Introduction
Abnormal changes in the phenotype and function of
vascular smooth muscle cells (VSMCs) have been implicated in
several vascular diseases, such as aneurysms and atherosclerosis
(1). VSMCs can have two
phenotypes: A contractile phenotype [markers: α-smooth muscle actin
(SMA), smooth muscle protein 22a (SM22a), myosin heavy chain 11
(MYH11)] and a synthetic phenotype [markers: Matrix
metalloproteinase (MMP) 2, tumor necrosis factor-α, osteopontin
(OPN)] (2,3). During disease development, the
phenotype of VSMCs can change from contractile to synthetic
(4,5).
Autophagy is a process wherein cells use lysosomes
to break down intracellular organelles and macromolecules (6). Alterations in the autophagy status of
VSMCs due to various external factors can lead to abnormal cell
proliferation, migration and matrix secretion (7). Cytokines activate signaling pathways
that affect autophagy and thereby influence phenotypic and
functional changes in VSMCs (8,9). The
transition from the contractile to the synthetic phenotype occurs
following autophagy of VSMCs (10). Therefore, identifying molecular
targets that can effectively regulate the phenotype and function of
VSMCs is crucial for the treatment of vascular diseases.
Long non-coding RNA (lncRNA) is a class of RNA
molecules that are >200 nt in length and do not encode proteins.
It has been discovered that lncRNAs play a crucial role in the
development of various diseases (11–13).
Small nucleolar RNA host gene 1 (SNHG1), a small ribosomal
housekeeping gene, is a lncRNA that originates from the U22 host
gene on chromosome 11 and is an important component of the 18 s
ribosomal RNA. It consists of ~3,927 bases and contains eight small
nucleotide RNAs (14,15). SNHG1 has been found to be
abnormally expressed in various diseases and regulates the
biological behavior of the disease via its target genes (16–20).
C-type lectin domain family 7 member A (CLEC7A), also known as
Dectin-1, is a member of the CLEC7 family and its gene encodes the
C-lectin/C-lectin domain-like domain. The encoded glycoprotein is a
small type II membrane receptor with an extracellular C-type
lectin-like domain fold and a cytoplasmic domain with an immune
receptor tyrosine activation motif (21). The functions of CLEC7A include
phagocytosis, production of reactive oxygen species and the
secretion of pro-inflammatory cytokines, which are essential for
antifungal defense (22,23). In addition, CLEC7A recognizes other
pathogens and endogenous ligands, suggesting a broader role in
immunity (24).
Aneurysms are closely related to the abnormality of
VSMCs. Overexpression of CLEC7A has been shown to be associated
with aneurysm healing following embolization (25). CLEC7A can also distinguish
intracranial aneurysms from normal samples (26).
Our previous study showed that the expression of
CLEC7A is significantly higher in intracranial aneurysms compared
with normal vascular tissue (27),
indicating the association of CLEC7A with VMSCs.
Based on the existing literature and previous
research, it was hypothesized that SNHG1 regulates CLEC7A to
modulate the status of autophagy in VSMCs, which in turn leads to
the development of vascular diseases. The present study used
rapamycin to induce autophagy in VSMCs. Following manipulation of
the expression of SNHG1 by silencing and overexpression, the
occurrence of autophagy, phenotypic changes and migratory ability
was assessed in VSMCs. VSMCs were then transduced with vectors
containing silenced or overexpressed SNHG1 to examine the
expression of CLEC7A. The interaction between SNHG1 and CLEC7A was
determined using a RNA-binding protein immunoprecipitation (RIP)
assay and a RNA pull-down assay. Finally, the phenotypic changes in
VSMCs were investigated by combining overexpression of CLEC7A with
silencing of SNHG1. The findings aimed to provide insights into the
mechanism by which SNHG1 regulates the phenotypic transition of
VSMCs by mediating autophagy via CLEC7A.
Materials and methods
Cell culture
Immortalized human aortic smooth muscle cells
(VSMCs) were purchased from Meisen Chinese Tissue Culture
Collections (cat. no. CTCC-001-0577) and seeded in complete medium
(cat. no. CTCC-001-0577-CM, Meisen Cell Technology Co., Ltd), which
consisted of F-12K medium containing 0.05 mg/ml ascorbic acid, 0.01
mg/ml insulin, 0.01 mg/ml transferrin, 10 ng/ml sodium selenite,
0.03 mg/ml endothelial cell growth supplement, 10 mM
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 10 mM TES, 10%
fetal bovine serum and 1% antibiotic-antimycotic medium under
humidified conditions (37°C; 5% CO2).
Cell transduction
Short hairpin RNAs (shRNAs) designed to target SNHG1
along with their corresponding empty vector (vector-sh) and the
plasmids for overexpression (OE) of SNHG1 or CLEC7A and the empty
vector (vector-OE) were procured from Hanbio Biotechnology Co.,
Ltd. The 2nd generation lentiviral system was used for
transfection. All recombinant lentivirus sequences were generated
as previously described (28).
VSMCs were transduced with lentiviral vectors containing SNHG1
silencing sequences, designated sh-SNHG1. Similarly, lentiviral
vectors containing human SNHG1 or CLEC7A overexpression sequences
were introduced into VSMCs, designated OE-SNHG1 and OE-CLEC7A,
respectively. The viral supernatant was used to infect target cells
at an MOI of 10. The VSMCs underwent a 72-h incubation period with
the recombinant lentiviruses and subsequently underwent puromycin
selection for 72 h using 3 µg/ml puromycin for selection of
transfected cells and 3 µg/ml for maintenance of the stable cell
lines. Finally, transduction efficacy was validated by assessing
mRNA expression levels using reverse transcription-quantitative
(RT-q) PCR. Experiments were conducted 48 h after the completion of
puromycin selection. The sequences of shRNA are detailed in
Table SI, whereas the sequences
utilized for overexpression are presented in Table SII.
RT-qPCR
RT-qPCR was used to quantitatively analyze gene
expression as previously described (29). Total RNA was extracted from VSMCs
with TRIzol® reagent (Thermo Fisher Scientific, Inc.).
The cDNA was synthesized from the RNA using a reverse transcriptase
kit (cat. no. RR036A; Takara Biotechnology Co., Ltd.) according to
the manufacturer's protocol. qPCR was performed using SYBR Green
qPCR Master Mix (cat. no. Q711-02; Nanjing Vazyme Biotech Co., Ltd)
following the manufacturer's protocol. The qPCR thermocycling
conditions were as follows: Initial denaturation at 95°C for 30
sec, followed by 40 cycles of denaturation at 95°C for 10 sec,
annealing at 56°C for 30 sec, and extension at 72°C for 60 sec.
GAPDH was used as an internal control. Relative expression levels
were calculated using the 2−ΔΔCq method (30). The experiment was performed as
three independent replicates. The primers used are listed in
Table SIII.
Western blotting
Western blot procedures were in accordance with
standard protocols as described in the earlier study by Yue et
al (31). Briefly, the
cellular proteins were extracted with radioimmunoprecipitation
assay buffer (cat. no. R0010; Beijing Solarbio Science &
Technology Co., Ltd.) and the protein concentration was determined
using the BCA Protein Assay Kit (cat. no. P0012S; Beyotime
Institute of Biotechnology) according to the manufacturer's
protocol. Proteins were then separated by SDS-PAGE on 12.5% gels
for LC3, SM22α and CLEC7A, and 10% gels for p62, BECN1, OPN, GAPDH
and β-actin. Following electrophoresis, the proteins were
transferred to a polyvinylidene membrane and subsequently blocked
with 5% skimmed milk in Tris-buffered saline with 1% Tween 20 for 1
h. Primary antibodies were incubated overnight at 4°C, while
treatment with secondary antibodies was performed at room
temperature for ~1 h. The antibodies used in this work were as
follows: Rabbit anti-LC3 (1:1,000; cat. no. 4108S; Cell Signaling
Technology, Inc.); Rabbit anti-P62 (1:1,000; cat. no. 16177S; Cell
Signaling Technology, Inc.); Rabbit anti-SM22a (1:1,000; cat. no.
CSB-PA00804A0Rb; Cusabio Technology, LLC); rabbit anti-α-smooth
muscle actin (α-SMA) (1:1,000; cat. no. R23450; Chengdu Zhengneng
Biotechnology Co., Ltd.); rabbit anti-osteopontin (OPN) (1:1,000;
cat. no. R26171; ZENBIO); rabbit anti-CLEC7A (1:1,000; cat. no.
PA5-34382; Thermo Fisher Scientific, Inc.); rabbit anti-GAPDH
(1:1,000; cat. no. GB15004-100; Wuhan Servicebio Technology Co.,
Ltd.); β-actin (1:1,000; cat. no. 380624; Chengdu Zhengneng
Biotechnology Co., Ltd.) anti-rabbit immunoglobulin (Ig) G,
horseradish peroxidase-linked antibody (1:10,000; cat. no. ab6721;
Abcam). The proteins were visualized using the ECL Western Blotting
Detection System (cat. no. 143237; Biosharp Life Sciences)
according to the manufacturer's instructions. Relative target
protein levels were quantified using ImageJ software (version
1.8.0; National Institutes of Health). GAPDH or β-actin served as
the internal control for normalization of target protein levels.
The experiment was performed in three independent replicates.
Cell counting kit-8 (CCK-8) assay
Rapamycin was first dissolved in a 100 mM dimethyl
sulfoxide (DMSO) solution (cat. no. D8371-50 ml; Beijing Solarbio
Science & Technology Co., Ltd.) and further diluted in fresh
medium to reach a concentration of 100 nM before use. Cells in the
logarithmic growth phase were dissociated using 0.25% trypsin to
prepare cell suspension with a density of 3×104
cells/ml, subsequently seeded into 96-well plates at 100 µl per
well. After a 48-h incubation period, the transduced cells were
detached using trypsin and reseeded at a concentration of
1×104 cells/ml. Subsequently, 100 ml of the cell
suspension was dispensed into the 96-well plates. Then three wells
were randomly selected at 0, 12 and 24 h after cultivation in a
controlled environment at 37°C. Each well was then treated with 10
ml of CCK-8 (cat. no. MA0218; MeilunBio) and further incubated for
2 h. Finally, the optical density (OD) was measured at 450 nm and a
curve was plotted to represent the dynamics of cellular growth.
Wound healing assays
VSMCs were seeded in a six-well plate, transduced
and subsequently treated. After a 24-h incubation period, a linear
scratch was meticulously created across the plate with a 200-µl
pipette tip and fresh serum-free medium was carefully added. Images
were then captured with an inverted fluorescence microscope (Axio
Vert.A1; Carl Zeiss AG) showing the progression of cell migration
after 0, 12 and 24 h. The wound closure was quantified using ImageJ
software (version 1.8.0). The width of the scratch was measured at
multiple points along the scratch, and the average width was
calculated. The wound closure rate was determined using the
formula: Wound closure rate (%)=(initial scratch width-current
scratch width)/initial scratch width ×100.
RIP assay
To investigate the interaction between the SNHG1
sequence and the CLEC7A protein, an RIP assay was performed
according to the manufacturer's protocol (cat. no. Bes5101;
Guangzhou BersinBio Biotechnology Co., Ltd.). Cells were washed
with phosphate-buffered saline, lysed in a buffer containing
protease and RNA enzyme inhibitors and divided into three groups:
Input, IP and IgG. Anti-CLEC7A antibody (cat. no. 60128; Cell
Signaling Technology, Inc.) and IgG antibody (cat. no. Bes5101;
Guangzhou BersinBio Biotechnology Co., Ltd.) were used for
incubation with the 0.8 ml lysates of the IP and IgG groups,
respectively. RNA-protein complexes were isolated with 20 µl
protein A/G magnetic beads and then digested with proteinase K.
Proteins bound to RNA were extracted with phenol/chloroform/isoamyl
alcohol (125:24:1) and analyzed by RT-qPCR. The percentage of input
and fold enrichment were calculated to assess the changes in
RNA-protein binding owing to CLEC7A stimulation.
RNA pull-down assay
Guangzhou BersinBio Biotechnology Co., Ltd.
developed a precise probe targeting SNHG1 to uncover potential
proteins that interact with SNHG1. Subsequently, an RNA pulldown
kit (cat. no. Bes5102; Guangzhou BersinBio Biotechnology Co., Ltd.)
was used to isolate proteins associated with SNHG1. For this
purpose, 40 µl magnetic beads were conjugated with the 100 µl SNHG1
probe, forming a complex. This complex of probe and magnetic beads
was subsequently introduced into the 0.8 ml cell lysate, where it
underwent a 2-h incubation that allowed interaction between the
SNHG1 probe and the specific proteins. Finally, 60 µl Protein
Elution Buffer and 0.6 µl dithiothreitol were added, and the
mixture was incubated at 37°C for 2 h with intermittent mixing.
After incubation, the mixture was placed on a magnet for 1 min and
the supernatant was collected. The eluted proteins then underwent
SDS-PAGE followed by western blotting using specific antibodies
against the proteins of interest. The probe sequence used for RNA
pull-down used is listed in Table
SIV.
RPI-Seq database prediction
We utilized the RPI-Seq database (Version 1.0, URL:
http://pridb.gdcb.iastate.edu/RPISeq/), a
computational tool designed to predict interactions between RNA and
proteins. The RPI-Seq database was accessed through its online
platform, and the sequences of SNHG1 and CLEC7A were submitted for
analysis. The prediction algorithm within RPI-Seq evaluates the
likelihood of interaction based on sequence and structural features
of the RNA and protein molecules.
Statistical analysis
Data are presented as mean ± standard deviation.
Statistical significance between two groups was determined using
Student's t-test, while comparisons between multiple groups were
performed using one-way analysis of variance followed by Tukey's
post hoc test. Statistical analyses were performed using GraphPad
Prism version 8 software (GraphPad; Dotmatics). P<0.05 was
considered to indicate a statistically significant difference.
Results
Autophagy is induced by rapamycin in
VSMCs
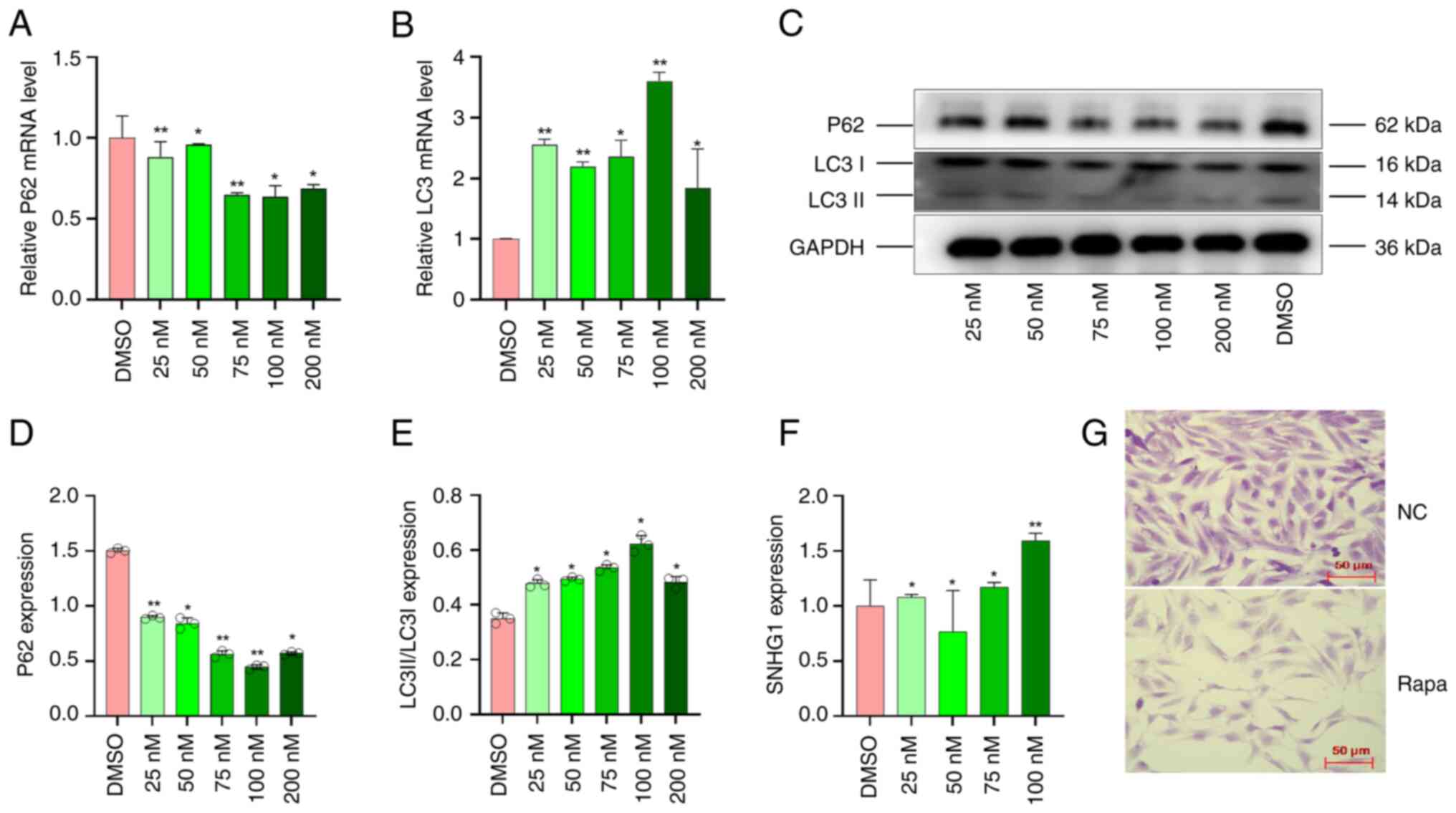
VSMCs were exposed to different concentrations of
rapamycin (25, 50, 75, 100 and 200 nM) for 24 h to induce
autophagy, while DMSO served as a control. The mRNA expression of
autophagy markers (32) LC3 (LC3 I
and LC3 II) and P62 was detected using RT-qPCR. Compared with the
control group, the mRNA expression of P62 was lowest and LC3 was
highest at the concentration of 100 nM (Fig. 1A and B). This indicated that the
concentration of 100 nM was the most suitable for inducing
autophagy. Western blot assay was used to measure the changes in
autophagy markers and showed that the protein concentrations of P62
decreased while those of LC3 I and LC3 II increased (Fig. 1C and E). These results indicated
that the optimal concentration of 100 nM can effectively induce
autophagy in VSMCs.
In addition, the expression of SNHG1 was measured by
RT-qPCR assay; SNHG1 expression increased significantly at a
concentration of 100 nM compared with other concentrations
(Fig. 1F). The morphological
changes in VSMCs treated with 100 nM rapamycin are shown. Normally,
VSMCs have a long spindle shape with clear boundaries and a smooth
surface. Following autophagy is induced, the number of cells
decreases and they become smaller, more irregular and lose their
spindle shape, as shown in Fig.
1G.
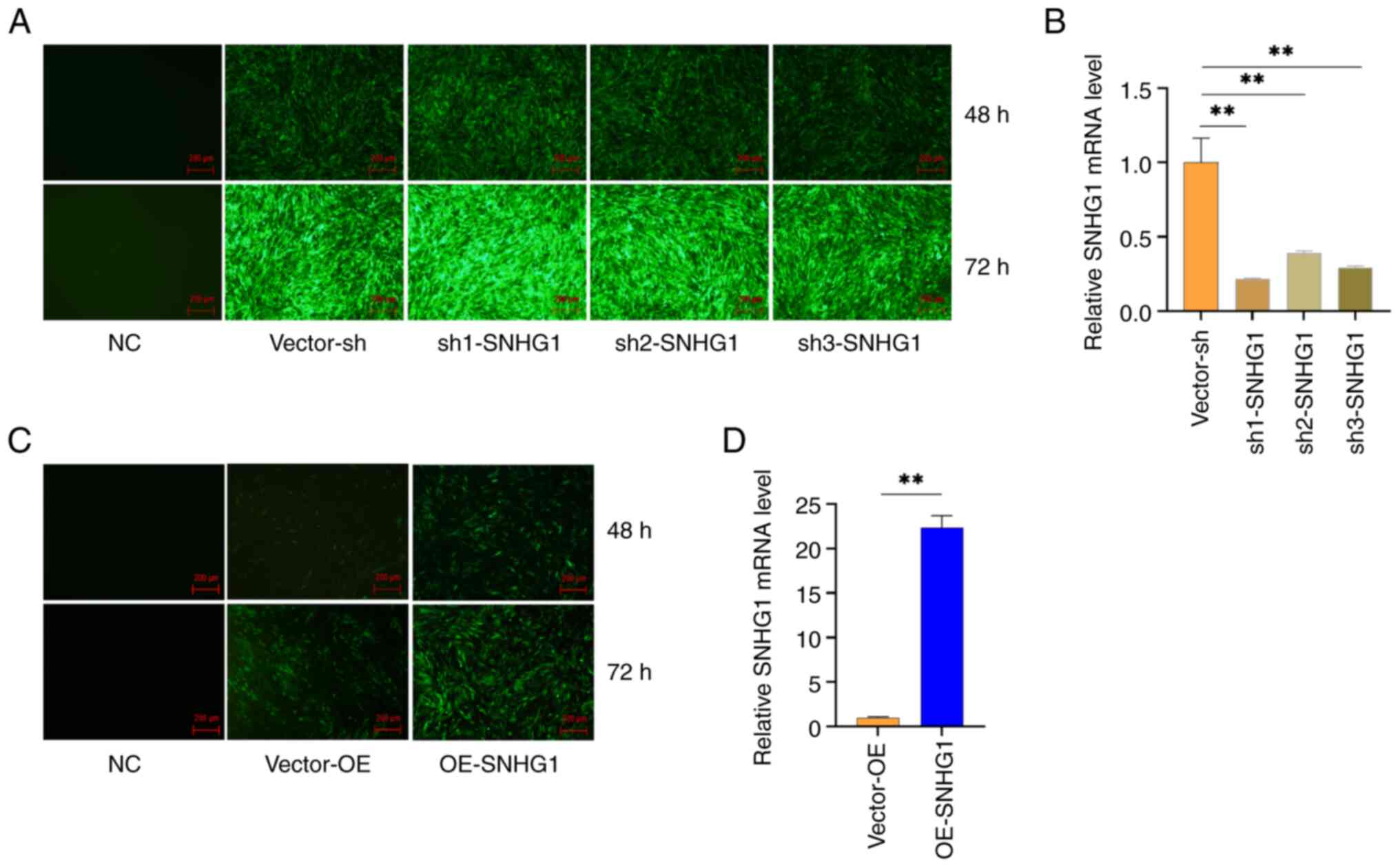
Establishment of stable expression of
SNHG1 in VSMCs
The present study introduced three interfering
sequences into VSMCs using lentiviruses over a period of 48 and 72
h. Subsequently, a fluorescence microscope was used to observe the
morphology and fluorescence expression of the cells. The results
showed that all three interfering sequences led to fluorescence
expression, with a fluorescence rate of >80%. By contrast, the
blank group showed no fluorescence expression, confirming the
successful transfer of the lentivirus into the cells (Fig. 2A). Therefore, a 72-h transduction
was chosen for the following experiment. RT-qPCR was performed to
measure the expression of SNHG1 in the cells. The results showed
that all three silencing sequences significantly inhibited the
expression of SNHG1 in the cells, with Sh1 showing the strongest
inhibition (Fig. 2B). The
overexpression sequence significantly increased the expression of
SNHG1 in the cells, reaching a level 22 times higher than that in
the control group (Fig. 2C and D).
These results indicated that the present study was able to
successfully manipulate SNHG1 levels in VSMCs.
SNHG1 facilitated transition of
phenotype VSMCs
α-SMA, SM22a and OPN are important markers for
phenotypic changes following autophagy. When the phenotype of VSMCs
shifts from contractile to synthetic, the expression of α-SMA and
SM22a decreases, while that of OPN increases (33–35).
The present study investigated the effects of SNHG1 on phenotypic
changes in VSMCs following autophagy induced by rapamycin at a
concentration of 100 nM. It first overexpressed or silenced SNHG1
and then combined treatment with rapamycin in the cells and found
that silencing SNHG1 led to an increase in SM22a and α-SMA
expression, while OPN expression decreased. Conversely,
overexpression of SNHG1 resulted in decreased SM22a and α-SMA
expression but increased OPN expression (Fig. 3A-C). Western blotting examination
of α-SMA, SM22a and OPN protein levels confirmed these results
(Fig. 3D-G). These results
indicated that silencing of SNHG1 in VSMCs can induce a shift from
a synthetic to a contractile phenotype during autophagy.
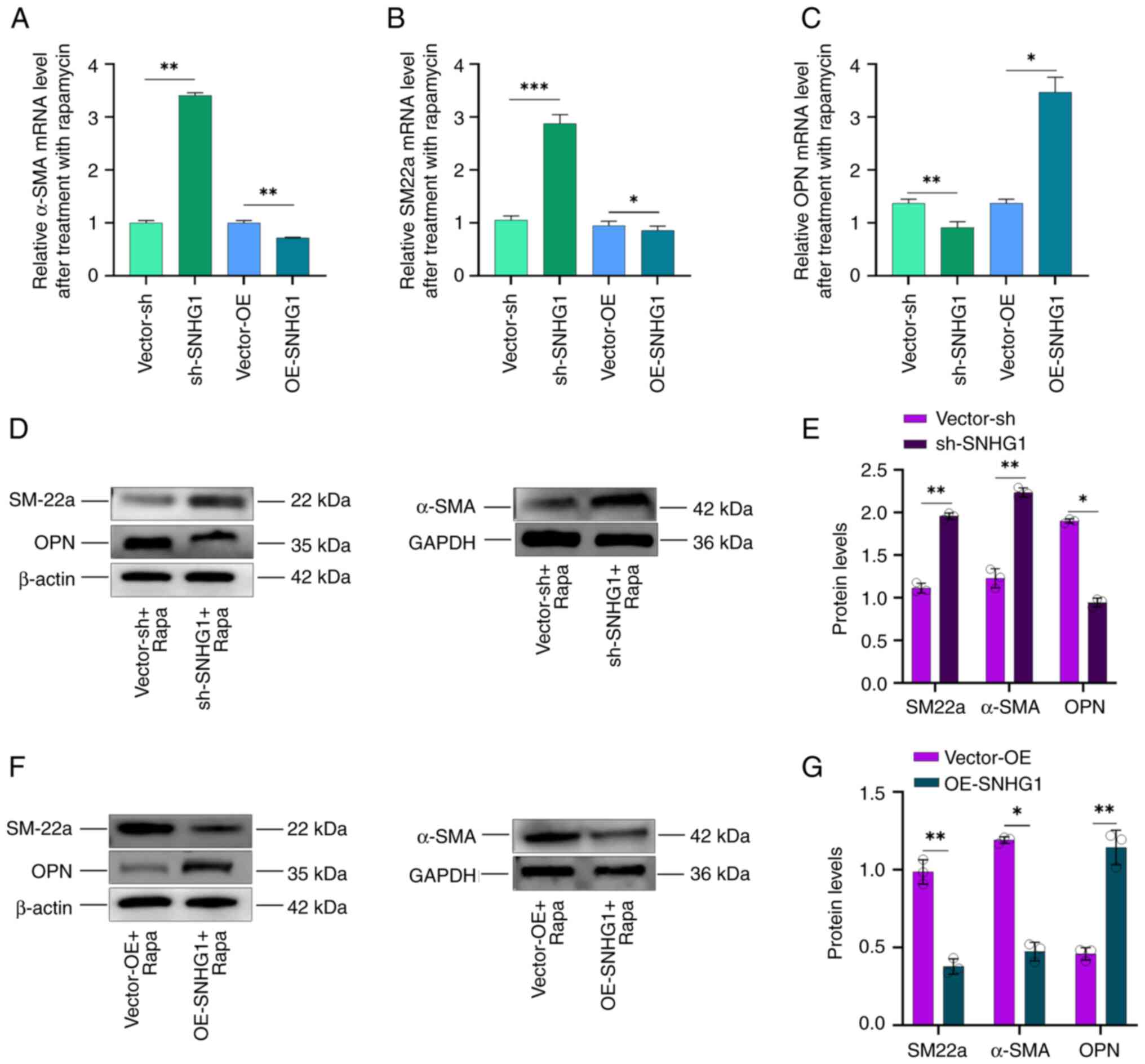 | Figure 3.Changes in VSMCs phenotype following
silencing and overexpression of SNHG1. (A) α-SMA, (B) SM22a and (C)
OPN mRNA expression following overexpressing or silencing of SNHG1
combining rapamycin treatment in VSMCs; (D-G) SM22a, α-SMA and OPN
protein levels following silencing or overexpression of SNHG1
combing with rapamycin treatment. Data were shown as mean ±
standard deviation of a representative experiment performed in
three independent replicates. *P<0.05, **P<0.01,
***P<0.001. VSMCs, vascular smooth muscle cells; SNHG1, small
ribosome housekeeping gene RNA1; α-SMA, α-smooth muscle actin;
SM22a, smooth muscle protein 22a; OPN, osteopontin; sh, short
hairpin; NC, negative control; OE, overexpression; Rapa,
rapamycin. |
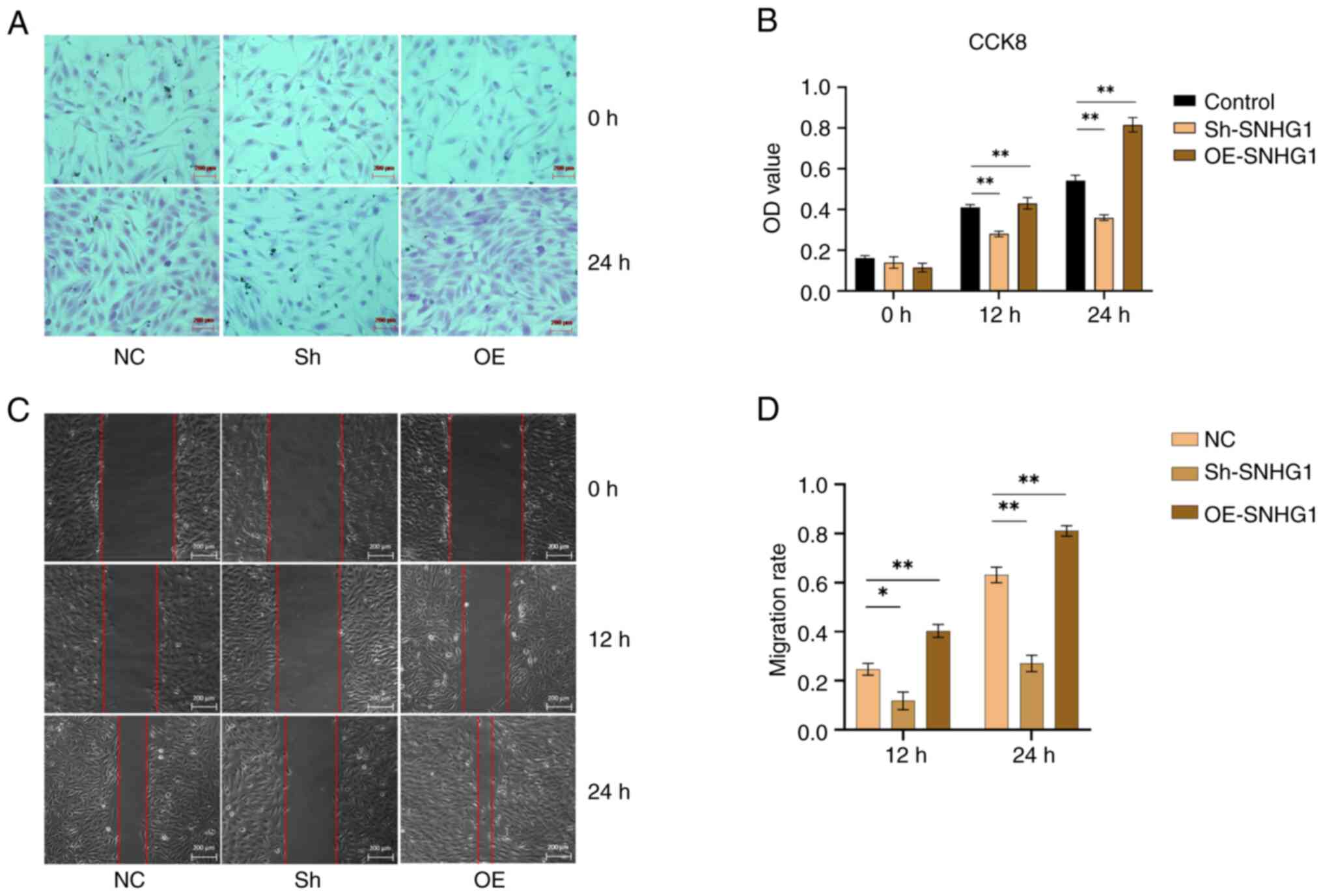
SNHG1 promotes migration and
proliferation of VSMCs
Following successful transduction of SNHG1, the cell
viability was tested using the CCK8 assay. The results showed that
overexpression of SNHG1 increased the proliferation of VSMCs.
Conversely, this effect was suppressed when SNHG1 was silenced,
leading to a decrease in the proliferation of VSMCs (Fig. 4A and B). Furthermore, wound healing
assays showed that silencing SNHG1 reduced the migration of VSMCs,
while overexpression of SNHG1 increasing their migration compared
with the control group (Fig. 4C and
D). Taken together, these results suggested that SNHG1 played a
role in promoting the proliferation and migration ability of
VSMCs.
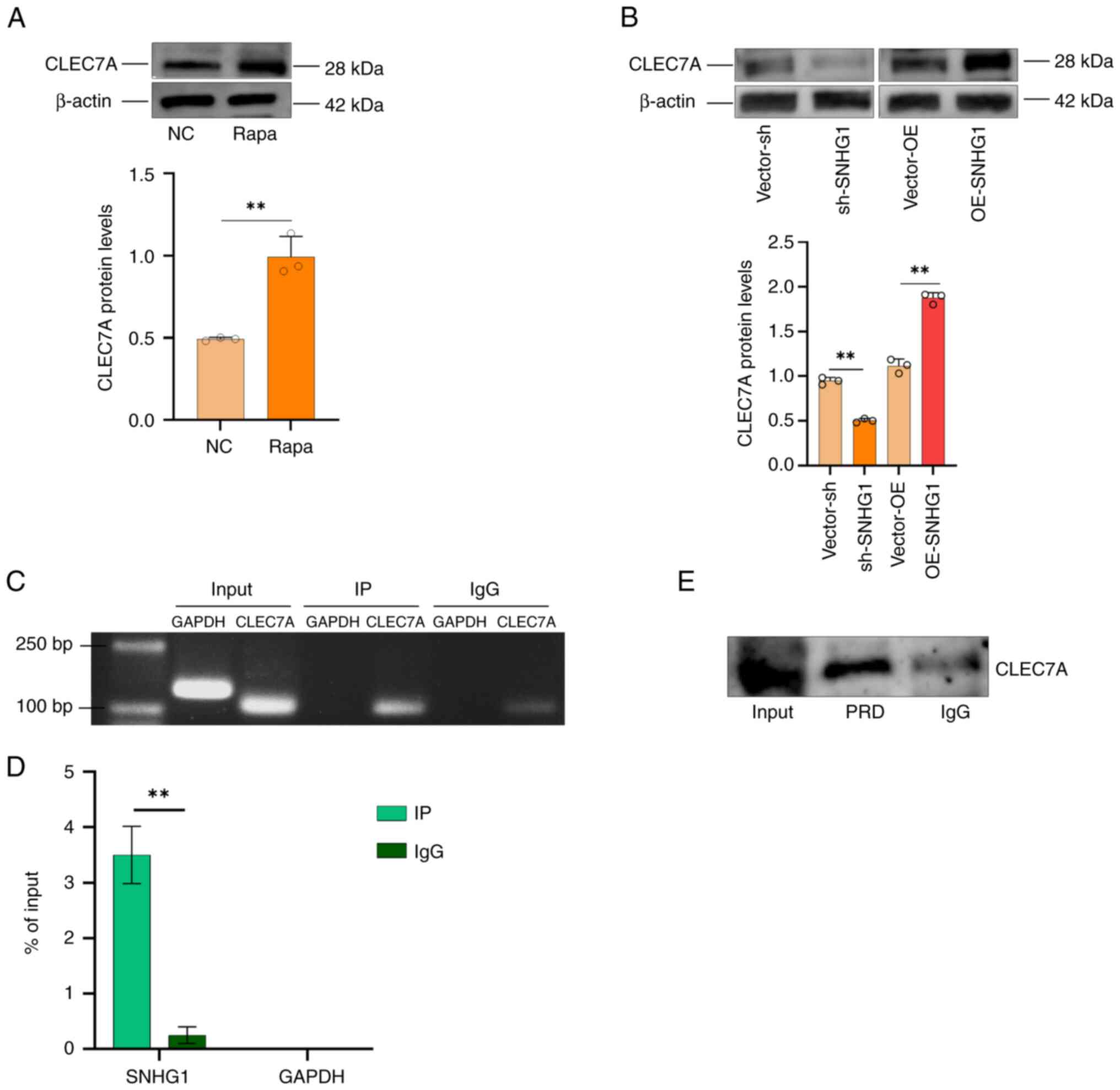
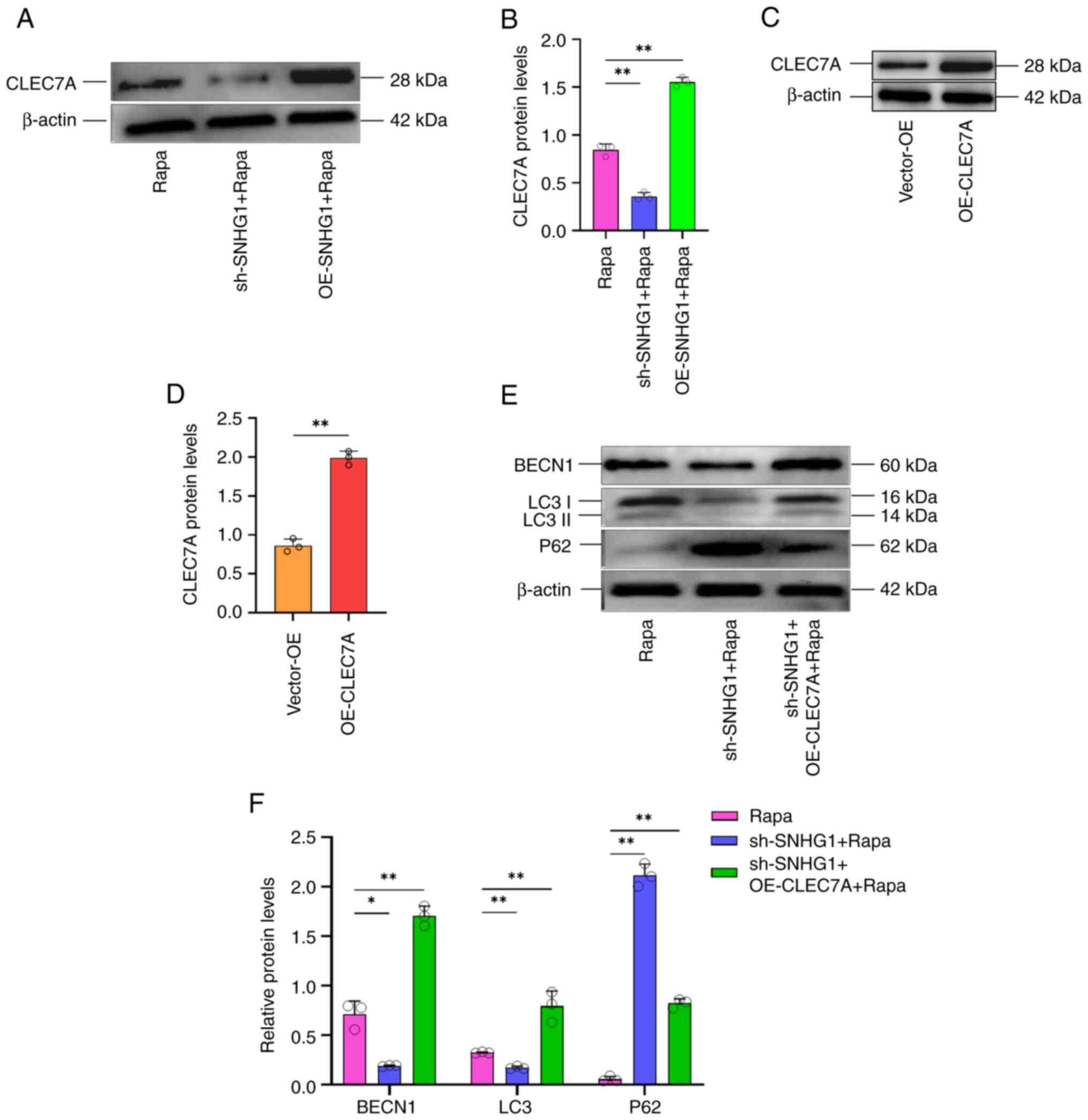
CLEC7A is a regulatory target of
SNHG1
In our previous study, an increase in the expression
of CLEC7A was observed in aneurysm tissues (27). In the present study, induction of
autophagy in VSMCs was found to result in an increase in CLEC7A
protein levels (Fig. 5A). This
suggested that CLEC7A played a role in aneurysm formation by
participating in autophagy in VSMCs. A possible relationship
between SNHG1 and CLEC7A was predicted using the RPI-Seq database
and the result indicated a strong association between them
(Fig. S1). This suggested that
CLEC7A is one of the targets regulated by SNHG1. To verify whether
CLEC7A is a direct regulatory target of SNHG1, CLEC7A protein
levels changed following silencing and overexpression of SNHG1 in
VSMCs (Fig. 5B); RIP and RNA
pull-down assays were used to verify the binding effect. As shown
in Fig. 5C and D, SNHG1 was pulled
down by an anti-CLEC7A antibody in the RIP assay. The RNA pull-down
assay then showed that CLEC7A proteins were bound to SNHG1 in VSMCs
(Fig. 5E). Taken together, these
results demonstrated that SNHG1 directly binds to CLEC7A in VSMCs.
SNGH1 and autophagy are interrelated and autophagy also regulated
CLEC7A via SNGH1.
Effect of SNHG1 on the expression of
CLEC7A on the phenotypic changes of VSMCs
To investigate the effect of SNHG1, which regulates
CLEC7A, on the phenotypic changes of VSMCs, autophagy was induced
following silencing or overexpressing of SNHG1. Silencing of SNHG1
decreased CLEC7A protein levels following induction of autophagy,
whereas overexpression of SNHG1 increased these levels (Fig. 6A and B). Next, an overexpression
vector for CLEC7A was successfully constructed and transduced into
VSMCs. Western blotting results confirmed the overexpression of
CLEC7A (Fig. 6C and D).
Subsequently, the silencing of SNHG1 was combined with the
overexpression of CLEC7A in these cells and they were treated with
rapamycin. As shown in Fig. 6E and
F, western blotting showed that autophagy markers changed
following silencing SNHG1, with LC 3II/LC 3I and Beclin1 decreasing
and P62 increasing. This effect was offset by combing with
overexpression of CLEC7A. Taken together, these results suggested
that SNHG1 regulated autophagy in VSMCs by enhancing CLEC7A.
Discussion
VSMCs undergo phenotypic transformation by switching
from a contractile phenotype to a synthetic phenotype characterized
by increased proliferation and migratory capacity. This
transformation is accompanied by a downregulation of contractile
proteins such as α-SMA and SM22a and an upregulation of MMPs and
inflammatory mediators (5,36,37).
This phenotypic transformation of VSMCs has been shown to
contribute to the development and progression of vascular diseases
(38). Autophagy is a cellular
process responsible for the degradation and recycling of damaged or
unnecessary cellular components, including proteins and organelles.
It plays a crucial role in maintaining cellular homeostasis and is
involved in various physiological and pathological processes.
Autophagy can influence the processes in VSMCs that are associated
with a change in phenotype and subsequently lead to the development
of disease (39). Genes related to
autophagy, such as ATG7 and ATG5, are implicated for VSMCs
(40). SNHG1 is associated with
the development of various diseases. In the case of ischemic
stroke, Zhang et al (41)
discovered that SNHG1, as a competitive endogenous RNA, can
influence pathological changes in cerebral blood vessels via the
hypoxia-inducible factor-1α/vascular endothelial growth factor
signaling pathway. Wang et al (42) also found that increased expression
of SNHG1 promotes angiogenesis in cerebral microvascular
endothelial cells following oxygen-glucose deprivation therapy and
this effect is achieved by targeting miR-199a. This suggests that
SNHG1 plays a critical role in vascular endothelial cell
proliferation and apoptosis. In addition, studies have shown that
SNHG1 regulates vascular endothelial cell proliferation and
angiogenesis through miR-196a (43), targeting the miR-340-5p/PIK3CA axis
in diabetic retinopathy (44). Li
et al (45) found that
SNHG1 attenuates high glucose-induced calcification/senescence of
VSMCs through post-transcriptional regulation of Bhlhe40 and
autophagy via Atg10.
These results suggest a close association between
SNHG1 and the development of vascular diseases. The present study
found that overexpression of SNHG1 also enhanced the proliferation
and migration of VSMCs. Furthermore, the results suggested that
silencing of SNHG1 in VSMCs following autophagy could induce a
transition from a synthetic phenotype to a contractile phenotype,
demonstrating that SNHG1 mediates autophagy of VSMCs.
CLEC7A has been implicated in several diseases.
Studies have shown that polymorphisms in CLEC7A could be promising
biomarkers for susceptibility to ulcerative colitis (46–48).
In addition, CLEC7A serves as a modifier gene in cytotoxic
T-lymphocyte associated protein 4 to maintain immune homeostasis
and tolerance (49). Another study
reported that CLEC7A drives intestinal fungus-mediated host lipid
deposition (50), involving
diabetic cardiomyopathy (51) and
Alzheimer's disease (52).
Importantly, the CLEC7A pathway has been shown to activate robust
autophagy-dependent unconventional protein secretion in human
macrophages (53).
The present study investigated the relationship
between SNHG1 and its target gene, CLEC7A, in regulating the
biological function of VSMCs. Our previous research has shown that
CLEC7A is involved in the development (27) and the present study showed that its
expression in VSMCs increased when the cells transitioned from a
contractile phenotype to a synthetic phenotype. The following
mechanistic study confirmed that CLEC7A was a direct target gene
regulated by SNHG1 in VSMCs by RIP and RNA pull-down assays. In
addition, it was observed that the expression of both SNHG1 and
CLEC7A increased following autophagy of VSMCs. When SNHG1 was
overexpressed, the contractile phenotype of VSMCs decreased while
the synthetic phenotype increased. Conversely, the trend was
reversed when SNHG1 was silenced. Furthermore, when CLEC7A was
overexpressed in VSMCs with silenced SNHG1 and induced autophagy,
an increase in autophagy markers was observed. These results
suggested that SNHG1 mediated the autophagy process in VSMCs by
targeting CLEC7A, which in turn triggered the phenotypic
transformation of VSMCs. Although the experiment was performed with
rapamycin treatment, resulting in increases in both CLEC7A and
SNHG1, an RIP assay for CLEC7A following rapamycin treatment would
be helpful to consolidate the role of CLEC7A in autophagy. In
addition, the subcellular localization of lncRNAs plays a crucial
role in determining their functional capabilities within cells.
These molecules exert an influence on different cellular
compartments and thus enable a spectrum of biological activities.
These activities range from the regulation of gene expression to
the modulation of cell structure and the response to stress
(54,55). For example, SNHG1 was previously
identified primarily in the nuclei of bladder cancer cells
(56); however, its distribution
in VSMCs remains unexplored. Therefore, to decipher the mechanism
of action of SNHG1 in the regulation of VSMCs, a fluorescence in
situ hybridization experiment needs to be performed to
determine precisely its subcellular localization in these cells.
While the findings of the present study emphasized the crucial
roles of SNHG1 and CLEC7A in regulating autophagy and VSMCs
phenotypic transformation, it is noteworthy that incorporating
MYH11 and Calponin as additional markers could have strengthened
the evidence. Future research on the expression patterns of MYH11
and Calponin related to VSMCs phenotype may offer valuable insights
for a comprehensive understanding of this process.
The present study demonstrated that SNHG1 promoted
proliferation and migration of VSMCs and increased expression
following autophagy of VSMCs. SNHG1 facilitated the transition of
VSMCs from a contractile to a synthetic phenotype by promoting
autophagy and this mechanism involves the regulation of CLEC7A.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was partly supported by research funding from
the National Natural Science Foundation of China (grant nos.
81860222 and 82060226); The Basic Ability Enhancement Program for
Young and Middle-aged Teachers of Guangxi (grant no. 2021KY0080);
Scientific research team incubation project of Guangxi Minzu
Hospital (grant no. FY202107) and Innovation Project of Guangxi
Graduate Education (grant no. YCSW2023218).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
CQ and RTH were responsible for study concept and
design. HWD, ZMD, HWD and ZMY performed the literature review. CQ,
RTH, HWD, WBT, SDZ, ZMD and ZMY were responsible for data analysis
and interpretation. HWD, WBT, SDZ, ZMY, ZMD, RTH and CQ were
responsible for manuscript writing and reviewing. CQ and RTH
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of
interest.
Glossary
Abbreviations
Abbreviations:
|
VSMCs
|
vascular smooth muscle cells
|
|
lncRNA
|
long non-coding RNA
|
|
SNHG1
|
small ribosome housekeeping gene
RNA1
|
|
CLEC7A
|
C-type lectin domain containing 7A
|
References
|
1
|
Penn DL, Witte SR, Komotar RJ and Connolly
E Jr: The role of vascular remodeling and inflammation in the
pathogenesis of intracranial aneurysms. J Clin Neurosci. 21:28–32.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liao XH, Wang N, Zhao DW, Zheng DL, Zheng
L, Xing WJ, Ma WJ, Bao LY, Dong J and Zhang TC: STAT3 protein
regulates vascular smooth muscle cell phenotypic switch by
interaction with myocardin. J Biol Chem. 290:19641–19652. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cao T, Zhang L, Yao LL, Zheng F, Wang L,
Yang JY, Guo LY, Li XY, Yan YW, Pan YM, et al: S100B promotes
injury-induced vascular remodeling through modulating smooth muscle
phenotype. Biochim Biophys Acta Mol Basis Dis. 1863:2772–2782.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhu LH, Huang L, Zhang X, Zhang P, Zhang
SM, Guan H, Zhang Y, Zhu XY, Tian S, Deng K and Li H: Mindin
regulates vascular smooth muscle cell phenotype and prevents
neointima formation. Clin Sci (Lond). 129:129–145. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bennett MR, Sinha S and Owens GK: Vascular
smooth muscle cells in atherosclerosis. Circ Res. 118:692–702.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhen Y and Stenmark H: Autophagosome
biogenesis. Cells. 12:6682023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tai S, Hu XQ, Peng DQ, Zhou SH and Zheng
XL: The roles of autophagy in vascular smooth muscle cells. Int J
Cardiol. 211:1–6. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Grootaert MOJ, Moulis M, Roth L, Martinet
W, Vindis C, Bennett MR and De Meyer GRY: Vascular smooth muscle
cell death, autophagy and senescence in atherosclerosis. Cardiovasc
Res. 114:622–634. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cheng CI, Lee YH, Chen PH, Lin YC, Chou MH
and Kao YH: Free fatty acids induce autophagy and LOX-1
upregulation in cultured aortic vascular smooth muscle cells. J
Cell Biochem. 118:1249–1261. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li T, Tan X, Zhu S, Zhong W, Huang B, Sun
J, Li F and Wang Y: SPARC induces phenotypic modulation of human
brain vascular smooth muscle cells via AMPK/mTOR-mediated
autophagy. Neurosci Lett. 712:1344852019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sabeena S: Role of noncoding RNAs with
emphasis on long noncoding RNAs as cervical cancer biomarkers. J
Med Virol. 95:e285252023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang PS, Wang Z and Yang C: Dysregulations
of long non-coding RNAs-The emerging ‘lnc’ in environmental
carcinogenesis. Semin Cancer Biol. 76:163–172. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Meybodi SM, Soleimani N, Yari A, Javadifar
A, Tollabi M, Karimi B, Meybodi ME, Seyedhossaini S, Milan PB and
Firoozabadi AD: Circulatory long noncoding RNAs
(circulatory-LNC-RNAs) as novel biomarkers and therapeutic targets
in cardiovascular diseases: Implications for cardiovascular
diseases complications. Int J Biol Macromol. 225:1049–1071. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cui Y, Zhang F, Zhu C, Geng L, Tian T and
Liu H: Upregulated lncRNA SNHG1 contributes to progression of
non-small cell lung cancer through inhibition of miR-101-3p and
activation of Wnt/β-catenin signaling pathway. Oncotarget.
8:17785–17794. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li J, Zhang Z, Xiong L, Guo C, Jiang T,
Zeng L, Li G and Wang J: SNHG1 lncRNA negatively regulates
miR-199a-3p to enhance CDK7 expression and promote cell
proliferation in prostate cancer. Biochem Biophys Res Commun.
487:146–152. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lu Y, Xi J, Zhang Y, Chen W, Zhang F, Li C
and Wang Z: SNHG1 inhibits ox-LDL-induced inflammatory response and
apoptosis of HUVECs via up-regulating GNAI2 and PCBP1. Front
Pharmacol. 11:7032020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li W, Dong X, He C, Tan G, Li Z, Zhai B,
Feng J, Jiang X, Liu C, Jiang H and Sun X: Correction to: LncRNA
SNHG1 contributes to sorafenib resistance by activating the Akt
pathway and is positively regulated by miR-21 in hepatocellular
carcinoma cells. J Exp Clin Cancer Res. 40:3772021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu J, Yang R, Hua X, Huang M, Tian Z, Li
J, Lam HY, Jiang G, Cohen M and Huang C: lncRNA SNHG1 promotes
basal bladder cancer invasion via interaction with PP2A catalytic
subunit and induction of autophagy. Mol Ther Nucleic Acids.
21:354–366. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhao J, Geng L, Chen Y and Wu C: SNHG1
promotes MPP+-induced cytotoxicity by regulating
PTEN/AKT/mTOR signaling pathway in SH-SY5Y cells via sponging
miR-153-3p. Biol Res. 53:12020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu Y, Zhu B, Yan Y, Bai S, Kang H, Zhang
J, Ma W, Gao Y, Hui B, Li R, et al: Long non-coding RNA SNHG1
stimulates ovarian cancer progression by modulating expression of
miR-454 and ZEB1. Mol Oncol. 15:1584–1596. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rizzetto L, De Filippo C, Rivero D,
Riccadonna S, Beltrame L and Cavalieri D: Systems biology of
host-mycobiota interactions: Dissecting Dectin-1 and Dectin-2
signalling in immune cells with DC-ATLAS. Immunobiology.
218:1428–1437. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang Y, Spatz M, Da Costa G, Michaudel C,
Lapiere A, Danne C, Agus A, Michel ML, Netea MG, Langella P, et al:
Deletion of both Dectin-1 and Dectin-2 affects the bacterial but
not fungal gut microbiota and susceptibility to colitis in mice.
Microbiome. 10:912022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Al Madhoun A, Kochumon S, Al-Rashed F,
Sindhu S, Thomas R, Miranda L, Al-Mulla F and Ahmad R: Dectin-1 as
a potential inflammatory biomarker for metabolic inflammation in
adipose tissue of individuals with obesity. Cells. 11:28792022.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Choraghe RP, Kolodziej T, Buser A, Rajfur
Z and Neumann AK: RHOA-mediated mechanical force generation through
Dectin-1. J Cell Sci. 133:jcs2361662020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rouchaud A, Johnson C, Thielen E,
Schroeder D, Ding YH, Dai D, Brinjikji W, Cebral J, Kallmes DF and
Kadirvel R: Differential gene expression in coiled versus
flow-diverter-treated aneurysms: RNA sequencing analysis in a
rabbit aneurysm model. AJNR Am J Neuroradiol. 37:1114–1121. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Turhon M, Maimaiti A, Gheyret D, Axier A,
Rexiati N, Kadeer K, Su R, Wang Z, Chen X, Cheng X, et al: An
immunogenic cell death-related regulators classification patterns
and immune microenvironment infiltration characterization in
intracranial aneurysm based on machine learning. Front Immunol.
13:10013202022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hu R, Huang L, Zhou M, Zhou S and Hu R:
Expression and clinical significance of CLEC7A in intracranial
aneurysm tissues and serum. J Minim Invasive Surg. 16:453–456.
2021.(In Chinese).
|
|
28
|
Su S, Shi YT, Chu Y, Jiang MZ, Wu N, Xu B,
Zhou H, Lin JC, Jin YR, Li XF and Liang J: Sec62 promotes gastric
cancer metastasis through mediating UPR-induced autophagy
activation. Cell Mol Life Sci. 79:1332022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Qiu WL, Zhang YW, Feng Y, Li LC, Yang L
and Xu CR: Deciphering pancreatic islet β cell and α cell
maturation pathways and characteristic features at the single-cell
level. Cell Metab. 25:1194–1205. e11942017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yue Y, Xu J, Li Y, Cheng K, Feng Q, Ma X,
Ma N, Zhang T, Wang X, Zhao X and Nie G: Antigen-bearing outer
membrane vesicles as tumour vaccines produced in situ by ingested
genetically engineered bacteria. Nat Biomed Eng. 6:898–909. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gomez-Sanchez R, Yakhine-Diop SM,
Rodriguez-Arribas M, Bravo-San Pedro JM, Martínez-Chacón G,
Uribe-Carretero E, Pinheiro de Castro DC, Pizarro-Estrella E,
Fuentes JM and González-Polo RA: MRNA and protein dataset of
autophagy markers (LC3 and p62) in several cell lines. Data Brief.
7:641–647. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ailawadi G, Moehle CW, Pei H, Walton SP,
Yang Z, Kron IL, Lau CL and Owens GK: Smooth muscle phenotypic
modulation is an early event in aortic aneurysms. J Thorac
Cardiovasc Surg. 138:1392–1399. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ding Y, Zhang M, Zhang W, Lu Q, Cai Z,
Song P, Okon IS, Xiao L and Zou MH: AMP-activated protein kinase
alpha 2 deletion induces VSMC phenotypic switching and reduces
features of atherosclerotic plaque stability. Circ Res.
119:718–730. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Horita H, Wysoczynski CL, Walker LA,
Moulton KS, Li M, Ostriker A, Tucker R, McKinsey TA, Churchill ME,
Nemenoff RA and Weiser-Evans MC: Nuclear PTEN functions as an
essential regulator of SRF-dependent transcription to control
smooth muscle differentiation. Nat Commun. 7:108302016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Owens GK, Kumar MS and Wamhoff BR:
Molecular regulation of vascular smooth muscle cell differentiation
in development and disease. Physiol Rev. 84:767–801. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Thyberg J: Phenotypic modulation of smooth
muscle cells during formation of neointimal thickenings following
vascular injury. Histol Histopathol. 13:871–891. 1998.PubMed/NCBI
|
|
38
|
Sawyer DM, Pace LA, Pascale CL, Kutchin
AC, O'Neill BE, Starke RM and Dumont AS: Lymphocytes influence
intracranial aneurysm formation and rupture: Role of extracellular
matrix remodeling and phenotypic modulation of vascular smooth
muscle cells. J Neuroinflammation. 13:1852016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jiang B, Li X, Wang M, Li GX, Ren PW, Wang
YQ, Xin SJ and Qin LF: Trehalose attenuates abdominal aortic
aneurysm formation by inducing autophagy in smooth muscle cells. J
Geriatr Cardiol. 20:214–222. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fang ZM, Feng X, Chen Y, Luo H, Jiang DS
and Yi X: Targeting autophagy in aortic aneurysm and dissection.
Biomed Pharmacother. 153:1135472022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang L, Luo X, Chen F, Yuan W, Xiao X,
Zhang X, Dong Y, Zhang Y and Liu Y: LncRNA SNHG1 regulates
cerebrovascular pathologies as a competing endogenous RNA through
HIF-1alpha/VEGF signaling in ischemic stroke. J Cell Biochem.
119:5460–5472. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang Z, Wang R, Wang K and Liu X:
Upregulated long noncoding RNA Snhg1 promotes the angiogenesis of
brain microvascular endothelial cells after oxygen-glucose
deprivation treatment by targeting miR-199a. Can J Physiol
Pharmacol. 96:909–915. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang L, Zhang Q, Lv L, Jianhua Z, Ting C
and Wu Y: LncRNA SNHG1 regulates vascular endothelial cell
proliferation and angiogenesis via miR-196a. J Mol Histol.
51:117–124. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
He FT, Fu XL, Li MH, Fu CY and Chen JZ:
LncRNA SNHG1 targets miR-340-5p/PIK3CA axis to regulate
microvascular endothelial cell proliferation, migration, and
angiogenesis in DR. Kaohsiung J Med Sci. 39:16–25. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li S, Ni Y, Li C, Xiang Q, Zhao Y, Xu H,
Huang W, Wang Y, Wang Y, Zhan J and Liu Y: Long noncoding RNA SNHG1
alleviates high glucose-induced vascular smooth muscle cells
calcification/senescence by post-transcriptionally regulating
Bhlhe40 and autophagy via Atg10. J Physiol Biochem. 79:83–105.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Legaki E, Koutouratsas T, Theocharopoulos
C, Lagkada V and Gazouli M: Polymorphisms in CLEC5A and CLEC7A
genes modify risk for inflammatory bowel disease. Ann
Gastroenterol. 37:64–70. 2024.PubMed/NCBI
|
|
47
|
Basson A, Trotter A, Rodriguez-Palacios A
and Cominelli F: Mucosal interactions between genetics, diet, and
microbiome in inflammatory bowel disease. Front Immunol. 7:2902016.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Iliev ID, Funari VA, Taylor KD, Nguyen Q,
Reyes CN, Strom SP, Brown J, Becker CA, Fleshner PR, Dubinsky M, et
al: Interactions between commensal fungi and the C-type lectin
receptor Dectin-1 influence colitis. Science. 336:1314–1317. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Turnbull C, Bones J, Stanley M, Medhavy A,
Wang H, Lorenzo AMD, Cappello J, Shanmuganandam S, Pandey A,
Seneviratne S, et al: DECTIN-1: A modifier protein in CTLA-4
haploinsufficiency. Sci Adv. 9:eadi95662023. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ma J, Zhou M, Song Z, Deng Y, Xia S, Li Y,
Huang X, Xiao D, Yin Y and Yin J: Clec7a drives gut fungus-mediated
host lipid deposition. Microbiome. 11:2642023. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yang N, Wang M, Lin K, Wang M, Xu D, Han
X, Zhao X, Wang Y, Wu G, Luo W, et al: Dectin-1 deficiency
alleviates diabetic cardiomyopathy by attenuating
macrophage-mediated inflammatory response. Biochim Biophys Acta Mol
Basis Dis. 1869:1667102023. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhao X, Sun J, Xiong L, She L, Li L, Tang
H, Zeng Y, Chen F, Han X, Ye S, et al: β-amyloid binds to microglia
Dectin-1 to induce inflammatory response in the pathogenesis of
Alzheimer's disease. Int J Biol Sci. 19:3249–3265. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Öhman T, Teirila L, Lahesmaa-Korpinen AM,
Cypryk W, Veckman V, Saijo S, Wolff H, Hautaniemi S, Nyman TA and
Matikainen S: Dectin-1 pathway activates robust autophagy-dependent
unconventional protein secretion in human macrophages. J Immunol.
192:5952–5962. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Deng J, Xu W, Jie Y and Chong Y:
Subcellular localization and relevant mechanisms of human
cancer-related micropeptides. FASEB J. 37:e232702023. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wei C, Xu Y, Shen Q, Li R, Xiao X, Saw PE
and Xu X: Role of long non-coding RNAs in cancer: From subcellular
localization to nanoparticle-mediated targeted regulation. Mol Ther
Nucleic Acids. 33:774–793. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Cai H, Xu H, Lu H, Xu W, Liu H, Wang X,
Zhou G and Yang X: LncRNA SNHG1 facilitates tumor proliferation and
represses apoptosis by regulating PPARgamma ubiquitination in
bladder cancer. Cancers (Basel). 14:47402022. View Article : Google Scholar : PubMed/NCBI
|