Introduction
Graves' orbitopathy (GO) is a type of orbital
inflammation that occurs in 15–30% of Graves' disease cases
(1,2). GO clinically presents with eyelid
retraction, exophthalmos and extraocular muscle enlargement;
notably, 3–5% of patients experience a vision-threatening severe
form, including compressive optic neuropathy and corneal exposure
(2,3).
Dysregulated orbital fibroblasts and infiltration of
orchestrated mononuclear cells, such as macrophage, T cells and B
cells, are pivotal to the pathogenesis of GO. Autoantibodies
targeting the thyrotropin and insulin-like growth factor 1
receptors initiate monocyte infiltration by T and B cells, as well
as macrophages (4). These
activated monocytes release cytokines that stimulate orbital
fibroblasts, which also produce proinflammatory cytokines that
exacerbate and maintain ocular inflammation (2,5).
Stimulated orbital fibroblasts proliferate to produce
glycosaminoglycans and differentiate into adipocytes or
myofibroblasts, eventually leading to orbital tissue swelling,
enlargement and fibrosis (2,4,5).
In a previous study on the pathogenesis and
potential therapies for GO, no significant genotypic differences
were found between patients with GO and without GO (6,7).
Current research has focused on epigenetic and environmental
factors (8,9), highlighting histone deacetylases
(HDACs) as promising epigenetic molecules. HDACs are classified
into four groups based on their enzymatic domain organization.
Zn2+-dependent HDACs, also known as classical HDACs,
include Class I (HDACs 1, 2, 3 and 8), Class IIa (HDACs 4, 5, 7 and
9), Class IIb (HDACs 6 and 10) and Class IV (HDAC11), whereas Class
III (sirtuins) molecules are NAD+-dependent. HDACs
mediate the deacetylation of lysine residues on the histone tail as
a post-translational modification and inhibit transcription factor
access to the DNA (10). This
modification regulates gene transcription, controlling the cell
cycle and immunological pathways (11). In addition to histone proteins,
deacetylation occurs in several non-histone proteins, including
transcription factors, and proteins involved in metabolism and the
cell cycle (12). HDAC inhibitors
(HDACis) have been approved by the US Food and Drug Administration
to treat hematological tumors, such as cutaneous or peripheral
T-cell lymphoma, and multiple myeloma, by inhibiting various
pathways involving cytokines, growth factors and protein kinases
(13).
Although extensive research has been conducted on
the role of HDACs in autoimmune diseases of the lungs and synovium
(14–21), their role in GO pathogenesis
remains largely unexplored (22).
Among the HDACis that have been approved for hematological
antitumor activity, panobinostat (LBH589), an orally available
pan-HDACi, was approved by the US Food and Drug Administration in
2015 for the treatment of multiple myeloma (13). Panobinostat has ≥10-fold higher
anticancer potency than vorinostat, another pan-HDACi, and its
half-maximal inhibitory concentration values are lower than those
of other pan-HDACis (23,24). Furthermore, panobinostat has been
shown to exert superior anti-fibrotic effects on primary idiopathic
pulmonary fibrosis compared with pirfenidone, the only currently
available treatment for the condition (20).
The present study aimed to evaluate the effects of
panobinostat on GO-induced inflammation, adipogenesis and fibrosis.
Additional experiments were performed to assess which HDACs exert
the therapeutic effects of panobinostat, with a particular focus on
HDAC7.
Materials and methods
Tissue and cell preparation
During orbital surgery performed at Severance
Hospital (Seoul, South Korea), orbital adipose tissue was collected
from 10 patients with GO and 8 controls as a surgical byproduct
(Table SI). The patients achieved
euthyroid status with a clinical activity score (CAS) (25) <3 for ≥6 months, and the control
individuals did not have GO or any other autoimmune diagnoses. The
present study followed the tenets of The Declaration of Helsinki
and was approved by the Institutional Review Board of Severance
Hospital, Yonsei University College of Medicine (IRB no.
4-2022-0244; Seoul, South Korea). Written informed consent was
obtained from all patients. Patient samples were collected between
May 2022 and December 2023.
Orbital fat tissues were stored in RNAlater
(Invitrogen; Thermo Fisher Scientific, Inc.) for RNA preparation.
For primary cell culture, orbital fat was minced and cultured in
Dulbecco's modified Eagle's medium/nutrient mixture F-12 (DMEM/F12;
Welgene, Inc.) supplemented with 20% fetal bovine serum (FBS;
Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin/streptomycin (Welgene, Inc.) in a humidified 5%
CO2 incubator at 37°C. As orbital fibroblasts
proliferated, the cells were serially passaged with
trypsin/ethylenediaminetetraacetic acid. The cell strains were
stored in liquid nitrogen (−196°C). Cells between passages three
and five were used for the subsequent experiments.
Peripheral blood mononuclear cell
(PBMC) preparation
Peripheral venous blood (10 ml) from healthy donors
(n=11) and patients with GO (n=32) was collected with ethics
approval (IRB no. 4-2022-0244) (Table
SII). Peripheral venous blood sampling was performed together
with orbital tissue sampling. As peripheral venous blood was drawn
prior to high-dose steroid therapy, the inclusion criteria were not
restricted to CAS <3. Consequently, more samples were obtained
from peripheral venous blood sampling than from orbital tissue
samples. PBMC isolation was performed via Ficoll-Paque (Cytiva)
density-gradient centrifugation at 1,000 × g for 20 min at 20°C.
PBMCs were preserved in TRIzol® (Invitrogen; Thermo
Fisher Scientific, Inc.) at −80°C in a freezer.
Cell viability assay
Orbital fibroblasts were seeded into 24-well culture
plates (1×105 cells/well) and exposed to various
concentrations of panobinostat (Selleck Chemicals) (untreated
control, 10, 30, 50, 70 and 100 nM) for 24 h at 37°C. Subsequently,
the cells were washed and incubated with MTT solution
(MilliporeSigma) for 2 h at 37°C. After solubilization with
dimethyl sulfoxide (MilliporeSigma), the dye absorbance was
measured using a microplate reader (BioTek Instruments; Agilent
Technologies, Inc.) at 560 and 630 nm. Cell viability was expressed
as a percentage of the untreated control cells.
Reverse transcription-quantitative PCR
(RT-qPCR)
Orbital fat tissues were homogenized using a tissue
homogenizer (Precellys 24; Bertin Instruments) and were lysed with
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.)
using a Precellys lysing kit (hard tissue homogenizing CK28; cat.
no. P000911-LYSK0-A; Bertin Instruments). RNA was also extracted
from primary cultured GO orbital fibroblasts cultured with or
without panobinostat (100 nM) for 24 h using TRIzol. The RNA
concentration was measured using a NanoDrop spectrophotometer
(Thermo Fisher Scientific, Inc.). cDNA was synthesized using a
SensiFAST cDNA Synthesis Kit (Meridian Bioscience, Inc.) according
to the manufacturer's protocol. qPCR amplification was performed
using a QuantStudio3 real-time PCR thermocycler (Applied
Biosystems; Thermo Fisher Scientific, Inc.) with specific primers
and SYBR green PCR master mix (Takara Bio, Inc.). The thermocycling
conditions were as follows: Activation at 50°C for 2 min and
incubation at 95°C for 10 min, followed by 40 cycles of
denaturation at 95°C for 15 sec and annealing at 60°C for 1 min,
and a final melting curve analysis at 95°C for 15 sec, 60°C for 15
sec and 95°C for 15 sec. The sequences of primers targeting
multiple HDAC genes (class I: HDAC1, 2 and 3; class IIa: HDAC4, 5
and 7; class IIb: HDAC6 and 10) are detailed in Table SIII. The results were normalized
to GAPDH and are displayed as fold change in cycle quantification
(Cq) value relative to that of the controls, using the
2−ΔΔCq method (26).
Western blotting
Orbital fibroblasts were treated with reagents, such
as recombinant IL-1β (10 ng/ml), TGF-β (5 ng/ml) (both from R&D
Systems, Inc.) and panobinostat (100 nM), for various durations in
a humidified 5% CO2 incubator at 37°C. The cells were
then washed with DPBS (Welgene, Inc.) and lysed using RIPA lysis
buffer (Welgene, Inc.) containing a Halt™ Protease
Inhibitor Cocktail (Thermo Fisher Scientific, Inc.). Protein
concentrations were determined using the BCA assay
(Pierce™ BCA Protein Assay Kit; cat. no. 23227, Thermo
Fisher Scientific, Inc.). Proteins were boiled in sample buffer and
equal amounts of protein (30 µg) were then resolved by SDS-PAGE on
8–15% gels, and were transferred to nitrocellulose membranes
(MilliporeSigma). Subsequently, the membranes were incubated with
5% skim milk blocking buffer for 1 h at room temperature (25°C).
After overnight incubation with primary antibodies at 4°C (Table SIV), the bands were incubated with
a secondary antibody [Pierce Goat Anti-Rabbit, (H+L), Peroxidase
Conjugated (1:5,000; cat. no. 31460; Thermo Fisher Scientific,
Inc.) and Pierce Goat Anti-Mouse, (H+L), Peroxidase Conjugated
(1:5,000; cat. no. 31430; Thermo Fisher Scientific, Inc)] for 1 h
at room temperature, after which, visualization was performed using
SuperSignal™ West Pico PLUS chemiluminescent substrates
(cat. no. 34580; Thermo Fisher Scientific, Inc.). Band intensities,
measured using ImageJ software ver. 1.54 g (National Institutes of
Health), were standardized to β-actin in the same sample.
Adipogenesis
Orbital fibroblasts were differentiated into
adipocytes for 14 days using adipogenic solution as previously
described (27). Briefly, the
adipogenic solution consisted of DMEM (Welgene, Inc.) supplemented
with 10% FBS (Thermo Fisher Scientific, Inc.), 33 µM biotin (Roche
Diagnostics GmbH), 17 µM pantothenic acid (Roche Diagnostics GmbH),
10 µg/ml transferrin (Roche Diagnostics GmbH), 0.2 nM T3 (Roche
Diagnostics GmbH), 1 µM insulin (Roche Diagnostics GmbH), 0.2 µM
carbaprostaglandin (Calbiochem; Merck KGaA) and 10 µM rosiglitazone
(Cayman Chemical Company), and the medium was replaced every 2–3
days. For the first 4 days, 10 µM dexamethasone (MilliporeSigma)
and 0.1 mM isobutylmethylxanthine (MilliporeSigma) were added to
induce adipogenesis. Cells were co-treated with panobinostat (10
nM) and/or IL-1β (10 ng/ml) for 14 days.
Oil Red O staining
Differentiated adipocytes treated with panobinostat
(10 nM) and/or IL-1β (10 ng/ml) for 14 days of adipogenesis were
washed with PBS, fixed with 10% formalin for 1 h at 4°C and stained
for 2 h at room temperature with Oil Red O (cat. no. O1516;
MilliporeSigma) working solution (Oil Red O:deionized water=6:4,
filtered). The plates were rinsed with PBS and the cells were
observed under an inverted light microscope (IX73; Olympus
Corporation) equipped with a charge-coupled device camera (DP71;
Olympus Corporation). Oil Red O stain was solubilized with 100%
isopropanol to quantify lipid accumulation, and the optical density
was measured using a spectrophotometer at 490 nm.
Silencing of HDACs
Small interfering (si)RNAs targeting HDAC6 and HDAC7
(si-HDAC6 and si-HDAC7) were obtained from Dharmacon Reagents;
Revvity, Inc. (SMARTpool format; cat. nos. L-003499-00 and
L-009330-00), and a non-targeting negative control No. 1 siRNA
(si-con) was obtained from Invitrogen; Thermo Fisher Scientific,
Inc. (Table SV). siRNAs (10 nM)
were transfected into 80% confluent orbital fibroblasts using
Lipofectamine® RNAiMAX (Invitrogen; Thermo Fisher
Scientific, Inc.). After 24 h in a humidified 5% CO2
incubator at 37°C, the medium was replaced with fresh complete
medium containing 10% fetal bovine serum and antibiotics, and the
cells were incubated for a further 48 h.
Statistical analysis
IBM SPSS Statistics for Windows v 29.0 (IBM Corp.)
was used for statistical analysis. All experiments were performed
twice using cells from 3 different patients. The results were
averaged and expressed as the mean ± SD. Normal distribution was
verified using the Kolmogorov-Smirnov test. For comparisons between
the experimental and control groups, either unpaired Student's
t-test or Mann-Whitney U-test was used. When comparing multiple
groups, Kruskal-Wallis test was used, followed by Dunn's post hoc
test. In the demographic analysis, Fisher's exact test was used to
compare two categorical variables. P<0.05 was considered to
indicate a statistically significant difference.
Results
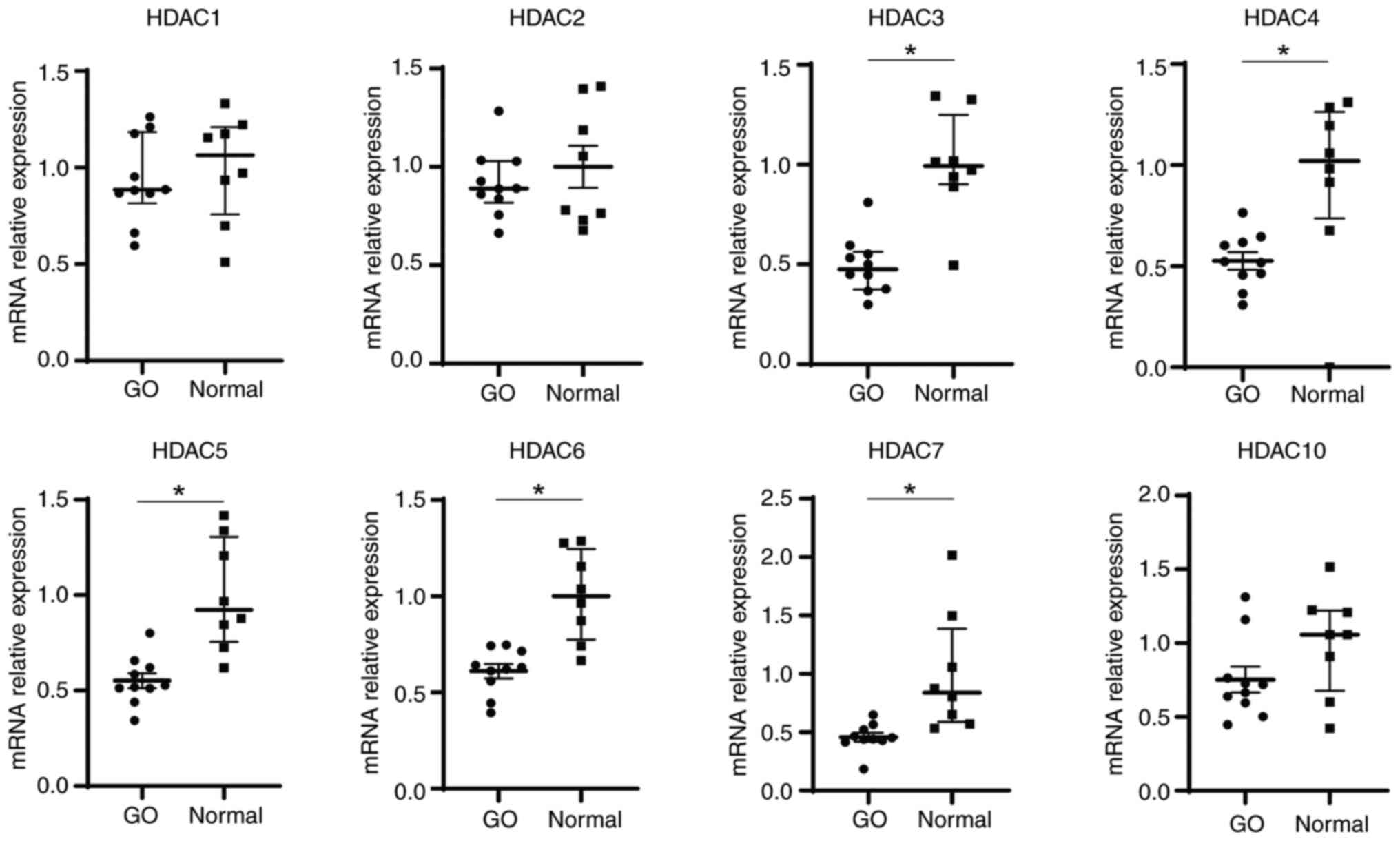
mRNA expression levels of HDACs in
orbital tissue and PBMC
Basal levels of HDAC transcripts were analyzed using
RT-qPCR. A total of eight HDAC genes were assessed: Class I (HDAC1,
2 and 3), class IIa (HDAC4, 5 and 7) and class IIb (HDAC6 and 10).
The median mRNA expression levels of HDAC3, 4, 5, 6 and 7 were
significantly lower in orbital tissues from patients with GO (n=10)
than in those from the control individuals (n=8) (Fig. 1). In addition, HDAC gene expression
patterns were detected in the PBMCs of patients with GO (n=32) and
normal controls (n=11). There were no significant differences in
the mRNA expression levels of HDACs between patients with GO and
normal controls (Fig. S1).
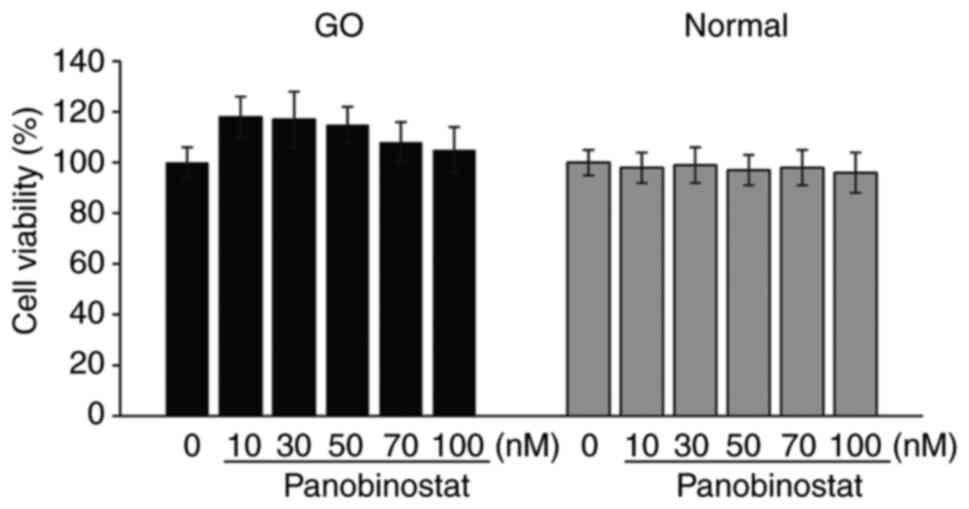
Effect of panobinostat on cell
viability
An MTT assay was performed to determine the
non-toxic concentration of panobinostat. In both normal and GO
orbital fibroblasts, panobinostat at concentrations between 10 and
100 nM did not reduce cell viability to <95% over the course of
a 24-h treatment (Fig. 2).
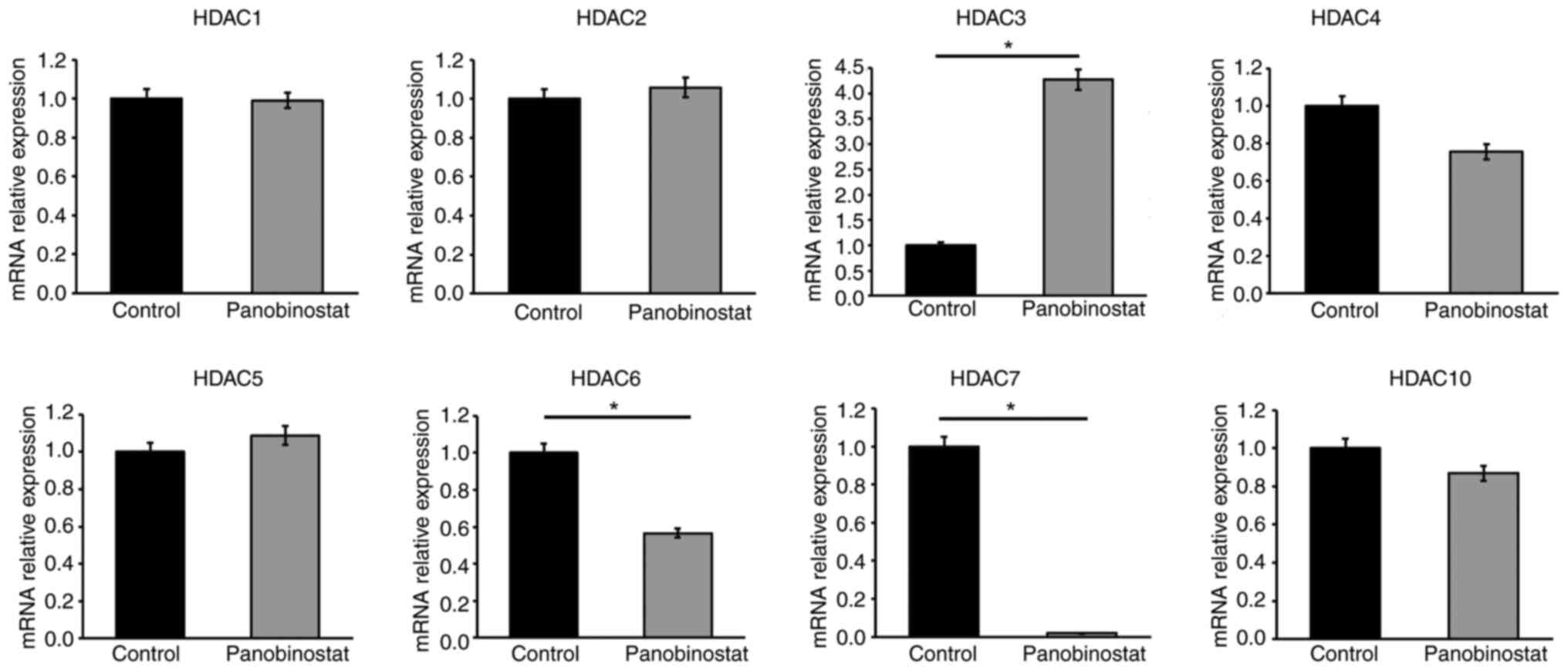
Suppression of HDACs by panobinostat
in GO orbital fibroblasts
Even with panobinostat, a non-selective HDACi, not
all HDAC genes were uniformly inhibited. Pan-HDACis can have
different effects on each HDAC depending on the cellular context
(28). To verify the selectivity
of panobinostat for GO orbital fibroblasts, the fibroblasts were
incubated for 24 h with and without panobinostat (100 nM), and the
expression levels of various HDAC transcripts were analyzed using
RT-qPCR. HDAC3 mRNA expression was found to be upregulated, whereas
HDAC6 and HDAC7 mRNA levels were significantly downregulated in
response to panobinostat (Fig.
3).
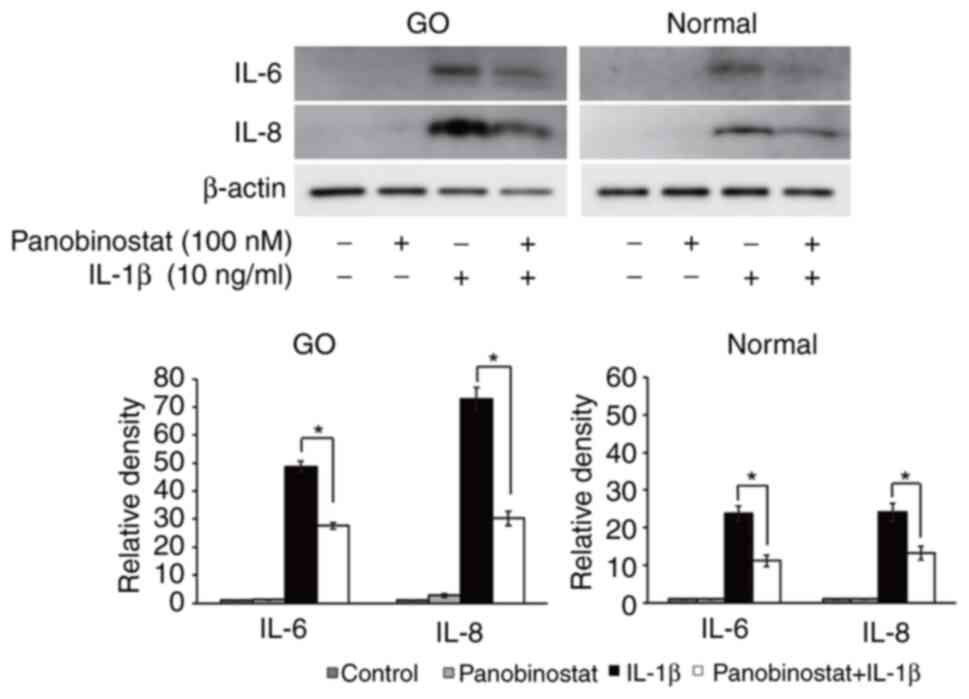
Therapeutic effect of panobinostat on
stimulated orbital fibroblasts
The present study evaluated the therapeutic effects
of panobinostat on multiple GO-related pathogenic processes
(inflammation, adipogenesis and fibrosis) in primary cultured
orbital fibroblasts. GO and normal orbital fibroblasts were
pretreated with panobinostat (100 nM) for 30 min, followed by
inflammatory stimulation using IL-1β (10 ng/ml) for 24 h. Western
blotting demonstrated that panobinostat treatment suppressed the
IL-1β-induced expression of the proinflammatory cytokines IL-6 and
IL-8 (Fig. 4).
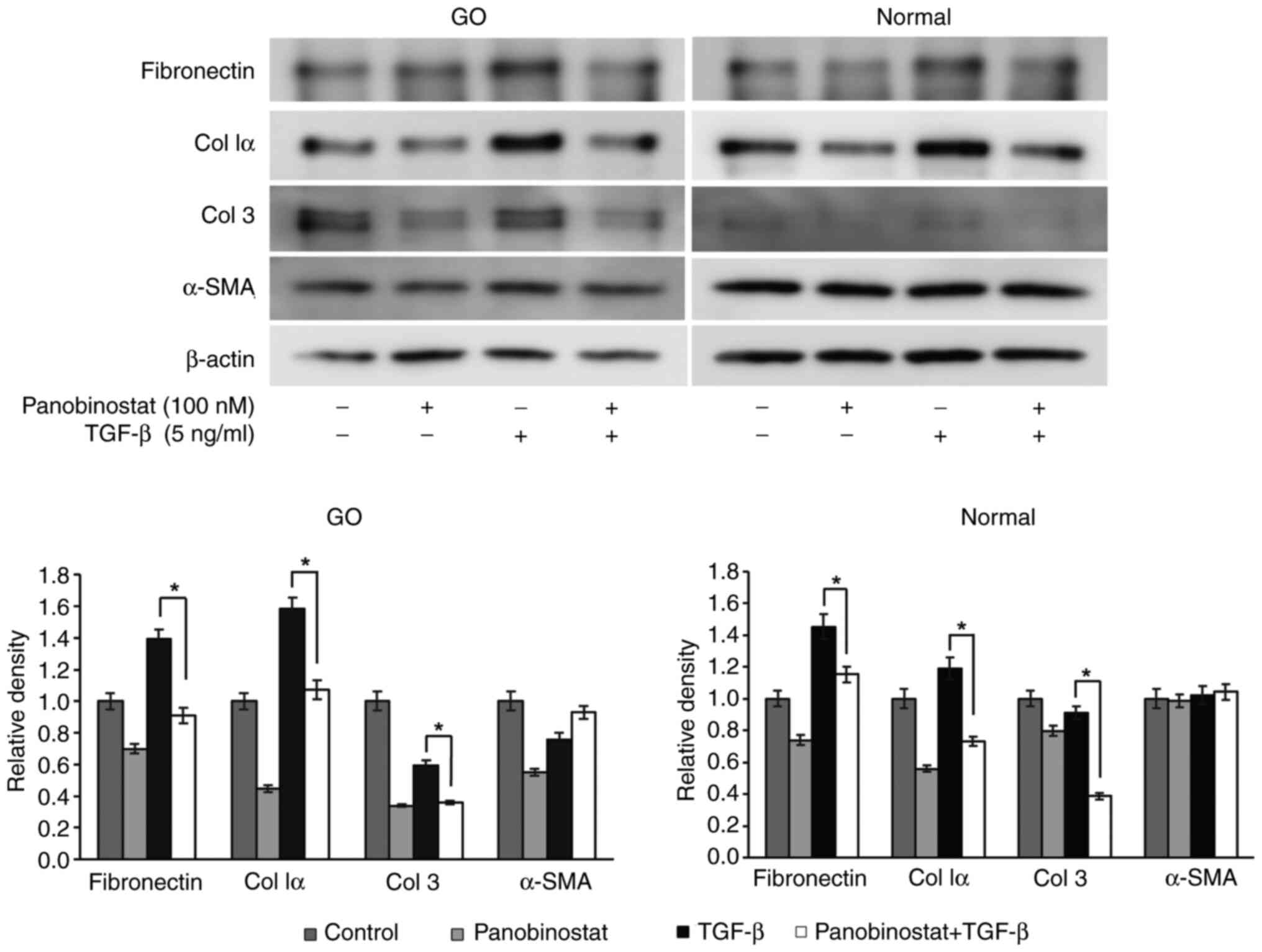
The stimulus was changed to TGF-β (5 ng/ml) under
the same experimental conditions to investigate the anti-fibrotic
effects of panobinostat. The increase in expression of the
profibrotic cytokines fibronectin, collagen Iα and collagen 3
induced by TGF-β was significantly attenuated after panobinostat
treatment (Fig. 5).
Finally, the effect of panobinostat on the adipocyte
differentiation of orbital fibroblasts was examined. During the 14
days of adipogenesis in an adipogenic medium, GO orbital
fibroblasts were also co-treated with panobinostat (10 nM) and/or
IL-1β (10 ng/ml). Panobinostat significantly mitigated
IL-1β-induced adipogenesis (Fig.
6A), as demonstrated by the spectrophotometric lipid
quantification (Fig. 6B). The
expression levels of adiponectin, leptin, HDAC7 and all the
investigated adipogenic transcription factors, including peroxisome
proliferator-activated receptor γ, CCAAT-enhancer-binding protein
(c/EBP)α, c/EBPβ and aP2, were reduced with panobinostat treatment
compared with in cells under adipogenic differentiation and IL-1β
stimulation (Fig. 6C and D).
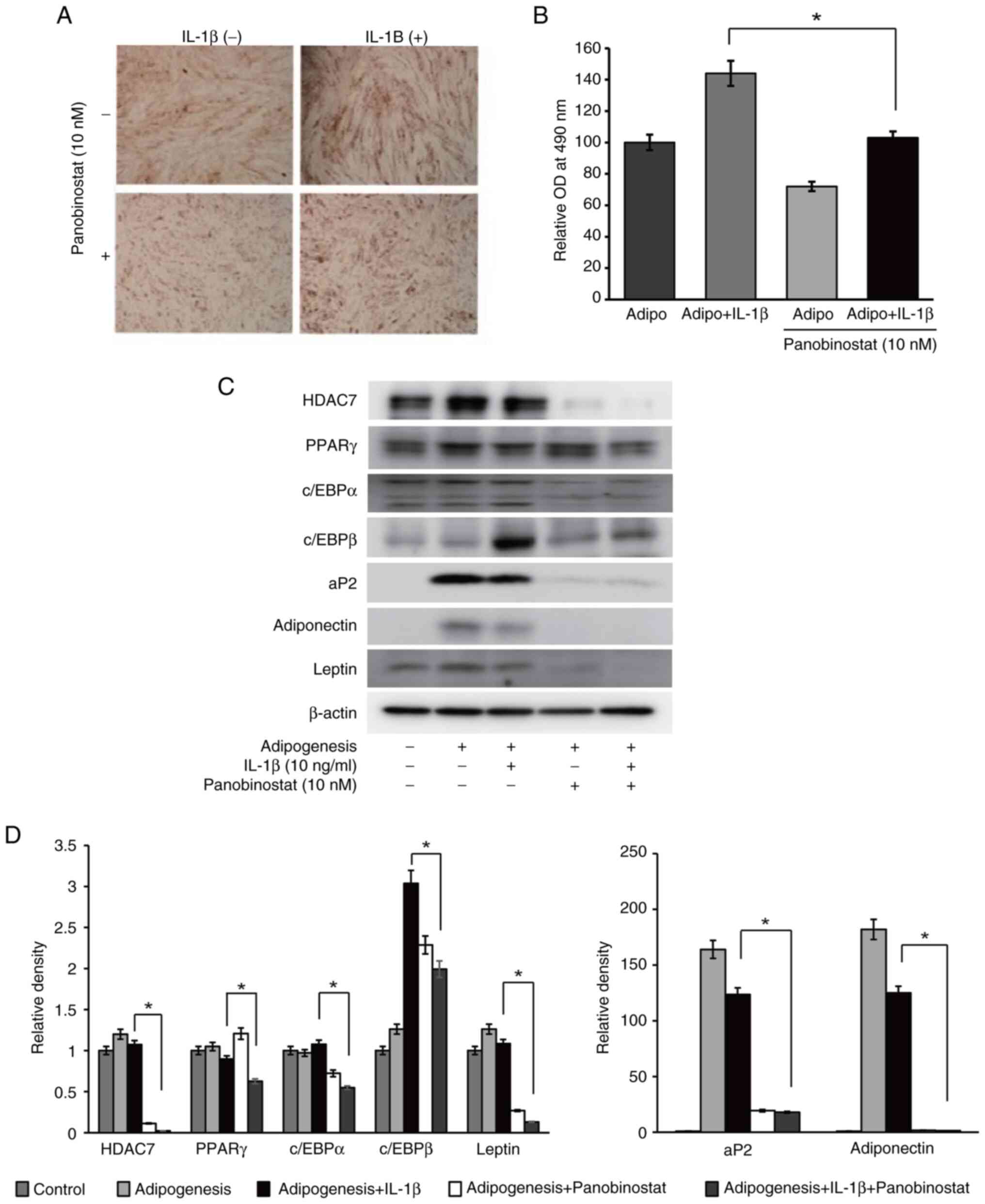 | Figure 6.Suppressive effect of panobinostat on
adipogenesis. Graves' orbitopathy orbital fibroblasts (n=3) were
differentiated into adipocytes after 14 days of incubation in
adipogenic medium. Panobinostat (10 nM) and/or IL-1β (10 ng/ml) was
added during the adipogenesis. All experiments were performed
twice. (A) To evaluate adipocyte differentiation, cells were
stained with Oil Red O, and cytoplasmic lipid droplets were
examined under a microscope (magnification, ×40). (B) Stained cell
lysates were solubilized, and absorbance was measured using a
spectrophotometer at 490 nm. Data are presented as the mean ± SD.
Statistical significance was determined using Mann-Whitney U-test
to compare the effect of panobinostat under adipogenic stimulation
with IL-1β. *P<0.05. (C) Adipogenic transcription factors,
including PPARγ, c/EBPα, c/EBPβ, aP2, adiponectin and leptin, and
HDAC7 were analyzed by western blotting after 14 days of adipogenic
differentiation of orbital fibroblasts. Representative gel images
are shown. (D) Densitometric analysis results of western blotting.
Data are presented as the mean ± SD normalized to β-actin.
Statistical significance was determined using Mann-Whitney U-test
to compare the effect of panobinostat under adipogenic stimulation
with IL-1β. *P<0.05. aP2, adipocyte protein 2; c/EBP,
CCAAT-enhancer-binding protein; HDAC7, histone deacetylase 7; OD,
optical density; PPARγ, peroxisome proliferator-activated receptor
γ. |
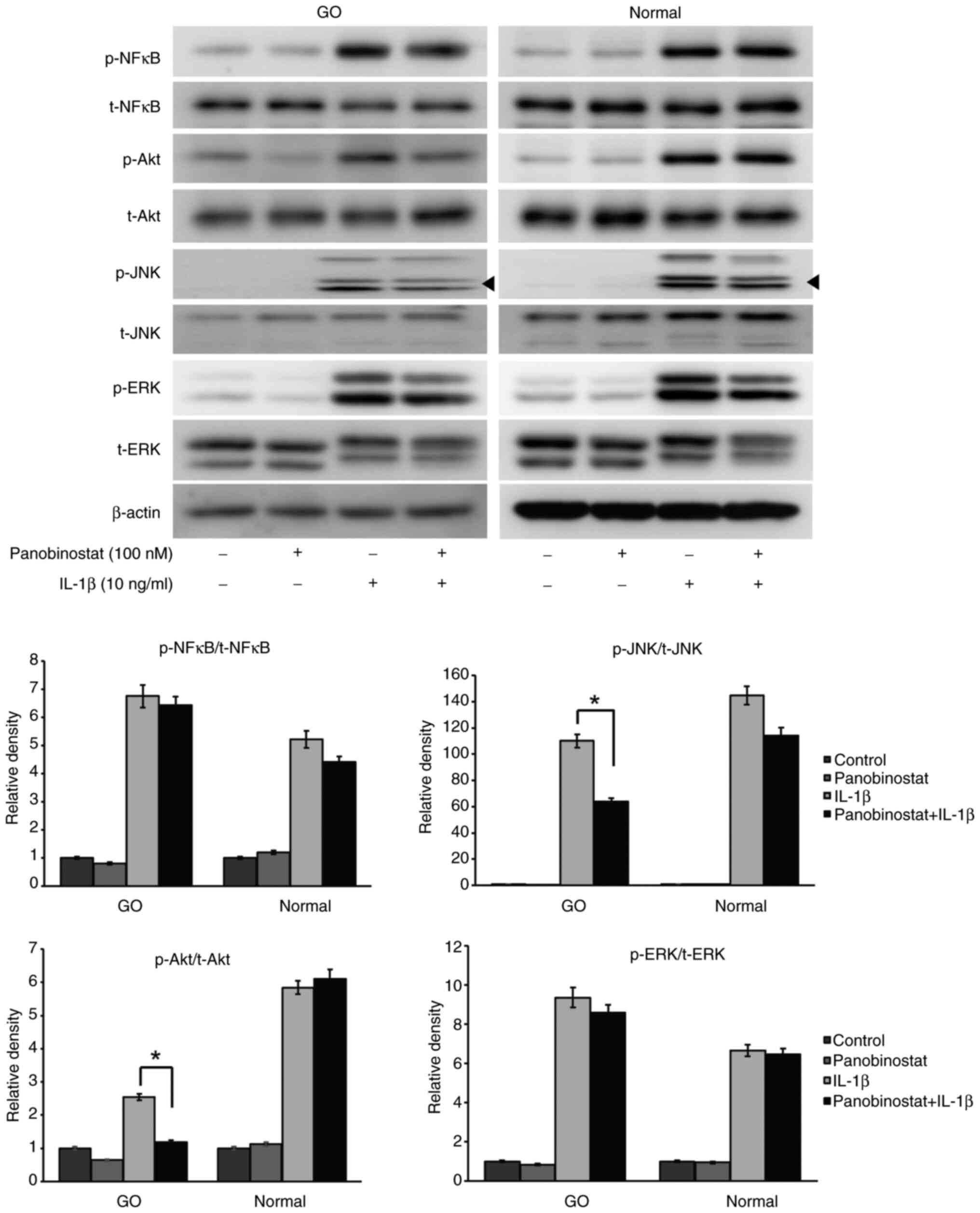
Effect of panobinostat on signaling
pathway molecules
The present study further investigated the
intracellular signaling pathway molecules affected by panobinostat
treatment. Orbital fibroblasts pretreated with panobinostat (100
nM) for 24 h were incubated with IL-1β (10 ng/ml) for 15 min.
IL-1β-induced JNK and Akt phosphorylation was significantly reduced
by panobinostat treatment in GO orbital fibroblasts, but not in
normal cells (Fig. 7).
As major transducers of TGF-β, the SMAD pathway
molecules SMAD1/5/9, SMAD2 and SMAD3 were investigated to determine
the effect of panobinostat. Treatment with 100 nM panobinostat for
3 h significantly reduced TGF-β (5 ng/ml; 1 h)-induced SMAD3
phosphorylation in normal orbital fibroblasts, whereas this
reduction was not significant in GO orbital fibroblasts (Fig. 8A). As non-SMAD pathway molecules,
Akt, JNK, p38 and ERK expression levels were examined under TGF-β
stimulation (5 ng/ml; 1 h) after panobinostat (100 nM) exposure for
3 h. Phosphorylation of Akt was attenuated in both GO and normal
cells, whereas phosphorylation of p38 was reduced in normal cells
(Fig. 8B).
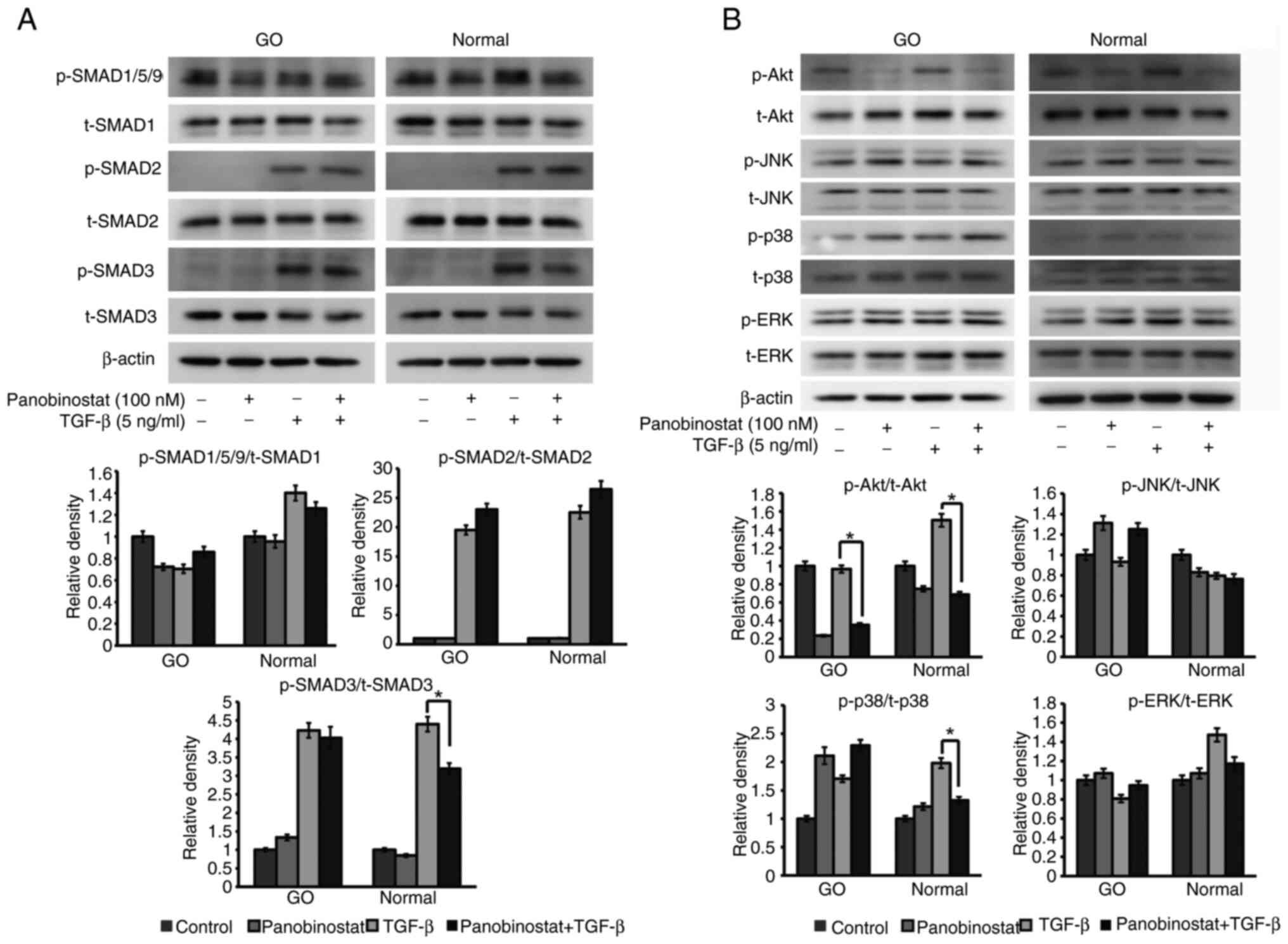 | Figure 8.Effect of panobinostat on signal
proteins under TGF-β stimulation. (A) Orbital fibroblasts were
exposed to 100 nM panobinostat for 3 h, and then stimulated with
TGF-β (5 ng/ml) for 1 h. The p-form of the SMAD signaling
transducers, SMAD1/5/9, SMAD2 and SMAD3, were analyzed by western
blotting, as were the total forms of SMAD1, SMAD2 and SMAD3. (B)
Non-SMAD pathways under TGF-β (5 ng/ml) stimulation for 1 h were
examined in orbital fibroblasts. Orbital fibroblasts were
pretreated with 100 nM panobinostat for 3 h before TGF-β treatment.
Western blot analysis of p- and t-Akt, JNK, p38 and ERK non-SMAD
pathway molecules was performed. Orbital fibroblasts from three
different GO samples and healthy subjects were used in the present
study. β-actin was used as a loading control. The ratio of
p-/t-proteins was measured and compared with that of the control,
and data are presented as the mean ± SD. Statistical significance
was determined using Mann-Whitney U-test to compare the effect of
panobinostat under TGF-β stimulation. *P<0.05. GO, Graves'
orbitopathy; p-, phosphorylated; t-, total. |
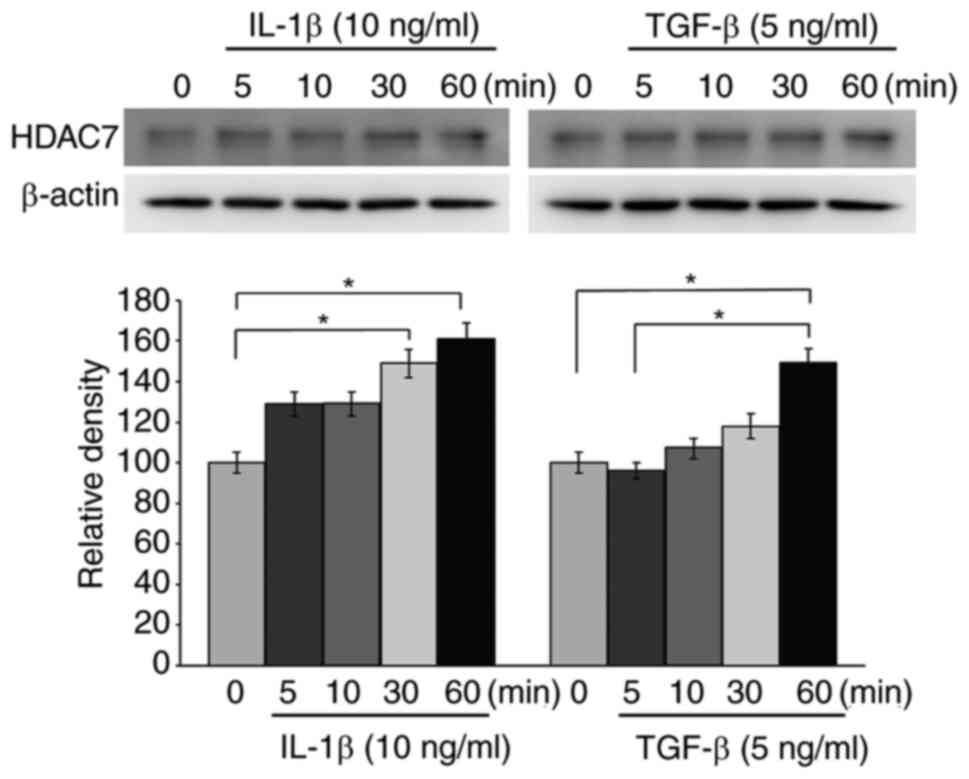
Role of HDAC7 in GO orbital
fibroblasts
Based on the results showing that HDAC7 was notably
suppressed by panobinostat (Fig.
3), the effects of HDAC7 on GO orbital fibroblasts were
examined. Similar to the results showing significantly lower HDAC
mRNA levels in GO tissues than in normal tissues (Fig. 1), western blot analysis revealed
that HDAC7 protein levels were also lower in GO tissues (n=7) than
in normal tissues (n=7) (Fig.
S2). GO orbital fibroblasts were then treated with IL-1β (10
ng/ml) and TGF-β (5 ng/ml) for 0, 5, 10, 30 and 60 min. Western
blot analysis revealed that HDAC7 protein levels were increased
after 30 min of IL-1β stimulation and after 60 min of TGF-β
stimulation (Fig. 9).
To elucidate the role of HDAC7, HDAC7 was silenced
using siRNA (Fig. 10A). si-HDAC7
and si-con were transfected into cells for 24 h, which were then
stimulated with IL-1β (10 ng/ml) or TGF-β (5 ng/ml) for 24 h to
evaluate the effects on inflammation and fibrosis. IL-6 levels,
which were increased following IL-1β treatment, were significantly
reduced in both GO and normal orbital fibroblasts transfected with
si-HDAC7, whereas IL-8 levels were only reduced in
si-HDAC7-transfected normal orbital fibroblasts (Fig. 10B). HDAC7 silencing also
significantly reduced the expression levels of the TGF-β-induced
profibrotic proteins fibronectin and α-smooth muscle actin (α-SMA)
in both GO and normal orbital fibroblasts, as well as collagen Iα
and collagen 3 in GO cells (Fig.
10C).
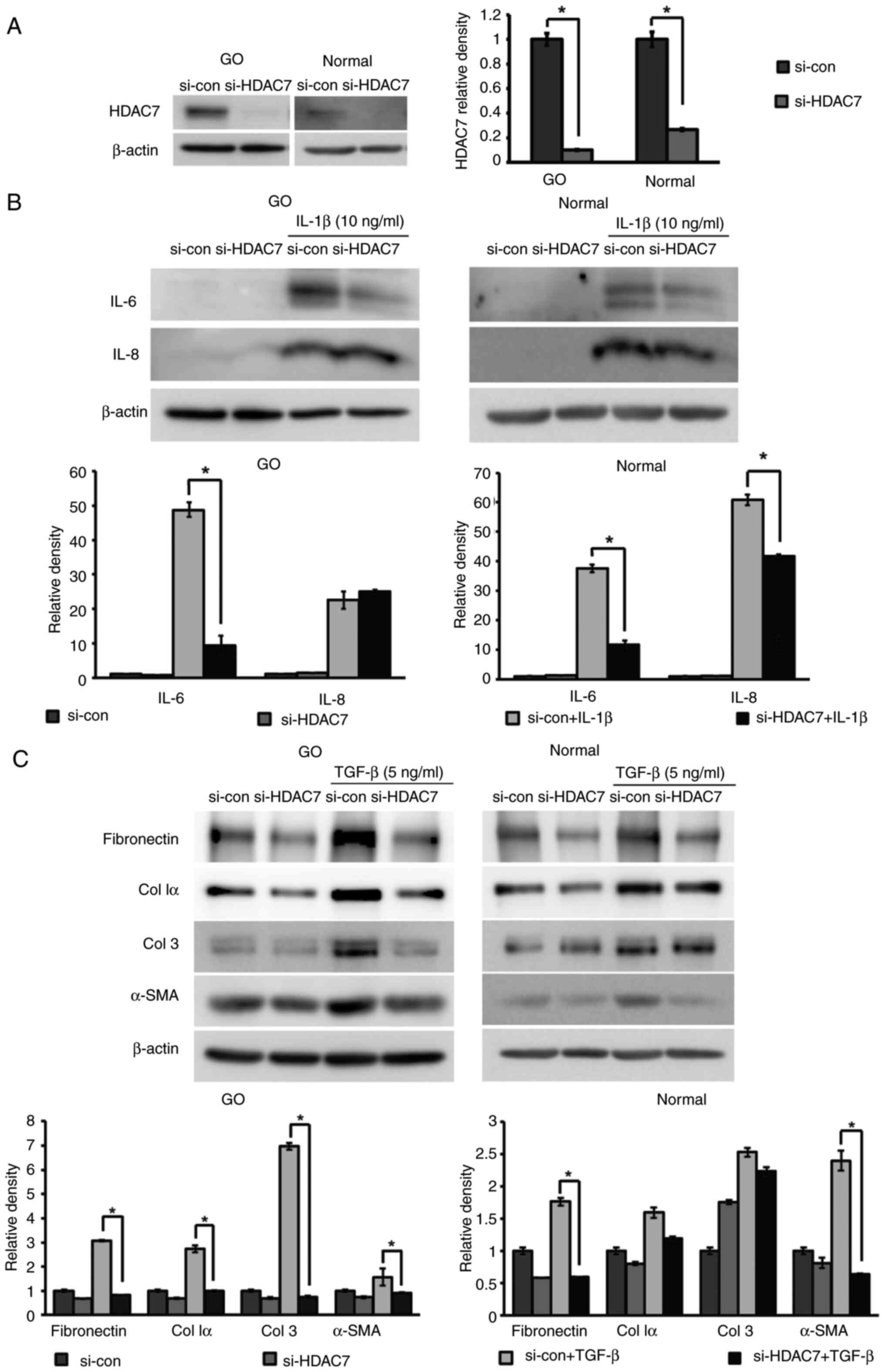 | Figure 10.Anti-inflammatory and anti-fibrotic
effects of silencing HDAC7 in orbital fibroblasts. HDAC7 was
knocked down via transfection with siRNA (10 nM) for 24 h and cells
were maintained for 48 h. (A) Silencing efficiency of si-HDAC7 was
demonstrated in both GO and normal orbital fibroblasts (n=3)
compared with si-con. (B) Cells were stimulated with IL-1β (10
ng/ml) for 24 h. Western blot analysis was performed to investigate
the expression levels of the proinflammatory cytokines IL-6 and
IL-8. (C) Orbital fibroblasts were treated with TGF-β (5 ng/ml) for
24 h, and profibrotic proteins, including fibronectin, Col Iα, Col
3 and α-SMA, were examined using western blot analysis. β-actin was
used as a loading control for normalization. Each experiment was
performed in duplicate. Data are presented as the mean ± SD.
Statistical significance was determined using Mann-Whitney U-test
to compare the effect of silencing HDAC7 under IL-1β or TGF-β
stimulation. *P<0.05. α-SMA, α-smooth muscle actin; Col,
collagen; con, control; GO, Graves' orbitopathy; HDAC7, histone
deacetylase 7; si/siRNA, small interfering RNA. |
Additional experiments were performed to determine
the effects of HDAC3 and HDAC6, which were significantly
upregulated and downregulated by panobinostat, respectively
(Fig. 3). Firstly, HDAC6 was
knocked down by siRNA transfection (Fig. S3A), and the orbital fibroblasts
were treated with IL-1β (10 ng/ml) or TGF-β (5 ng/ml). The protein
expression levels of the IL-1β-induced proinflammatory cytokines,
IL-6 and IL-8, did not differ between cells transfected with
si-HDAC6 and si-con (Fig. S3B).
Upon TGF-β stimulation, the expression levels of profibrotic
proteins, fibronectin and α-SMA, were downregulated by si-HDAC6
transfection in normal cells; however, the expression levels of
Col1α and Col3 in normal cells and of all profibrotic proteins in
GO cells showed no difference in response to si-HDAC6 transfection
(Fig. S3C).
Secondly, as shown in Fig. 3, HDAC7 mRNA expression was
decreased, whereas HDAC3 mRNA expression was significantly
upregulated by panobinostat treatment. As the enzymatic activity of
HDAC7 in the nucleus is influenced by HDAC3 (29), the effect of siHDAC7 on HDAC3
expression was investigated. The effects of panobinostat on the
protein expression levels of HDAC3 and HDAC7 in orbital fibroblasts
were confirmed by western blotting, demonstrating downregulation of
HDAC7 and upregulation of HDAC3, in accordance with the mRNA
results (Fig. S4A). However,
silencing of HDAC7 did not affect the protein expression levels of
HDAC3 in orbital fibroblasts (Fig.
S4B).
Discussion
In the present study, the pan-HDACi panobinostat was
identified as a potent attenuator of orbital fibroblast activation.
Panobinostat suppressed the expression of proinflammatory,
profibrotic and adipogenic proteins induced by specific stimulators
in orbital fibroblasts. Furthermore, it selectively inhibited
different HDAC genes in GO orbital fibroblasts. Notably, blockage
of HDAC7 was revealed to be associated with the therapeutic effect
of panobinostat on GO pathogenesis.
The orbital tissues analyzed were primarily derived
from patients undergoing orbital decompression surgery, typically
performed during the inactive phase of GO. Most HDAC genes in GO
orbital tissues exhibited reduced mRNA expression compared with
that in normal orbital tissues, possibly due to negative feedback
after the active phase. Additionally, HDAC7 protein expression was
increased in orbital fibroblasts following stimulation with IL-1β
and TGF-β. However, the present study revealed no significant
differences in the mRNA expression levels of HDAC genes in PBMCs
between patients with GO and healthy controls. This is in contrast
to a previous study that reported elevated mRNA expression levels
of HDAC1 and HDAC2 in PBMCs from patients with Graves' disease,
leading to histone H4 hypoacetylation (30). This discrepancy could be because
PBMCs are a mixed collection of different cells, such as T cells, B
cells, macrophages and mast cells, which might obscure specific
changes in HDAC expression. Furthermore, a number of systemic
inflammatory diseases, such as rheumatoid arthritis and chronic
obstructive pulmonary disease, can influence HDAC mRNA expression
in PBMCs (31,32), while GO may represent a more
localized inflammatory manifestation in the orbit. Therefore,
despite the lack of differences in PBMC HDAC mRNA expression, HDACs
may serve as specific biomarkers for tissue and localized
therapeutic targets in GO pathogenesis, as the present study
identified marked differences in HDAC expression in GO orbital
tissue, and HDAC inhibition in orbital fibroblasts suppressed the
expression of pro-inflammatory, adipogenic and pro-fibrotic
proteins.
A recent report showed that HDAC4 expression was
increased upon stimulation with platelet-derived growth factor-BB
(PDGF-BB) in GO orbital fibroblasts, whereas HDAC4 silencing
reduced collagen 1α1, α-SMA, hyaluronan synthase 2 and hyaluronic
acid production (22). This aligns
with the present finding that HDAC inhibition could be a potential
therapy for alleviating GO-related tissue remodeling. However,
unlike previous research that focused on hyaluronan, the present
study investigated proinflammatory cytokines, which are the more
fundamental causes of GO, including IL-6 and IL-8 (2,33–36).
Another focus of the current study was fibrosis, which could
potentially result in long-lasting impairment. Lastly, although
PDGF-BB serves a pivotal role in GO pathogenesis, it stems from
immune cells activated by numerous cytokines, including IL-1β,
IFN-γ and TGF-β (37); therefore,
Therefore, the inhibitory effects of panobinostat on broader
regulators, such as IL-1β and TGF-β, which act upstream of PDGF-BB,
may have a more significant impact on treating GO.
Despite their pan-inhibitory nature, the sensitivity
of each pan-HDACi varies in different cellular contexts (28). In myeloma cells, panobinostat has
been shown to predominantly upregulate HDAC6, uniquely reducing
HDAC7 (38). In the present study,
panobinostat exhibited a selective inhibitory effect on the mRNA
expression levels of HDACs in GO orbital fibroblasts, reducing the
expression of both HDAC6 and 7, while elevating HDAC3 expression.
Similarly, another pan-HDACi, trichostatin A (TSA), has been
reported to increase HDAC3 expression and decrease HDAC7 expression
in the skin fibroblasts of patients with systemic sclerosis
(39). The present study explored
the therapeutic mechanism of panobinostat by targeting specific
HDACs to mitigate the adverse effects associated with pan-HDAC
inhibition. Specific inhibition using si-HDAC6 and si-HDAC7
confirmed that the downregulation of HDAC7 was associated with the
anti-inflammatory and anti-fibrotic effects of panobinostat, in
contrast to HDAC6 silencing, which showed no effect on the
expression of inflammatory and fibrotic proteins. As the enzymatic
activity of HDAC7 in the nucleus is influenced by HDAC3, which
binds to silencing mediator of retinoic acid and thyroid hormone
receptor and nuclear receptor corepressor (29), the present study examined whether
the therapeutic effect of panobinostat could occur directly through
HDAC7 reduction, not mediated by elevated HDAC3. Since HDAC7
reduction did not induce HDAC3 elevation, it was concluded that
HDAC7 may independently mediate the therapeutic effect of
panobinostat.
HDAC7 serves a crucial role in the pathogenesis of
inflammation-related diseases associated with fibrosis. One study
demonstrated that cytokine-induced type I collagen and fibronectin
levels were reduced by TSA treatment through the inhibition of SMAD
transcription factors in systemic sclerosis skin fibroblasts
(40). In a subsequent study, it
was confirmed that TGF-β-induced type I and type III collagen
expression was reduced by HDAC7 knockdown (39). In fibroblasts derived from
Peyronie's plaques, HDAC7 silencing has been shown to curtail
fibroblast differentiation into myofibroblasts and decrease
fibrotic protein production (41).
In the lung tissue from a mouse model of ovalbumin-induced
fibrosis, nuclear-translocated HDAC7 upon endothelin-1 stimulation
promoted connective tissue growth factor production (42). Another study deemed HDAC7 pivotal
for HDAC-dependent deacetylation in the promotor region of the
anti-fibrotic gene PPARGC1A under TGF-β stimulation (19).
Previous studies have suggested that HDAC inhibition
may affect the SMAD-dependent canonical TGF-β signaling pathway
(40,41). However, the findings of the present
study regarding the downstream target of panobinostat in orbital
fibroblasts suggested that Akt was a possible signaling pathway
molecule that may be present during both TGF-β and IL-1β
stimulation. In a few previous studies on idiopathic pulmonary
fibrosis, the PI3K-Akt signaling pathway was observed. Tubastatin
A, a selective inhibitor of HDAC6, was shown to abrogate
TGF-β-induced type I collagen expression by suppressing Akt
phosphorylation in lung fibroblasts (43,44).
Additionally, TSA treatment reduced α-SMA expression in response to
TGF-β, which was dependent on HDAC4 mediated by phosphorylation of
Akt (45). Taken together, Akt
signaling may contribute to the therapeutic effects of HDAC
inhibition not only on inflammation but also on GO fibrosis.
The present study has several limitations. First,
the sample size for each experiment was limited and varied. The
discrepancy in sample numbers arose due to differences in the
invasiveness of collecting different types of samples, tissue or
blood. To address the limitations associated with pooled sample
analysis, subgroup analysis in patients with GO according to CAS
was conducted in PBMCs, which did not reveal any significant
differences (data not shown). Additionally, in the in vitro
study using orbital fibroblasts, primary orbital fibroblasts were
cultured from three different tissue samples in a randomized manner
and the experiments were performed twice to obtain average results.
This approach helped to minimize the limitations of pooled sample
analysis by incorporating controlled variability and ensuring more
representative results. Second, the experiments were limited to
orbital fibroblasts. The present study is a preliminary in
vitro study using orbital fibroblasts. Since a mouse model for
GO is currently under investigation and shows promising results
(46), HDACis could potentially be
applied to in vivo systems. Lastly, unlike pan-HDACis, a
specific HDACi could be used. Using a specific HDACi is desirable
to reduce the effects on other HDACs and to minimize the toxicity
associated with pan-HDAC inhibition. To date, only a few potent
HDAC7is have been identified. They exhibit high potency as HDAC7is;
however, they also affect other class IIa HDACs (HDAC4, 5 and 9)
(47). Notably, class IIa HDACs
are associated with various immune cells, including macrophages and
lymphocytes (48). Furthermore,
given that HDAC4, one of the class IIa HDACs, has been recognized
as a target for hyaluronan production in GO (22), HDAC7is still have therapeutic
potential against GO even though they inhibit other class IIa HDACs
in addition to HDAC7. Further investigation is necessary when
considering the use of HDAC7is in vivo.
In conclusion, the present study on GO highlights
the therapeutic potential of HDAC inhibition. Specifically,
panobinostat was identified as a potent inhibitor of
proinflammatory, adipogenic and profibrotic stimulation in GO
orbital fibroblasts, predominantly via the Akt signaling pathway.
The anti-inflammatory and anti-fibrotic actions of panobinostat
were evident through its downregulation of HDAC7 mRNA expression.
Considering the crucial role of HDACs in autoimmune processes and
fibrosis, the present findings demonstrate the potential of
HDAC7-targeted interventions. As the understanding of GO
pathogenesis advances, the current study proposes epigenetic
modulation, especially by targeting HDAC7, as a promising
therapeutic option. Further studies should focus on elucidating
broader molecular mechanisms and validating these findings in
vivo.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Portions of this study were presented at the ASOPRS
2022 Fall Scientific Symposium (Chicago, IL, USA).
Funding
This work was supported by the National Research Foundation of
Korea grant funded by the Korean government (Ministry of Science
and ICT; grant no. RS-2023-00208570).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
JSY and HJB designed the present study. SHC
performed the experiments, and JSY, HJB and JK confirmed the
authenticity of all the raw data. JSY, HJB, JK and DOK analyzed and
interpreted the data. JSY and JK provided the laboratory, reagents
and materials, and JK procured the funding. HJB, SHC and DOK wrote
the manuscript, and JSY and JK edited it. All authors have read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
The present study was conducted according to the
guidelines of The Declaration of Helsinki, and was approved by the
Institutional Review Board of the Severance Hospital, Yonsei
University College of Medicine (IRB no. 4-2022-0244; Seoul, South
Korea). Written informed consent was obtained from all subjects
involved in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lazarus JH: Epidemiology of Graves'
orbitopathy (GO) and relationship with thyroid disease. Best Pract
Res Clin Endocrinol Metab. 26:273–279. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bahn RS: Graves' ophthalmopathy. N Engl J
Med. 362:726–738. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bartley GB, Fatourechi V, Kadrmas EF,
Jacobsen SJ, Ilstrup DM, Garrity JA and Gorman CA: Clinical
features of Graves' ophthalmopathy in an incidence cohort. Am J
Ophthalmol. 121:284–290. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rotondo Dottore G, Torregrossa L, Lanzolla
G, Mariotti S, Menconi F, Piaggi P, Cristofani Mencacci L,
Posarelli C, Maglionico MN, Dallan I, et al: Role of the
mononuclear cell infiltrate in Graves' orbitopathy (GO): Results of
a large cohort study. J Endocrinol Invest. 45:563–572. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huang Y, Fang S, Li D, Zhou H, Li B and
Fan X: The involvement of T cell pathogenesis in thyroid-associated
ophthalmopathy. Eye (Lond). 33:176–182. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yin X, Latif R, Bahn R and Davies TF:
Genetic profiling in Graves' disease: Further evidence for lack of
a distinct genetic contribution to Graves' ophthalmopathy. Thyroid.
22:730–736. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hadj-Kacem H, Rebuffat S, Mnif-Féki M,
Belguith-Maalej S, Ayadi H and Péraldi-Roux S: Autoimmune thyroid
diseases: Genetic susceptibility of thyroid-specific genes and
thyroid autoantigens contributions. Int J Immunogenet. 36:85–96.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang Y, Ma XM, Wang X, Sun X, Wang LJ, Li
XQ, Liu XY and Yu HS: Emerging insights into the role of
epigenetics and gut microbiome in the pathogenesis of Graves'
ophthalmopathy. Front Endocrinol (Lausanne). 12:7885352022.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rotondo Dottore G, Bucci I, Lanzolla G,
Lanzolla G, Dallan I, Sframeli A, Torregrossa L, Casini G, Basolo
F, Figus M, et al: Genetic profiling of orbital fibroblasts from
patients with Graves' orbitopathy. J Clin Endocrinol Metab.
106:e2176–e2190. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kouzarides T: Chromatin modifications and
their function. Cell. 128:693–705. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shakespear MR, Halili MA, Irvine KM,
Fairlie DP and Sweet MJ: Histone deacetylases as regulators of
inflammation and immunity. Trends Immunol. 32:335–343. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Narita T, Weinert BT and Choudhary C:
Functions and mechanisms of non-histone protein acetylation. Nat
Rev Mol Cell Biol. 20:156–174. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gatla HR, Muniraj N, Thevkar P, Yavvari S,
Sukhavasi S and Makena MR: Regulation of chemokines and cytokines
by histone deacetylases and an update on histone decetylase
inhibitors in human diseases. Int J Mol Sci. 20:11102019.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen H, Pan J, Wang JD, Liao QM and Xia
XR: Suberoylanilide hydroxamic acid, an inhibitor of histone
deacetylase, induces apoptosis in rheumatoid arthritis
fibroblast-like synoviocytes. Inflammation. 39:39–46. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bahn RS: Thyrotropin receptor expression
in orbital adipose/connective tissues from patients with
thyroid-associated ophthalmopathy. Thyroid. 12:193–195. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang Y and Zhang B: Trichostatin A, an
inhibitor of histone deacetylase, inhibits the viability and
invasiveness of hypoxic rheumatoid arthritis fibroblast-like
synoviocytes via PI3K/Akt signaling. J Biochem Mol Toxicol.
30:163–169. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Oh BR, Suh DH, Bae D, Ha N, Choi YI, Yoo
HJ, Park JK, Lee EY, Lee EB and Song YW: Therapeutic effect of a
novel histone deacetylase 6 inhibitor, CKD-L, on collagen-induced
arthritis in vivo and regulatory T cells in rheumatoid arthritis in
vitro. Arthritis Res Ther. 19:1542017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
de Zoeten EF, Wang L, Butler K, Beier UH,
Akimova T, Sai H, Bradner JE, Mazitschek R, Kozikowski AP, Matthias
P and Hancock WW: Histone deacetylase 6 and heat shock protein 90
control the functions of Foxp3(+) T-regulatory cells. Mol Cell
Biol. 31:2066–2078. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jones DL, Haak AJ, Caporarello N, Choi KM,
Ye Z, Yan H, Varelas X, Ordog T, Ligresti G and Tschumperlin DJ:
TGFβ-induced fibroblast activation requires persistent and targeted
HDAC-mediated gene repression. J Cell Sci. 132:jcs2334862019.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Korfei M, Stelmaszek D, MacKenzie B,
Skwarna S, Chillappagari S, Bach AC, Ruppert C, Saito S, Mahavadi
P, Klepetko W, et al: Comparison of the antifibrotic effects of the
pan-histone deacetylase-inhibitor panobinostat versus the IPF-drug
pirfenidone in fibroblasts from patients with idiopathic pulmonary
fibrosis. PLoS One. 13:e02079152018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sanders YY, Hagood JS, Liu H, Zhang W,
Ambalavanan N and Thannickal VJ: Histone deacetylase inhibition
promotes fibroblast apoptosis and ameliorates pulmonary fibrosis in
mice. Eur Respir J. 43:1448–1458. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ekronarongchai S, Palaga T, Saonanon P,
Pruksakorn V, Hirankarn N, van Hagen PM, Dik WA and Virakul S:
Histone deacetylase 4 controls extracellular matrix production in
orbital fibroblasts from graves' ophthalmopathy patients. Thyroid.
31:1566–1576. 2021.PubMed/NCBI
|
|
23
|
Atadja P: Development of the pan-DAC
inhibitor panobinostat (LBH589): Successes and challenges. Cancer
Lett. 280:233–241. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shao W, Growney J, Feng Y, Wang P,
Yan-Neale Y, O'Connor G, Kwon P, Yao YM, Fawell S and Atadja P:
Potent anticancer activity of a pan-deacetylase inhibitor
panobinostat (LBH589) as a single agent in in vitro and in vivo
tumor models. Cancer Res. 68 (9 Suppl):S7352008.
|
|
25
|
Mourits MP, Koornneef L, Wiersinga WM,
Prummel MF, Berghout A and van der Gaag R: Clinical criteria for
the assessment of disease activity in Graves' ophthalmopathy: A
novel approach. Br J Ophthalmol. 73:639–644. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Byeon HJ, Chae MK, Ko J, Lee EJ, Kikkawa
DO, Jang SY and Yoon JS: The role of adipsin, complement factor D,
in the pathogenesis of Graves' orbitopathy. Invest Ophthalmol Vis
Sci. 64:132023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dokmanovic M, Perez G, Xu W, Ngo L, Clarke
C, Parmigiani RB and Marks PA: Histone deacetylase inhibitors
selectively suppress expression of HDAC7. Mol Cancer Ther.
6:2525–2534. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fischle W, Dequiedt F, Fillion M, Hendzel
MJ, Voelter W and Verdin E: Human HDAC7 histone deacetylase
activity is associated with HDAC3 in vivo. J Biol Chem.
276:35826–35835. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yan N, Zhou JZ, Zhang JA, Cai T, Zhang W,
Wang Y, Muhali FS, Guan L and Song RH: Histone hypoacetylation and
increased histone deacetylases in peripheral blood mononuclear
cells from patients with Graves' disease. Mol Cell Endocrinol.
414:143–147. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tan C, Xuan L, Cao S, Yu G, Hou Q and Wang
H: Decreased histone deacetylase 2 (HDAC2) in peripheral blood
monocytes (PBMCs) of COPD patients. PLoS One. 11:e01473802016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li Y, Zhou M, Lv X, Song L, Zhang D, He Y,
Wang M, Zhao X, Yuan X, Shi G and Wang D: Reduced activity of HDAC3
and increased acetylation of histones H3 in peripheral blood
mononuclear cells of patients with rheumatoid arthritis. J Immunol
Res. 2018:73135152018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xie K: Interleukin-8 and human cancer
biology. Cytokine Growth Factor Rev. 12:375–391. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sampson AP: The role of eosinophils and
neutrophils in inflammation. Clin Exp Allergy. 30 (Suppl
1):S22–S27. 2000. View Article : Google Scholar
|
|
35
|
Gu LQ, Jia HY, Zhao YJ, Liu N, Wang S, Cui
B and Ning G: Association studies of interleukin-8 gene in Graves'
disease and Graves' ophthalmopathy. Endocrine. 36:452–456. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Antonelli A, Fallahi P, Elia G, Ragusa F,
Paparo SR, Ruffilli I, Patrizio A, Gonnella D, Giusti C, Virili C,
et al: Graves' disease: Clinical manifestations, immune
pathogenesis (cytokines and chemokines) and therapy. Best Pract Res
Clin Endocrinol Metab. 34:1013882020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Virakul S, van Steensel L, Dalm VASH,
Paridaens D, van Hagen PM and Dik WA: Platelet-derived growth
factor: A key factor in the pathogenesis of graves' ophthalmopathy
and potential target for treatment. Eur Thyroid J. 3:217–226. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cheng T, Kiser K, Grasse L, Iles L,
Bartholomeusz G, Samaniego F, Orlowski RZ and Chandra J: Expression
of histone deacetylase (HDAC) family members in
bortezomib-refractory multiple myeloma and modulation by
panobinostat. Cancer Drug Resist. 4:888–902. 2021.PubMed/NCBI
|
|
39
|
Hemmatazad H, Rodrigues HM, Maurer B,
Brentano F, Pileckyte M, Distler JH, Gay RE, Michel BA, Gay S,
Huber LC, et al: Histone deacetylase 7, a potential target for the
antifibrotic treatment of systemic sclerosis. Arthritis Rheum.
60:1519–1529. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Huber LC, Distler JHW, Moritz F,
Hemmatazad H, Hauser T, Michel BA, Gay RE, Matucci-Cerinic M, Gay
S, Distler O and Jüngel A: Trichostatin A prevents the accumulation
of extracellular matrix in a mouse model of bleomycin-induced skin
fibrosis. Arthritis Rheum. 56:2755–2764. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kang DH, Yin GN, Choi MJ, Song KM, Ghatak
K, Minh NN, Kwon MH, Seong DH, Ryu JK and Suh JK: Silencing histone
deacetylase 7 alleviates transforming growth factor-β1-induced
profibrotic responses in fibroblasts derived from peyronie's
plaque. World J Mens Health. 36:139–146. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hua HS, Wen HC, Weng CM, Lee HS, Chen BC
and Lin CH: Histone deacetylase 7 mediates endothelin-1-induced
connective tissue growth factor expression in human lung
fibroblasts through p300 and activator protein-1 activation. J
Biomed Sci. 28:382021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Saito S, Zhuang Y, Shan B, Danchuk S, Luo
F, Korfei M, Guenther A and Lasky JA: Tubastatin ameliorates
pulmonary fibrosis by targeting the TGFβ-PI3K-Akt pathway. PLoS
One. 12:e01866152017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu Y, Wang R, Han H and Li L: Tubastatin
A suppresses the proliferation of fibroblasts in epidural fibrosis
through phosphatidylinositol-3-kinase/protein kinase B/mammalian
target of rapamycin (PI3K/AKT/mTOR) signalling pathway. J Pharm
Pharmacol. 74:426–434. 2022. View Article : Google Scholar
|
|
45
|
Guo W, Shan B, Klingsberg RC, Qin X and
Lasky JA: Abrogation of TGF-beta1-induced fibroblast-myofibroblast
differentiation by histone deacetylase inhibition. Am J Physiol
Lung Cell Mol Physiol. 297:L864–L870. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bao Y, Kim D, Cho YH, Ku CR, Yoon JS and
Lee EJ: Cre-loxP system-based mouse model for investigating Graves'
disease and associated orbitopathy. Thyroid. 33:1358–1367. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang Y, Abrol R, Mak JYW, Das Gupta K,
Ramnath D, Karunakaran D, Fairlie DP and Sweet MJ: Histone
deacetylase 7: A signalling hub controlling development,
inflammation, metabolism and disease. FEBS J. 290:2805–2832. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liu L, Dong L, Bourguet E and Fairlie DP:
Targeting class IIa HDACs: Insights from phenotypes and inhibitors.
Curr Med Chem. 28:8628–8672. 2021. View Article : Google Scholar : PubMed/NCBI
|