL2 prevents cardiac hypertrophy
in mice with cardiac-specific deletion of ROCK2. FASEB J.
27:1439–1449. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Shimizu T, Narang N, Chen P, Yu B, Knapp
M, Janardanan J, Blair J and Liao JK: Fibroblast deletion of ROCK2
attenuates cardiac hypertrophy, fibrosis and diastolic dysfunction.
JCI Insight. 2:e931872017. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Ikeda S, Satoh K, Kikuchi N, Miyata S,
Suzuki K, Omura J, Shimizu T, Kobayashi K, Kobayashi K, Fukumoto Y,
et al: Crucial role of rho-kinase in pressure overload-induced
right ventricular hypertrophy and dysfunction in mice. Arterioscler
Thromb Vasc Biol. 34:1260–1271. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Pracyk JB, Tanaka K, Hegland DD, Kim KS,
Sethi R, Rovira II, Blazina DR, Lee L, Bruder JT, Kovesdi I, et al:
A requirement for the rac1 GTPase in the signal transduction
pathway leading to cardiac myocyte hypertrophy. J Clin Invest.
102:929–937. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Elnakish MT, Moldovan L, Khan M, Hassanain
HH and Janssen PM: Myocardial Rac1 exhibits partial involvement in
thyroxin-induced cardiomyocyte hypertrophy and its inhibition is
not sufficient to improve cardiac dysfunction or contractile
abnormalities in mouse papillary muscles. J Cardiovasc Pharmacol.
61:536–544. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Li PL, Liu H, Chen GP, Li L, Shi HJ, Nie
HY, Liu Z, Hu YF, Yang J, Zhang P, et al: STEAP3 (Six-Transmembrane
Epithelial Antigen of Prostate 3) Inhibits Pathological Cardiac
Hypertrophy. Hypertension. 76:1219–1230. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Clerk A, Pham FH, Fuller SJ, Sahai E,
Aktories K, Marais R, Marshall C and Sugden PH: Regulation of
mitogen-activated protein kinases in cardiac myocytes through the
small G protein Rac1. Mol Cell Biol. 21:1173–1184. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Sawada N, Li Y and Liao JK: Novel aspects
of the roles of Rac1 GTPase in the cardiovascular system. Curr Opin
Pharmacol. 10:116–121. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Cacciapuoti F: Molecular mechanisms of
left ventricular hypertrophy (LVH) in systemic hypertension
(SH)-possible therapeutic perspectives. J Am Soc Hypertens.
5:449–455. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Hauck L, Harms C, Grothe D, An J, Gertz K,
Kronenberg G, Dietz R, Endres M and von Harsdorf R: Critical role
for FoxO3a-dependent regulation of p21CIP1/WAF1 in response to
statin signaling in cardiac myocytes. Circ Res. 100:50–60. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Moradi A, Maroofi A, Hemati M, Hashemzade
T, Alborzi N and Safari F: Inhibition of GTPase Rac1 expression by
vitamin D mitigates pressure overload-induced cardiac hypertrophy.
Int J Cardiol Heart Vasc. 37:1009222021.PubMed/NCBI
|
|
79
|
Zhang C, Jin DD, Wang XY, Lou L and Yang
J: Key enzymes for the mevalonate pathway in the cardiovascular
system. J Cardiovasc Pharmacol. 77:142–152. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Zeidan A, Gan XT, Thomas A and Karmazyn M:
Prevention of RhoA activation and cofilin-mediated actin
polymerization mediates the antihypertrophic effect of adenosine
receptor agonists in angiotensin II- and endothelin-1-treated
cardiomyocytes. Mol Cell Biochem. 385:239–248. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Fan S, Xiong Q, Zhang X, Zhang L and Shi
Y: Glucagon-like peptide 1 reverses myocardial hypertrophy through
cAMP/PKA/RhoA/ROCK2 signaling. Acta Biochim Biophys Sin (Shanghai).
52:612–619. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Tawara S and Shimokawa H: Progress of the
study of rho-kinase and future perspective of the inhibitor.
Yakugaku Zasshi. 127:501–514. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Zhang Y and Wu S: Effects of fasudil on
pulmonary hypertension in clinical practice. Pulm Pharmacol Ther.
46:54–63. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Bock JB, Matern HT, Peden AA and Scheller
RH: A genomic perspective on membrane compartment organization.
Nature. 409:839–841. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Stenmark H and Olkkonen VM: The Rab GTPase
family. Genome Biol. 2:REVIEWS30072001. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Pereira-Leal JB and Seabra MC: The
mammalian Rab family of small GTPases: Definition of family and
subfamily sequence motifs suggests a mechanism for functional
specificity in the Ras superfamily. J Mol Biol. 301:1077–1087.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Yang XZ, Li XX, Zhang YJ,
Rodriguez-Rodriguez L, Xiang MQ, Wang HY and Zheng XF: Rab1 in cell
signaling, cancer and other diseases. Oncogene. 35:5699–5704. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Schonn JS, van Weering JR, Mohrmann R,
Schlüter OM, Südhof TC, de Wit H, Verhage M and Sørensen JB: Rab3
proteins involved in vesicle biogenesis and priming in embryonic
mouse chromaffin cells. Traffic. 11:1415–1428. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Filipeanu CM, Zhou F, Lam ML, Kerut KE,
Claycomb WC and Wu G: Enhancement of the recycling and activation
of beta-adrenergic receptor by Rab4 GTPase in cardiac myocytes. J
Biol Chem. 281:11097–11103. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Xu W, Fang F, Ding J and Wu C:
Dysregulation of Rab5-mediated endocytic pathways in Alzheimer's
disease. Traffic. 19:253–262. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Dornan LG and Simpson JC: Rab6-mediated
retrograde trafficking from the Golgi: The trouble with tubules.
Small GTPases. 14:26–44. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Stypulkowski E, Feng Q, Joseph I, Farrell
V, Flores J, Yu S, Sakamori R, Sun J, Bandyopadhyay S, Das S, et
al: Rab8 attenuates Wnt signaling and is required for mesenchymal
differentiation into adipocytes. J Biol Chem. 296:1004882021.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Wilson B, Flett C, Gemperle J, Lawless C,
Hartshorn M, Hinde E, Harrison T, Chastney M, Taylor S, Allen J, et
al: Proximity labelling identifies pro-migratory endocytic
recycling cargo and machinery of the Rab4 and Rab11 families. J
Cell Sci. 136:jcs2604682023. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Banworth MJ and Li G: Consequences of Rab
GTPase dysfunction in genetic or acquired human diseases. Small
GTPases. 9:158–181. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Banworth MJ, Liang Z and Li G: A novel
membrane targeting domain mediates the endosomal or Golgi
localization specificity of small GTPases Rab22 and Rab31. J Biol
Chem. 298:1022812022. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Izumi T: In vivo Roles of Rab27 and its
effectors in exocytosis. Cell Struct Funct. 46:79–94. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Neumann AJ and Prekeris R: A Rab-bit hole:
Rab40 GTPases as new regulators of the actin cytoskeleton and cell
migration. Front Cell Dev Biol. 11:12689222023. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Moore I, Schell J and Palme K:
Subclass-specific sequence motifs identified in Rab GTPases. Trends
Biochem Sci. 20:10–12. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Filipeanu CM, Zhou F and Wu G: Analysis of
Rab1 function in cardiomyocyte growth. Methods Enzymol.
438:217–226. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Etzion S, Etzion Y, DeBosch B, Crawford PA
and Muslin AJ: Akt2 deficiency promotes cardiac induction of Rab4a
and myocardial β-adrenergic hypersensitivity. J Mol Cell Cardiol.
49:931–940. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Seachrist JL and Ferguson SS: Regulation
of G protein-coupled receptor endocytosis and trafficking by Rab
GTPases. Life Sci. 74:225–235. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Del Calvo G, Baggio Lopez T and
Lymperopoulos A: The therapeutic potential of targeting cardiac
RGS4. Ther Adv Cardiovasc Dis. 17:175394472311993502023. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Lymperopoulos A, Borges JI and Stoicovy
RA: RGS proteins and cardiovascular Angiotensin II Signaling: Novel
opportunities for therapeutic targeting. Biochem Pharmacol.
218:1159042023. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Magalhaes AC, Dunn H and Ferguson SS:
Regulation of GPCR activity, trafficking and localization by
GPCR-interacting proteins. Br J Pharmacol. 165:1717–1736. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Rogers JH, Tamirisa P, Kovacs A,
Weinheimer C, Courtois M, Blumer KJ, Kelly DP and Muslin AJ: RGS4
causes increased mortality and reduced cardiac hypertrophy in
response to pressure overload. J Clin Invest. 104:567–576. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Chidiac P, Sobiesiak AJ, Lee KN, Gros R
and Nguyen CH: The eIF2B-interacting domain of RGS2 protects
against GPCR agonist-induced hypertrophy in neonatal rat
cardiomyocytes. Cell Signal. 26:1226–1234. 2014. View Article : Google Scholar : PubMed/NCBI
|