Introduction
According to global clinical data, renal cell
carcinoma (RCC) is the most lethal form of urogenital cancer,
characterized by a mortality rate of 30–40% (1,2).
Kidney renal clear cell carcinoma (KIRC) accounts for 70–80% of all
RCC cases worldwide (3). Effective
therapeutic targets and molecular drugs for KIRC are limited,
rendering it difficult to treat. Notably, the incidence of KIRC has
been steadily increasing in recent years. From 2000 to 2016,
countries such as Japan, Italy and the USA experienced an increase
in age-standardized rate of RCC, rising from 5.3, 12 and 10.7 per
100,000 to 7.8, 13.7 and 13.3 per 100,000, respectively (1,4). And
KIRC displays resistance to radiotherapy, chemotherapy and
immunotherapy, underscoring surgery as the primary treatment
modality (5). Among patients with
KIRC, 60% exhibit a survival duration of 1–2 years post-diagnosis,
with 30% manifesting distant metastasis at the time of diagnosis
globally (2). Therefore, the
identification of effective therapeutic targets is key for early
diagnosis and intervention in KIRC.
Pleckstrin homology domain-containing family A
member 4 (PLEKHA4) is as a key molecular player in cancer biology,
particularly in glioma (6,7). It serves multiple roles in cancer
progression, operating through diverse mechanisms. In glioblastoma,
PLEKHA4 is involved in the regulation of apoptotic regulators,
exerting an inhibitory effect on apoptosis. Moreover, PLEKHA4
triggers dishevelled accumulation and promotes the upregulation of
Wnt signaling in cultured mammalian cells, including human HeLa,
293T and 293, and mouse C57MG and MV7 Rat2a cells (8).
Dysregulated Wnt signaling has been associated with
a spectrum of diseases, encompassing embryonic malformations,
degenerative disorders and cancer. Perturbations in β-catenin
function can stem from diverse origins, including extracellular
cues, cytoplasmic components and nuclear factors (9). Aberrations in β-catenin activities
are linked to the development of various human degenerative
disorders and numerous types of cancer, including those affecting
the breast, colon and kidney (10–12).
Consequently, the involvement of β-catenin in these mechanisms has
spurred investigation, including in the context of KIRC (4,13).
In oncogenesis, aberrant Wnt/β-catenin signaling amplifies the
activation of its target genes, such as cyclin D1, which serve a
crucial role in the pathway (14).
Despite the high expression of PLEKHA4 observed in various types of
cancer, such as glioma and melanoma (7,8), the
precise mechanisms by which it influences KIRC progression remain
inadequately elucidated. The present study investigated the role of
PLEKHA4 in KIRC to improve understanding of the molecular
mechanisms underlying KIRC and to facilitate the development of
novel therapeutic approaches.
Materials and methods
Bioinformatics analysis
Pan-cancer analysis and gene set variation analysis
(GSVA) were performed using Gene Set Cancer Analysis (GSCA;
http://guolab.wchscu.cn/GSCA/#/). GSVA
was performed to estimate the integrated level of gene set
expression for each The Cancer Genome Atlas (TCGA) sample. The gene
expression profiles GSE15641 (15)
and GSE40435 (16), which were
obtained with the GPL96 platform, were retrieved from Gene
Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/). The GSE15641
profile encompassed data from 49 RCC tumors, 20 non-RCC renal
tumors and 23 normal kidney samples. The GSE40435 profile included
information from 101 pairs of KIRC and adjacent non-tumor renal
samples. The data from the GEO database were processed using
GEOquery (2.64.2) (https://bioconductor.org/packages/release/bioc/html/GEOquery.html)
and subsequent normalization was performed using the
normalizeBetweenArrays function from the limma package (3.52.2)
(https://www.bioconductor.org/packages/release/bioc/html/limma.html).
The outcomes were visualized using ggplot2 (3.3.6) (https://cran.r-project.org/src/contrib/Archive/ggplot2/)
and ComplexHeatmap (2.13.1) (https://github.com/jokergoo/ComplexHeatmap).
Calibration, standardization and log2 transformation were applied
to all gene expression data. Kyoto Encyclopedia of Genes and
Genomes (KEGG) and Gene Ontology (GO) analyses were performed using
clusterProfiler [4.4.4] (https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html)
and GOplot [1.0.2] (https://cran.r-project.org/web/packages/GOplot/index.html).
The expression patterns of PLEKHA4 across pathological and
histological grades were assessed using R (version 4.2.1)
(https://www.r-project.org/) and
visualized utilizing ggplot2 (3.3.6). RNA sequencing data were
obtained from TCGA-KIRC project through the STAR pipeline of TCGA
database (https://www.cancer.gov/ccg/research/genome-sequencing/tcga),
with the data extracted in TPM format. Gene Mania (https://genemania.org/) was also used to explore the
interaction between genes.
Cell culture
Human KIRC cell lines 769-P, 786-O and CAKI1 were
procured from the National Collection of Authenticated Cell
Cultures. According to the depmap portal (The Cancer Dependency Map
Project at Broad Institute; http://depmap.org/portal/), PLEKHA4 is highly
expressed in 769-P and 786-O cell lines, and is expressed at lower
levels in CAKI1 cell lines. The 769-P and 786-O cell lines were
used for knockdown assays, and the CAKI1 cell line for
overexpression assays.
Briefly, 769-P and 786-O cells were maintained in
RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS and 1% penicillin-streptomycin, and CAKI1
cells were cultured in DMEM (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS (Biological Industries; Sartorius AG) and
1% penicillin-streptomycin (Biological Industries; Sartorius AG).
Cell cultures were maintained in a 37°C incubator with 5%
CO2. For drug treatment, lithium chloride (LiCl, Selleck
Chemicals) at a concentration of 5 mM was added to the medium and
incubated for 12 h in a 37°C incubator with 5% CO2 to
activate the Wnt/β-catenin signaling pathway.
Cell transduction and
transfection
769-P and 786-O cells were transduced with short
hairpin RNA (shRNA) lentiviruses targeting PLEKHA4. The two shRNAs
used were Sh1 [K7453 LV3(H1/GFP&Puro)-PLEKHA4-Homo-2851; target
sequence: 5′-GCGAGTCACTCTGCTACAATC-3′] and Sh2 [K7451
LV3(H1/GFP&Puro)-shPLEKHA4#2; target sequence:
5′-AGCTACAATATTAGACCAGA-3′], along with a negative control (NC)
lentivirus (LV3-shNC; target sequence: 5′-GTTCTCCGAACGTGTCACGT-3′).
These lentiviruses were produced by Shanghai GenePharma Co.,
Ltd.
The 3rd generation system was used for lentiviral
transduction. Briefly, 293T cells (National Collection of
Authenticated Cell Cultures) were used to produce the viruses using
the recombinant shuttle and packaging plasmids pGag/Pol, pRev and
pVSV-G. The concentration and purity of the plasmids were measured
using UV absorption, ensuring that the A260/A280 ratio of the
extracted plasmid DNA was between 1.8 and 2.0. The recombinant
plasmid and packaging plasmids were mixed in a ratio of 8:4:4:4 µg
(sh1,sh2 or NC:pGag/Pol:pRev:pVSV-G), and were added to the 293T
cells and incubated in a 37°C incubator containing 5%
CO2 for 4–6 h. After removing the mixture, culture
medium was added and incubation was continued for 72 h in a 37°C
incubator containing 5% CO2. The supernatant was then
collected from the dishes and cell debris was removed by passing it
through a 0.45-µm filter. Viral particles were concentrated by
ultracentrifugation at 70,000 × g for 2 h at 20°C using conical
tubes and a swinging bucket rotor. The pellets were then
resuspended in 100 µl 1X HBSS (cat. no. 14025-092; Invitrogen;
Thermo Fisher Scientific, Inc.). An additional 100 µl 1X HBSS was
added to the tubes, bringing the final volume to 200 µl, which was
transferred to a screw-cap microfuge tube, wrapped in parafilm and
vortex at low speed for 15–30 min. After resuspension, the tube was
briefly spun for 10 sec, the supernatant was transferred to a fresh
tube and 20-µl aliquots were generated. The aliquots were stored at
−20°C for up to 1 month or at −80°C for longer, avoiding more than
three freeze-thaw cycles. Lentiviral infection was carried out
immediately after the virus preparation. The virus titers were as
follows; sh1 (LV3-PLEKHA4-Homo-2851), 7×108 TU/ml, sh2
(shPLEKHA4#2), 7×108 TU/ml and shNC (LV3-shNC),
9×108 TU/ml. The multiplicity of infection (MOI) values
of sh1, sh2 or shNC in 769-P cells were 5, 10 and 10; and the MOI
values of sh1, sh2 or shNC in 786-O cells was 10, 20 and 15. After
72 h of transfection and virus collection, the lentiviruses were
added to the cells and were incubated for 24 h for gene
transduction. Polybrene (2 µg/ml) was mixed with the virus prior to
adding it to the cells to enhance infection efficiency. Following
transduction, cells were subjected to selection with 1 µg/ml
puromycin for 7 days; the same concentration (1 µg/ml) was used for
maintenance. The infection efficacy was assessed through western
blot analysis. Western blotting and other experiments were
performed immediately after the 7-day puromycin selection.
To induce PLEKHA4 overexpression in CAKI1 cells,
pIRES2-EGFP-PLEKHA4 or control vector (pIRES2-EGFP-empty) plasmids
were obtained from Shanghai GenePharma Co., Ltd. Plasmids were
transfected into cells at 70% confluence using EndoFectin MAX
(GeneCopoeia, Inc.). For transfection in a 6-well plate, 2.5 µg
plasmids and 5–12.5 µl EndoFectin MAX were diluted with serum-free
DMEM, each in 125 µl and were left for 5 min. Subsequently, they
were mixed gently and the mixture was left for 5–20 minto form the
DNA-EndoFectin complex, which was added to cells in the wells of a
6-well plate. The cells were incubated in a CO2
incubator at 37°C. Gene expression could be detected 48 h after
transfection.
Cell proliferation assay
The 769-P and 786-O cells were plated in 96-well
plates at a density of 3,000 cells/well and cultured for 0, 24, 48,
72 and 96 h. Subsequently, cells were treated with 10 µl Cell
Counting Kit-8 (Shanghai Yeasen Biotechnology Co., Ltd.) solution
and incubated for 1 h at 37°C. The absorbance values were measured
at 450 nm. Each experiment was replicated ≥3 times.
Colony formation assay
The transduced 769-P and 786-O cells were seeded in
6-well plates at a concentration of 5×103 cells/well and
incubated for 7 days. At room temperature, cells were washed twice
with PBS, fixed with 4% paraformaldehyde for 15 min and
subsequently stained with Giemsa (both Beijing Solarbio Science
& Technology Co., Ltd.) for 20 min, before being washed twice
again with PBS. A colony was defined as containing a minimum of 50
cells and colonies were quantified using ImageJ, bundled with
64-bit Java 8 (https://imagej.net/ij/index.html; National Institutes
of Health).
Wound healing assay
The 769-P and 786-O cells were seeded in 6-well
plates at a density of 1×106 cells/well and grown to 90%
confluence. Subsequently, the cells were incubated overnight in
serum-free medium. The cell monolayers were then mechanically
wounded using a 10-µl pipette tip. Images of the wounds were
captured at 0 and 24 h using a Nikon Eclipse Ti-S/L100 Inverted
Phase Contrast Fluorescent Microscope (Nikon Corporation) with a
10X objective. The wound was measured using ImageJ, bundled with
64-bit Java 8 and statistical analysis was performed using SPSS 29
(IBM Corp.). Each experiment was conducted a minimum of three
times.
Flow cytometric analysis of cell cycle
progression
After trypsinizing the cells without EDTA, cells
were centrifuged at 300 × g for 5 min at 4°C. Cells were then
washed twice with pre-cooled PBS and resuspended in 100 µl 1X
Binding Buffer (Shanghai Yeasen Biotechnology Co., Ltd.). The cells
were then incubated with RNase A and PI Staining Solution (Shanghai
Yeasen Biotechnology Co., Ltd.) in the dark at room temperature for
10–15 min. Finally, 400 µl 1X Binding Buffer (Shanghai Yeasen
Biotechnology Co., Ltd.) was added on ice. The samples were
analyzed using a flow cytometer CytoFLEX SRT (Beckman Coulter,
Inc.) within 1 h. FlowJo V8 (FlowJo, LLC) was used for
analysis.
Western blotting
Briefly, 769-P and 786-O cells, and genetically
transduced 769-P, 786-O and CAKI1 cells were seeded in 6-well
plates at a density of 1×106 cells/well and cultured in
RPMI-1640 or DMEM containing 10% FBS until they reached ~90%
confluence. Protein lysate was extracted using RIPA buffer (Beijing
Solarbio Science & Technology Co., Ltd.). Nuclear protein
extraction was performed using the Nuclear and Cytoplasmic
Extraction kit (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. Protein concentration
was determined using the BCA Protein Assay Kit (Beyotime Institute
of Biotechnology). Proteins (30 µg/lane) were separated by SDS-PAGE
on 8–10% gels, transferred onto PVDF membranes and blocked with 5%
non-fat dry milk (MilliporeSigma) at room temperature for 1 h, then
incubated with primary antibodies overnight. The following primary
antibodies were used: PLEKHA4 (cat. no. NBP1-56679; Novus
Biologicals; Bio-Techne), β-actin (cat. no. GB12001-100; Wuhan
Servicebio Technology Co., Ltd.), β-catenin (cat. no. AF6266;
Affinity Biosciences), phosphorylated (p-)GSK3β (cat. no.
sc-373800; Santa Cruz Biotechnology, Inc.), GSK3β (cat. no.
sc-377213; Santa Cruz Biotechnology, Inc.), cyclin D1 (cat. no.
AF0931; Affinity Biosciences) and Lamin B1 (cat. no. sc-374015;
Santa Cruz Biotechnology, Inc.). The primary antibodies were
diluted at 1:1,000 in Primary Antibody Dilution Buffer (Beyotime
Institute of Biotechnology) and were incubated with the membranes
at 4°C overnight. Subsequently, the blots were probed with
HRP-conjugated anti-rabbit and anti-mouse secondary antibodies
(cat. nos. RGAR001 and RGAM001; Proteintech Group, Inc.). The
secondary antibodies were diluted at 1:5,000 in Tris-buffered
saline-0.1% Tween 20 solution and were incubated with the membranes
at room temperature for 2 h. ECL Western Blotting Substrate
(Beijing Solarbio Science & Technology Co., Ltd.) was used to
visualize the blots using the Shenhua Science Technology Co., Ltd.
system. The grayscale values of proteins were measured using
ImageJ, bundled with 64-bit Java 8. Each experiment was conducted
≥3 times.
Animal studies
The animal experiments were approved by the
Laboratory Animal Ethics Committee Yanbian University (approval no.
YD20230911020; Yanji, China). Briefly, 4-week-old male nude mice
(average weight, 14 g), were procured from the Experimental Animal
Center of Yanbian University and were randomly divided into two
groups (n=5/group): The shNC group and the shPLEKHA4 group. Nude
mice were housed in a specific pathogen-free environment with a
temperature of 25°C and humidity of 30%. The mice were exposed to a
12-h light/dark cycle and were fed adult mouse feed sterilized with
cobalt-60. The water provided was sterilized by autoclaving. Access
to food and water was free. The mice were subcutaneously injected
with a 200-µl solution containing 5×106 769-P cells
transduced with sh-NC or sh-PLEKHA4 in the right flank. Tumor size
was monitored every 2 days. After 15 days, the animals were
humanely euthanized for tissue collection. The following humane
endpoints were applied: Tumor weight must not exceed 10% of the
normal body weight of the mice, and the diameter of the tumor in
any direction on the body surface of adult mice must not exceed 20
mm. All mice were euthanized by cervical dislocation following
anesthesia with intravenous injection of sodium pentobarbital at a
dose of 70 mg/kg body weight. Death was confirmed by the absence of
respiration and heartbeat for >5 min.
Hematoxylin and eosin (H&E)
staining and immunohistochemistry (IHC)
The subcutaneous tumors were fixed in 10% formalin
at room temperature for 24 h, dehydrated in graded ethanol and
embedded in paraffin. The paraffin-embedded tumors were sectioned
into 4-µm slices. The slices were then oven-baked at 56°C overnight
and stained using the H&E Stain kit (Beijing Solarbio Science
& Technology Co., Ltd.), according to the manufacturer's
protocol.
For IHC, the slides were placed in 10 mM sodium
citrate buffer (pH 6.0) and boiled, then simmered for 10 min, for
antigen retrieval. Subsequently, the slides were cooled on the
bench for 30 min. To remove endogenous peroxidase activity, slides
were incubated with 3% hydrogen peroxide aqueous solution, limiting
the exposure time to ~10 min at room temperature. Furthermore,
blocking was performed using TBS- 0.1% Tween with 5% normal goat
serum (Cell Signaling Technology, Inc.). The sections were then
incubated overnight at 4°C with primary antibodies against PLEKHA4
(1:200 dilution; cat. no. NBP1-56679; Novus Biologicals;
Bio-Techne). The next day, a biotinylated Goat Anti-Rabbit IgG
H&L (Biotin) secondary antibody (1:100; cat. no. ab207995;
Abcam) was added, and the slides were incubated at room temperature
for 30 min. Then streptavidin-horseradish peroxidase (cat. no.
SA10001; Invitrogen; Thermo Fisher Scientific, Inc.) was then used
to incubate the slides at room temperature for 30 min and 200 µl
DAB was added to each slice as the chromogen. Hematoxylin was used
to counterstain the slices at room temperature for 1 min. Finally,
images of the slides were captured using a Nikon Eclipse Ti-S/L100
Inverted Phase Contrast Fluorescent Microscope with a 10X
objective.
Statistical analysis
Statistical analysis was conducted using unpaired
Student's t-test to compare the differences between two groups,
while one-way ANOVA with Tukey's post hoc analysis was performed
for comparisons among multiple groups. Data analysis was performed
using SPSS 26.0 (IBM Corp.). The data are presented as the mean ±
SD. Each experiment was replicated ≥3 times. P<0.05 was
considered to indicate a statistically significant difference.
Results
PLEKHA4 is upregulated in KIRC and is
associated with β-catenin signaling
GSCA was used to perform a comprehensive pan-cancer
analysis to investigate the expression patterns of PLEKHA4 in human
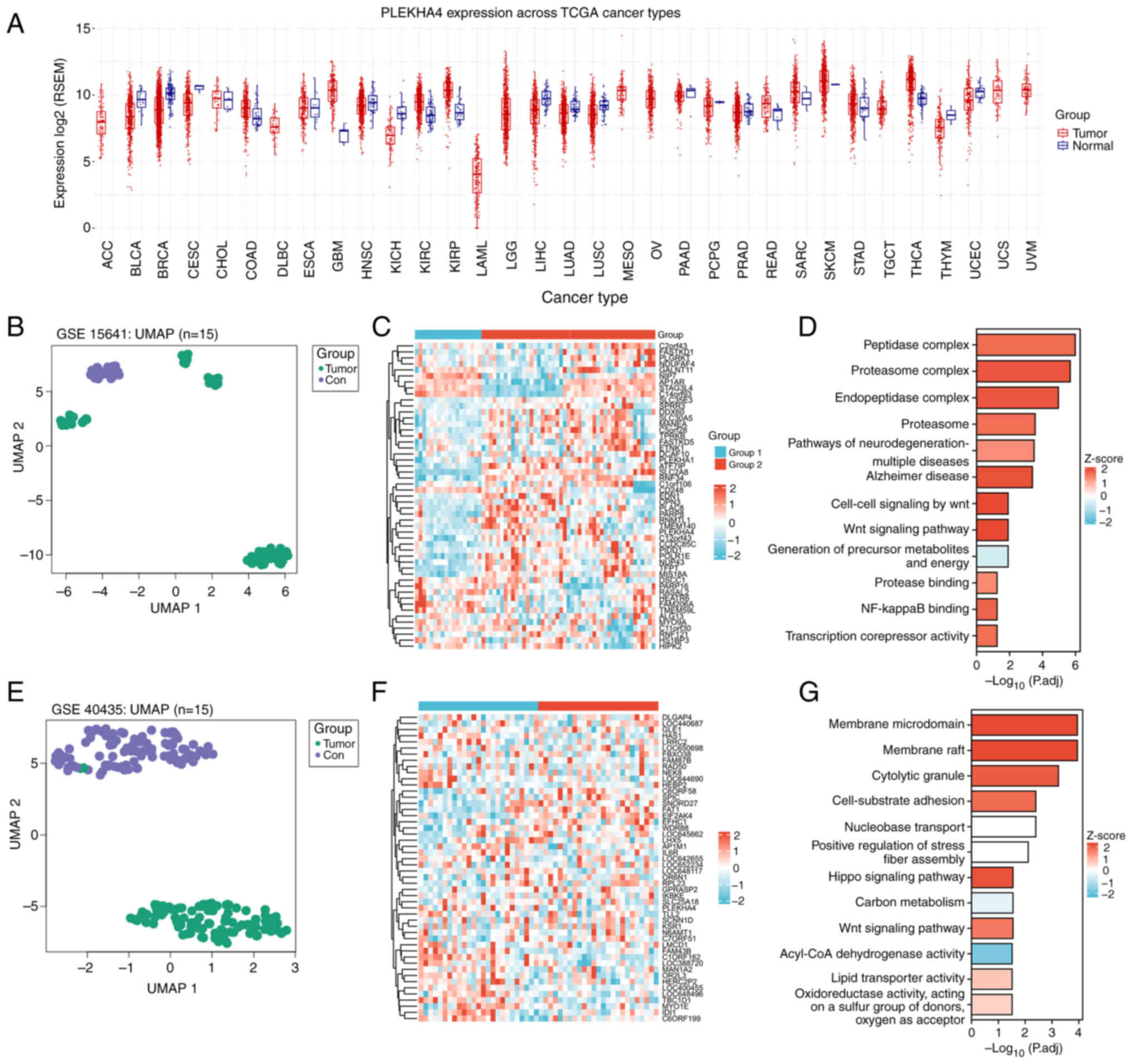
cancer tissues. PLEKHA4 was upregulated in numerous types of
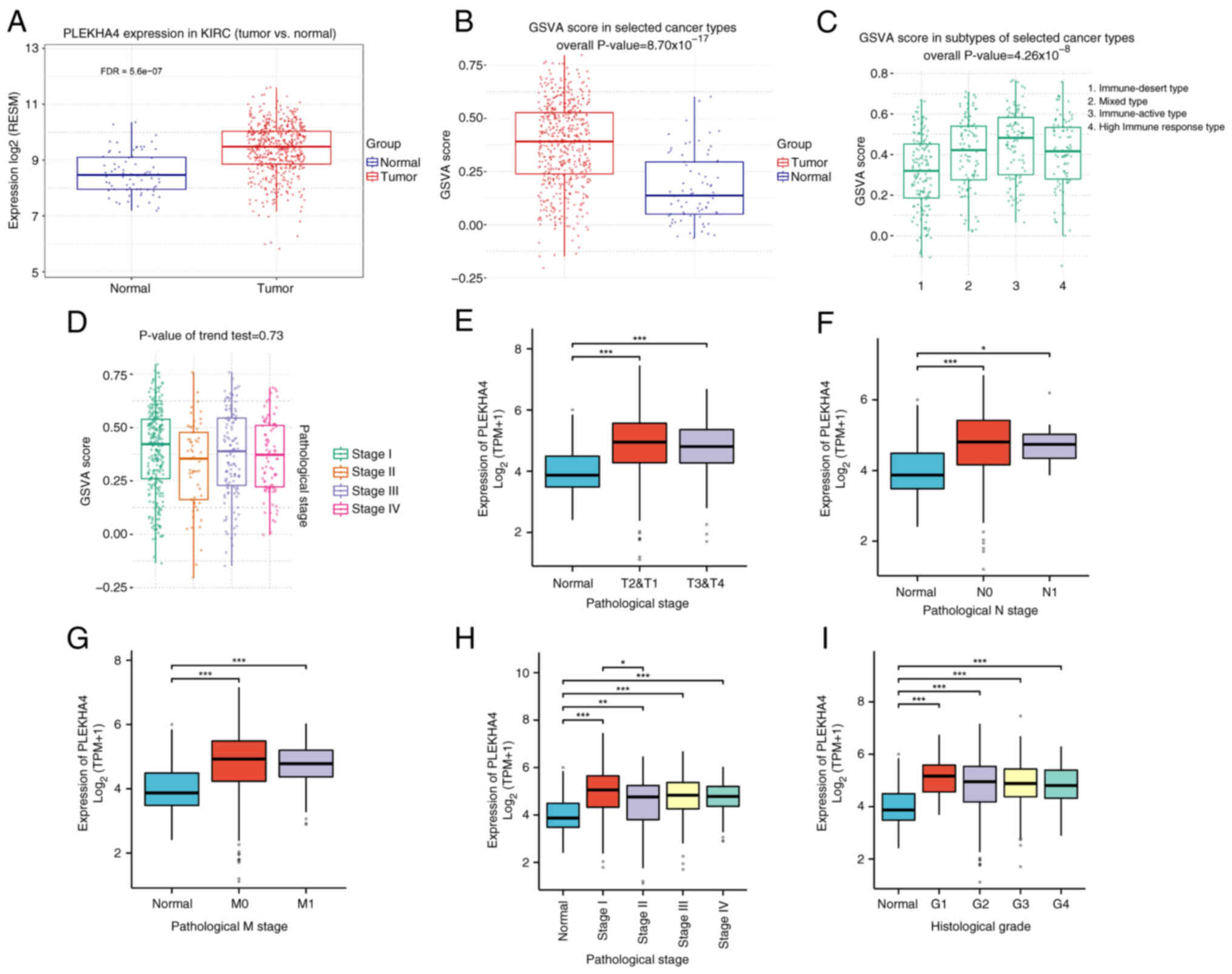
cancer, including KIRC (Figs. 1A
and 2A). Based on data from GEO
datasets, tumor tissues exhibited distinctive gene expression
profiles compared with normal tissue (a mix of healthy control
tissues and normal tissues adjacent to the cancer tissues)
(Fig. 1B and E). The heatmaps
illustrate the elevated expression of PLEKHA4 in renal tumors and
KIRC tissue (Fig. 1C and F).
Furthermore, GO and KEGG analyses revealed the upregulation of
genes enriched in the ‘Wnt signaling pathway’ in both renal tumors
and KIRC tissue (Fig. 1D and G).
The results of both GO and KEGG analyses are presented as combined
outcomes. To elucidate PLEKHA4 expression in KIRC, GSVA was used;
there was a higher GSVA score in KIRC compared with in normal
kidney tissue (Fig. 2B). GSVA
scores represent the degree to which the genes in the gene set are
coordinately up- or downregulated within that sample. In addition,
GSVA scores were evaluated across various subtypes and stages of
KIRC (Fig. 2C and D). PLEKHA4
expression in different pathological and histological grades of
KIRC demonstrated significantly elevated levels in comparison with
normal kidney tissue (Fig. 2E-I).
Across various pathological stages, PLEKHA4 expression was
consistently higher in cancerous tissues compared with in normal
tissue (Fig. 2E). Specifically,
when splitting patients according to pathological T stage, both
T2&T1 and T3&T4 stage tissues exhibited elevated PLEKHA4
levels compared with in normal tissues from healthy individuals
(Fig. 2F). Similarly, when
splitting patients according to pathological N stage, tissues from
patients with both N0 and N1 cancer showed increased PLEKHA4
expression (Fig. 2G) compared with
in normal tissues from healthy individuals. Regarding pathological
M stage, tissues from patients with both M0 and M1 cancer
demonstrated higher PLEKHA4 levels (Fig. 2H) compared with in normal tissues
from healthy individuals. Additionally, all stages (I, II, III, IV)
and histologic grades (G1, G2, G3, G4) of KIRC exhibited elevated
PLEKHA4 expression compared with that in normal tissues from
healthy people (Fig. 2I).
PLEKHA4 knockdown inhibits KIRC cell
proliferation
In order to assess PLEKHA4 expression in KIRC and
normal tissues, the Human Protein Atlas (https://www.proteinatlas.org/) was used, which aims to
map all human proteins in cells, tissues and organs using
technologies such as antibody-based imaging, proteomics,
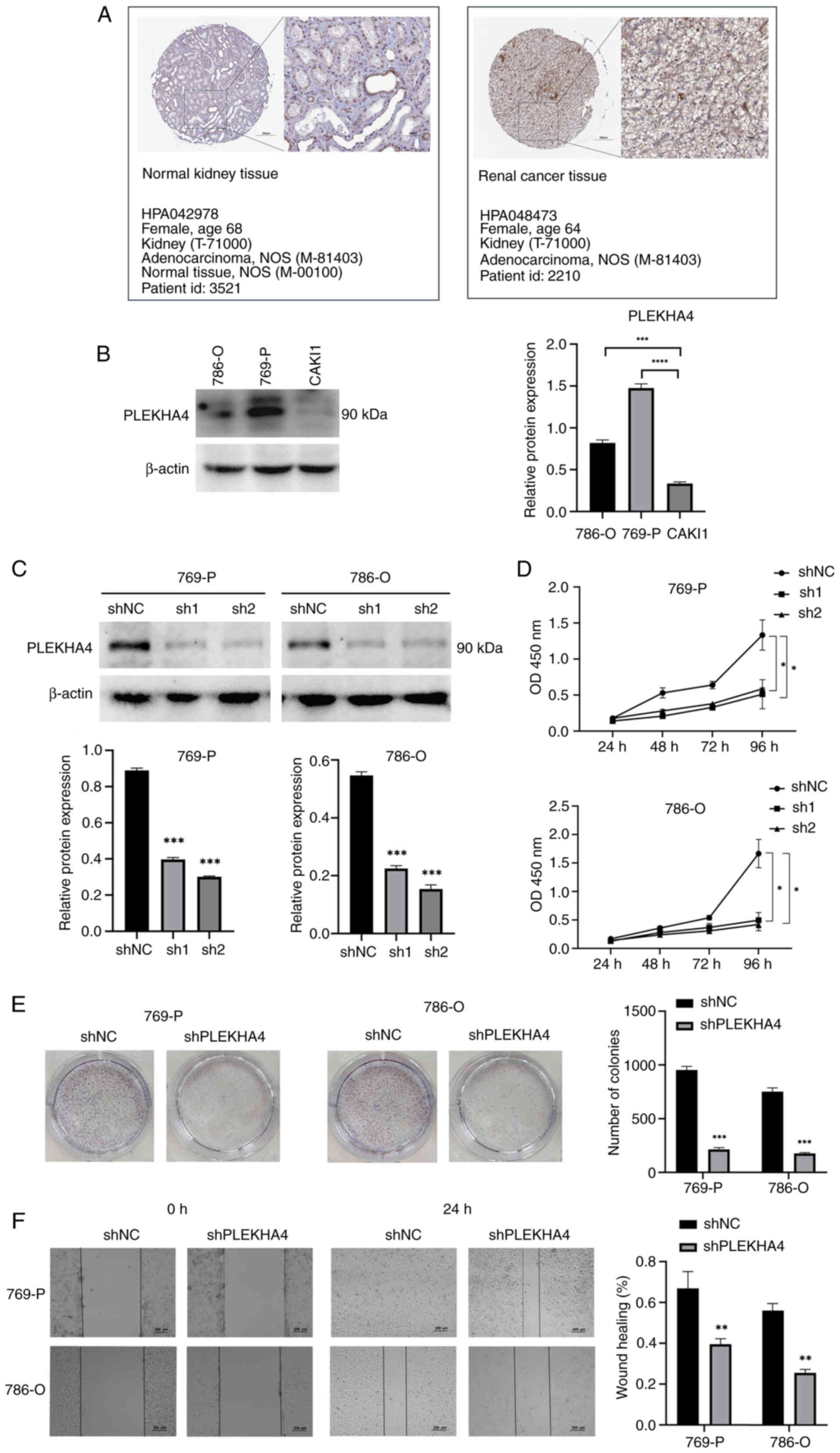
transcriptomics and systems biology. High expression levels of
PLEKHA4 were observed in KIRC tissue (Fig. 3A), and its levels in were also
examined KIRC cells (Fig. 3B);
786-O and 769-P cells had higher PLEKHA4 levels than CAKI1 cells.
Subsequently, PLEKHA4 knockdown experiments were conducted in 769-P
and 786-O cells. The virus infection efficiencies of K7453 in 769-P
and 786-O cells were 68 and 41%, respectively, while those of K7451
were 73 and 57%, respectively. For LV3-shNC, the transfection
efficiencies in 769-P and 786-O cells were 63 and 54%,
respectively. Protein levels in normal KIRC and PLEKHA4 knockdown
cells were evaluated using western blot analysis. The results
demonstrated a significant decrease in PLEKHA4 protein expression
in 769-P and 786-O cells following knockdown (Fig. 3C). Proliferation, colony formation
and migration assays indicated that PLEKHA4 knockdown inhibited
cell proliferation (Fig. 3D),
colony formation (Fig. 3E) and
migration (Fig. 3F).
Knockdown of PLEKHA4 inhibits KIRC
cell malignancy
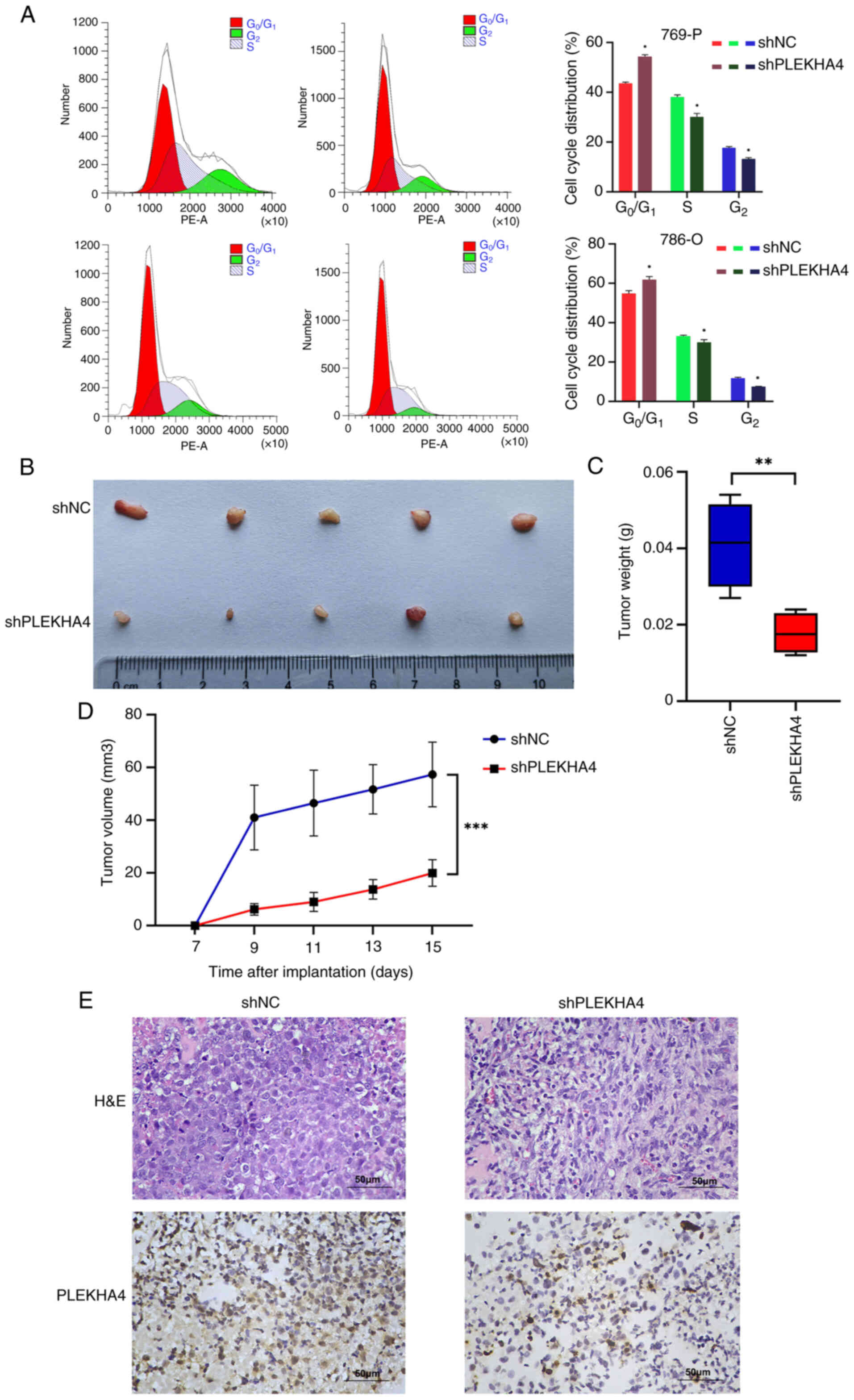
Flow cytometric analysis was conducted to examine
the cell cycle following PLEKHA4 knockdown. The results revealed
G1/S phase arrest in the two KIRC cell lines (Fig. 4A). Subsequently, a subcutaneous
tumor growth model was established to investigate the impact of
PLEKHA4 on tumor growth. The knockdown of PLEKHA4 attenuated tumor
growth (Fig. 4B-D). Tumor samples
were collected and subjected to H&E staining and IHC to assess
PLEKHA4 expression in vivo. Cells within the SHPLEKHA4 group
exhibited lighter staining and were a smaller size with increased
heterogeneity compared with those in the shNC group (Fig. 4E).
PLEKHA4 regulates Wnt/β-catenin
signaling in KIRC cells
Given the observed upregulation of Wnt signaling in
KIRC, Gene Mania was used to explore the interaction between
PLEKHA4 and proteins associated with β-catenin (Fig. 5A). The results indicated that
PLEKHA4 may have interactions with GSK3β and β-catenin. Following
PLEKHA4 knockdown in 769-P and 786-O cells, western blotting
revealed a reduction in the expression levels of p-GSK3β, β-catenin
and cyclin D1 (Fig. 5B). The
translocation of β-catenin into the nucleus is crucial for
initiating gene transcription and driving tumorigenesis (14). PLEKHA4 knockdown resulted in
decreased β-catenin levels in the nuclear fraction compared with in
shNC cells, while cytosolic β-catenin levels were increased
(Fig. 5C). In 786-O and 769-P
cells, cytoplasmic β-catenin expression was significantly lower
than nuclear expression in the shNC group. Conversely, in sh1- and
sh2-transfected cells, cytoplasmic β-catenin expression was
significantly higher than nuclear expression. This indicates that
PLEKHA4 may facilitate the nuclear translocation of β-catenin,
since knockdown of PLEKHA4 reduced the nuclear translocation of
β-catenin. Lamin B1 was used as a marker to compare β-catenin
expression levels in the nucleus with those in the cytoplasm
(Fig. 5C), suggesting that PLEKHA4
promoted the nuclear translocation of β-catenin. To investigate the
impact of PLEKHA4 on the Wnt/β-catenin pathway, KIRC cells were
also treated with the Wnt signaling activator LiCl following
PLEKHA4 knockdown. Western blotting demonstrated that LiCl reversed
alterations induced by PLEKHA4 knockdown (Fig. 5D). Furthermore, overexpression was
conducted to examine the role of PLEKHA4 on CAKI1 cells. PLEKHA4
overexpression led to an upregulation of proteins associated with
Wnt/β-catenin signaling (Fig. 5E).
These findings indicated that PLEKHA4 may serve a regulatory role
in Wnt/β-catenin signaling in KIRC cells.
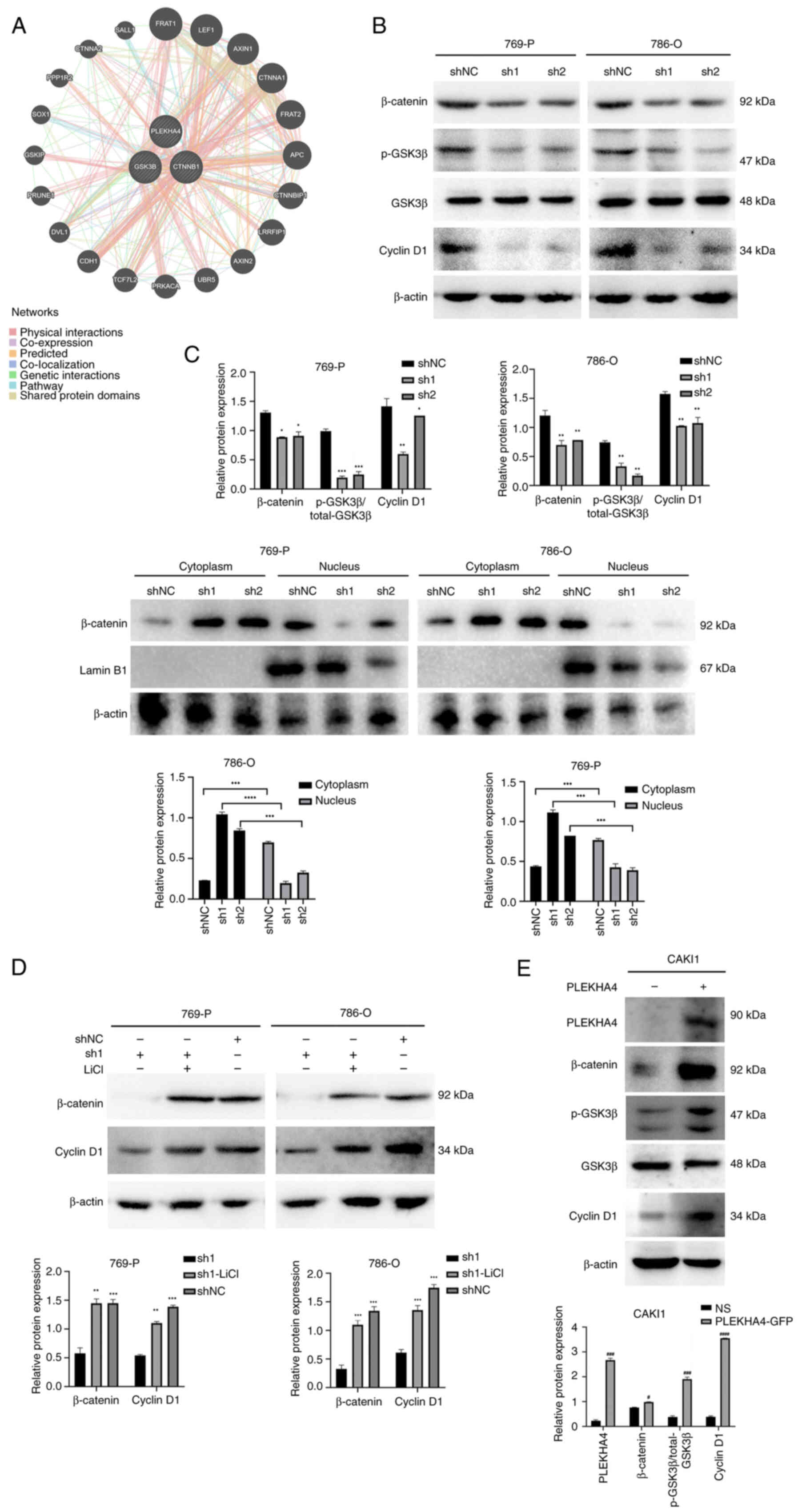 | Figure 5.PLEKHA4 knockdown inhibits the
Wnt/β-catenin signaling pathway. (A) Functional association
networks of PLEKHA4 and β-catenin-related genes. (B) Western
blotting showed that knockdown of PLEKHA4 decreased the expression
of β-catenin, p-GSK3β and cyclin D1. (C) Effects of PLEKHA4
knockdown on subcellular localization of β-catenin in 769-P and
786-O cells. (D) LiCl reverses the changes induced by PLEKHA4
knockdown in related proteins. (E) PLEKHA4 overexpression increases
expression of β-catenin, p-GSK3β and cyclin D1. *P<0.05,
**P<0.01, ***P<0.001, ****P<0.0001 vs. shNC or as
otherwise indicated. #P<0.05,
###P<0.001, ####P<0.0001 vs. NS.
PLEKHA4, pleckstrin homology domain-containing family A member 4;
sh, short hairpin; NC, negative control; NS, empty vector; p-,
phosphorylated. |
Discussion
Kidney cancer accounts for 2–3% of all cancer cases
worldwide and is considered one of the most common types of
urological malignancy, with >330,000 new cases reported each
year (4,7). Renal cancer is a complex disease
comprising different subtypes, with KIRC being the most predominant
form. This subtype is associated with a widespread and deadly
condition, with 431,288 new cases and 179,368 deaths reported
globally in 2020 (6), underscoring
the importance of improving the understanding of its biological
attributes (7). A key challenge is
a lack of clear understanding regarding the underlying mechanisms
of KIRC (4). PLEKHA4, also known
as PEPP1, encodes a protein featuring a pleckstrin homology domain
positioned near the N-terminus and harbors a putative
phosphatidylinositol 3,4,5-trisphosphate binding motif (6). PLEKHA4 has been shown to be
upregulated in glioblastoma and inhibits apoptosis by modulating
apoptotic regulators. The present study focused on the role of
PLEKHA4 in KIRC, but did not specifically explore apoptosis. The
present results indicated that PLEKHA4 was also upregulated in KIRC
and knocking down PLEKHA4 inhibited cell proliferation, which may
lead to increased apoptosis. The aforementioned data highlight the
role of PLEKHA4 in promoting tumor progression, potentially via
modulation of apoptotic pathways and cell proliferation.
Previous studies have highlighted the role of
PLEKHA4 in promoting aberrant Wnt signaling in mouse cells (C57MG,
C57/MV7) and human melanoma proliferation (6–8).
Furthermore, PLEKHA4 has been implicated in modulating chemokines
and JAK/STAT pathways, along with impacting the cell cycle in
glioma, suggesting its potential use as a prognostic biomarker for
glioma (7). Nevertheless, further
data is needed to confirm its utility in different grades of KIRC,
given the current limitations of bioinformatics analysis, due to
the limited availability of clinical tissue RNA sequencing data.
Moreover, more clinical data must be collected due to the shortage
of clinical tissue samples. Studies have identified Von
Hippel-Lindau (VHL) tumor suppressor gene as being closely linked
to RCC (17,18), highlighting its association with
the pathogenesis of this malignancy. Notably, VHL has been
identified as a target of β-catenin, indicating the potential
importance of Wnt/β-catenin signaling in RCC development (18). In the present study, the knockdown
of PLEKHA4 in KIRC cells notably influenced the expression of
classical Wnt/β-catenin targets, such as cyclin D1, leading to cell
cycle arrest at the G1/S phase. This aligns with
findings from previous research (19). An aberrant cell cycle is a
recognized hallmark of cancer, with cyclin D1 playing a key role in
regulating the G1-S transition (9). Numerous studies have reported
elevated cyclin D1 expression in RCC (10,17,19).
Cyclin D1 can be transcriptionally activated by β-catenin within
the nucleus (18,20).
The canonical Wnt signaling pathway is a highly
conserved regulatory mechanism key for embryonic development and
the maintenance of adult tissue homeostasis. Perturbations in this
pathway can contribute to a spectrum of diseases, including
congenital malformations, neurodegenerative disorders, diabetes and
diverse forms of cancer (21,22).
A key event in the pathway entails the nuclear translocation of
β-catenin. Upon binding of Wnt ligands, inhibition of the β-catenin
destruction complex in the cytoplasm occurs. This leads to the
accumulation of β-catenin, facilitating its translocation into the
nucleus (21,22). The translocation of β-catenin into
the nucleus results in increased expression of c-Myc and MMPs,
thereby promoting the advancement of cancer (23,24).
c-Myc serves a vital role in tumorigenesis across various human
tissues (25), while MMPs are
linked to metastasis and angiogenesis, further driving cancer
progression (24). The present
study conducted a rescue assay to counteract the effects of PLEKHA4
knockdown. Notably, the Wnt/β-catenin activator LiCl effectively
reversed the alterations induced by PLEKHA4 knockdown.
Additionally, the overexpression of PLEKHA4 in a KIRC cell line
with low PLEKHA4 expression (CAKI1) activated the Wnt/β-catenin
signaling pathway. These findings provide crucial supplementary
evidence supporting the regulatory role of PLEKHA4 in Wnt/β-catenin
signaling in KIRC cells. Targeted inhibition of Wnt signaling may
be a promising strategy for the development of novel anticancer
therapy if it can be accomplished selectively to minimize adverse
effects on healthy tissue (26–28).
The present study used 769-P, 786-O and CAKI1 cells, which have
been widely used in renal cancer studies, and there is a
substantial body of literature reporting their biological
characteristics and experimental responses (29–32).
By using the 769-P, 786-O and CAKI1 cell lines, more representative
and broadly applicable experimental results can be obtained.
In conclusion, PLEKHA4 was upregulated in KIRC and
was associated with cell proliferation. Knockdown of PLEKHA4
suppressed β-catenin signaling and impeded its nuclear
translocation. However, the specific mechanism by which PLEKHA4
interacts with β-catenin remains unexplored. It is unclear whether
PLEKHA4 directly binds to β-catenin or facilitates its
translocation through intermediary molecules. Future investigations
are warranted to elucidate this potential interaction.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Project of Education
Department of the Jilin Province of China (grant no.
JJKH20180910KJ).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YY, GA and SC performed the experiments. DX made
substantial contributions to data analysis. XL and LD made
substantial contributions to the bioinformatics analysis, drafted
the manuscript, critically reviewed the manuscript for important
intellectual content and constructed the figures. LL and TJ made
substantial contributions to the conception or design of the work.
LL and TJ confirm the authenticity of all the raw data. All authors
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Laboratory
Animal Ethics Committee Yanbian University (approval no.
YD20230911020). All methods were performed in accordance with
Yanbian University Laboratory Animal Management Rules.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bahadoram S, Davoodi M, Hassanzadeh S,
Bahadoram M, Barahman M and Mafakher L: Renal cell carcinoma: An
overview of the epidemiology, diagnosis, and treatment. G Ital
Nefrol. 39:2022–vol3. 2022.PubMed/NCBI
|
|
2
|
Bex A, Albiges L, Ljungberg B, Bensalah K,
Dabestani S, Giles RH, Hofmann F, Hora M, Kuczyk MA, Lam TB, et al:
Updated European association of urology guidelines for
cytoreductive nephrectomy in patients with synchronous metastatic
clear-cell renal cell carcinoma. Eur Urol. 74:805–809. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Botrugno OA, Fayard E, Annicotte JS, Haby
C, Brennan T, Wendling O, Tanaka T, Kodama T, Thomas W, Auwerx J
and Schoonjans K: Synergy between LRH-1 and beta-catenin induces G1
cyclin-mediated cell proliferation. Mol Cell. 15:499–509. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chow WH, Dong LM and Devesa SS:
Epidemiology and risk factors for kidney cancer. Nat Rev Urol.
7:245–257. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gobbo S, Eble JN, Grignon DJ, Martignoni
G, MacLennan GT, Shah RB, Zhang S, Brunelli M and Cheng L: Clear
cell papillary renal cell carcinoma: A distinct histopathologic and
molecular genetic entity. Am J Surg Pathol. 32:1239–1245. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gao X, Liu Y, Hong S, Yang H, Guan B and
Ma X: PLEKHA4 is associated with tumour microenvironment, stemness,
proliferation and poor prognosis of gliomas. J Integr Neurosci.
22:1352023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shami Shah A, Batrouni AG, Kim D, Punyala
A, Cao W, Han C, Goldberg ML, Smolka MB and Baskin JM:
PLEKHA4/kramer attenuates dishevelled ubiquitination to modulate
Wnt and planar cell polarity signaling. Cell Rep. 27:2157–2170.e8.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tanton H, Sewastianik T, Seo HS, Remillard
D, Pierre RS, Bala P, Aitymbayev D, Dennis P, Adler K, Geffken E,
et al: A novel β-catenin/BCL9 complex inhibitor blocks oncogenic
Wnt signaling and disrupts cholesterol homeostasis in colorectal
cancer. Sci Adv. 8:eabm31082022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nusse R and Clevers H: Wnt/β-catenin
signaling, disease, and emerging therapeutic modalities. Cell.
169:985–999. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu Q, Krause M, Samoylenko A and Vainio S:
Wnt signaling in renal cell carcinoma. Cancers (Basel). 8:572016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu X, Zhang M, Xu F and Jiang S: Wnt
signaling in breast cancer: Biological mechanisms, challenges and
opportunities. Mol Cancer. 19:1652020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li Y, Xiao X, Chen H, Chen Z, Hu K and Yin
D: Transcription factor NFYA promotes G1/S cell cycle transition
and cell proliferation by transactivating cyclin D1 and CDK4 in
clear cell renal cell carcinoma. Am J Cancer Res. 10:2446–2463.
2020.PubMed/NCBI
|
|
14
|
Ji J, Xu Y, Xie M, He X, Ren D, Qiu T, Liu
W, Chen Z, Shi W, Zhang Z, et al: VHL-HIF-2α axis-induced SEMA6A
upregulation stabilized β-catenin to drive clear cell renal cell
carcinoma progression. Cell Death Dis. 14:832023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jones J, Out H, Spentzos D, Kolia S, Inan
M, Beecken WD, Fellbaum C, Gu X, Joseph M, Pantuck AJ, et al: Gene
signatures of progression and metastasis in renal cell cancer. Clin
Cancer Res. 11:5730–5739. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wozniak MB, Le Calvez-Kelm F,
Abedi-Ardekani B, Byrnes G, Durand G, Carreira C, Michelon J,
Janout V, Holcatova I, Foretova L, et al: Integrative genome-wide
gene expression profiling of clear cell renal cell carcinoma in
Czech Republic and in the United States. PLoS One. 8:e578862013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
MacDonald BT, Tamai K and He X:
Wnt/beta-catenin signaling: Components, mechanisms, and diseases.
Dev Cell. 17:9–26. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Maretzky T, Reiss K, Ludwig A, Buchholz J,
Scholz F, Proksch E, de Strooper B, Hartmann D and Saftig P: ADAM10
mediates E-cadherin shedding and regulates epithelial cell-cell
adhesion, migration, and beta-catenin translocation. Proc Natl Acad
Sci USA. 102:9182–9187. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shami Shah A, Cao X, White AC and Baskin
JM: PLEKHA4 promotes Wnt/β-catenin signaling-mediated
G1-S transition and proliferation in melanoma. Cancer
Res. 81:2029–2043. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hedberg Y, Davoodi E, Roos G, Ljungberg B
and Landberg G: Cyclin-D1 expression in human renal-cell carcinoma.
Int J Cancer. 84:268–272. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Albrecht LV, Tejeda-Muñoz N and De
Robertis EM: Cell Biology of canonical Wnt signaling. Annu Rev Cell
Dev Biol. 37:369–389. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Niehrs C: The complex world of WNT
receptor signalling. Nat Rev Mol Cell Biol. 13:767–779. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bian J, Dannappel M, Wan C and Firestein
R: Transcriptional regulation of Wnt/β-catenin pathway in
colorectal cancer. Cells. 9:21252020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hashemi M, Hasani S, Hajimazdarany S,
Ghadyani F, Olyaee Y, Khodadadi M, Ziyarani MF, Dehghanpour A,
Salehi H, Kakavand A, et al: Biological functions and molecular
interactions of Wnt/β-catenin in breast cancer: Revisiting
signaling networks. Int J Biol Macromol. 232:1233772023. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang Y and Wang X: Targeting the
Wnt/β-catenin signaling pathway in cancer. J Hematol Oncol.
13:1652020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Krishnamurthy N and Kurzrock R: Targeting
the Wnt/beta-catenin pathway in cancer: Update on effectors and
inhibitors. Cancer Treat Rev. 62:50–60. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Neiheisel A, Kaur M, Ma N, Havard P and
Shenoy AK: Wnt pathway modulators in cancer therapeutics: An update
on completed and ongoing clinical trials. Int J Cancer.
150:727–740. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang N, Wei P, Gong A, Chiu WT, Lee HT,
Colman H, Huang H, Xue J, Liu M, Wang Y, et al: FoxM1 promotes
β-catenin nuclear localization and controls Wnt target-gene
expression and glioma tumorigenesis. Cancer Cell. 20:427–442. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li P, Chen T, Kuang P, Liu F, Li Z, Liu F,
Wang Y, Zhang W and Cai X: Aurora-A/FOXO3A/SKP2 axis promotes tumor
progression in clear cell renal cell carcinoma and dual-targeting
Aurora-A/SKP2 shows synthetic lethality. Cell Death Dis.
13:6062022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Swiatek M, Jancewicz I, Kluebsoongnoen J,
Zub R, Maassen A, Kubala S, Udomkit A, Siedlecki JA, Sarnowski TJ
and Sarnowska E: Various forms of HIF-1α protein characterize the
clear cell renal cell carcinoma cell lines. IUBMB Life.
72:1220–1232. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
He W, Cong Z, Niu C, Cheng F, Yi T, Yao Z,
Zhang Y, Jiang X, Sun X, Niu Z and Fu Q: A prognostic signature
based on genes associated with m6A/m5C/m1A/m7G modifications and
its immunological characteristics in clear cell renal cell
carcinoma. Sci Rep. 14:187082024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yin X, Wang J and Zhang J: Identification
of biomarkers of chromophobe renal cell carcinoma by weighted gene
co-expression network analysis. Cancer Cell Int. 18:2062018.
View Article : Google Scholar : PubMed/NCBI
|