Introduction
Chronic obstructive pulmonary disease (COPD) is a
prevalent chronic respiratory condition associated with high
morbidity, mortality and disability rates, which result in death of
approximately 3.2 million individuals annually (1,2). By
2030, COPD is projected to become the third leading cause of
mortality worldwide (3).
Currently, the clinical treatment for COPD aims to alleviate the
clinical symptoms, typically through the use of bronchodilators and
oxygen therapy (4). However, COPD
faces issues, such as under-diagnosis and misdiagnosis, and there
is currently no treatment method to halt the progression of COPD
(5). Therefore, it is imperative
to study potential mechanisms responsible for the occurrence of
COPD and develop novel predictive and therapeutic targets.
Inflammation is the key mechanism in the development
of COPD (6). Alveolar macrophages
(AMs) are the first line of defense in the lung and play a key role
in lung inflammation (7). Studies
have shown that in COPD, there is an increase in number of lung
macrophages and a decrease in antigen presentation ability
(8,9). Smoking is a common risk factor for
the development of COPD (10); M1
polarization of lung macrophages is increased in smokers and
patients with COPD (11).
Therefore, inhibiting cigarette smoke (CS)-induced M1 polarization
of AMs may offer a novel approach for anti-inflammatory treatment
of COPD.
Silent information regulator 1 (SIRT1) is the
mammalian homolog of the yeast silent information regulator 2
protein and is expressed in low levels in the lung of patients with
COPD; SIRT1 is associated with a decline in lung function,
indicating it is a potential biological marker for the severity of
COPD (12). Studies have shown
that SIRT1 serves a key role in COPD by regulating oxidative
stress, inflammatory responses, autophagy and apoptosis (13–15).
The NOD-like receptor thermal protein domain associated protein 3
(NLRP3) signal is a classic inflammatory signaling pathway and
NLRP3 expression is increased in CS-induced AMs (16). SIRT1 can inhibit the expression of
TNF receptor associated factor 6 (TRAF6) (17), which is involved in promoting
polarization of M1 macrophages; its expression is also increased in
serum of patients with COPD (18).
Notably, TRAF6 can bind to NLRP3 and activate its signaling
(19,20).
The present study aimed to explore whether SIRT1 can
inhibit CS extract (CSE)-induced AM damage via the TRAF6/NLRP3
signaling pathway, thus contributing to its protective role in
COPD. The aim of the present study was to elucidate the role and
mechanisms of action of SIRT1 in the occurrence and development of
COPD and to provide an experimental basis for understanding the
pathogenesis COPD. The present results may aid in the development
of novel drug and clinical treatments for COPD.
Materials and methods
Preparation of CSE
CSE was prepared by burning three cigarettes [11.0
tar, 1.1 nicotine, 17.0 mg carbon monoxide; Chongqing Hongsheng
Industrial (Group) Co., Ltd.]. The smoke was dissolved in
serum-free F-12K medium (20 ml; cat. no. 21127022, Gibco; Thermo
Fisher Scientific, Inc) and filtered through a 0.22-µm filter. The
resulting 100% CSE was used within 1 h of preparation. F-12K medium
was used to adjust the CSE working concentration.
Cell culture and treatment
The rat alveolar macrophage cell line NR8383
(American Type Culture Collection), was cultured in F-12K medium
(cat. no. 21127022, Gibco; Thermo Fisher Scientific, Inc) with 15%
FBS (Thermo Fisher Scientific, Inc) and 1% penicillin-streptomycin
at 37°C in a humidified atmosphere of 5% CO2. The cells
were treated as aforementioned (21) with CSE (5, 10 and 20%) for 24, 48
and 72 h at 37°C, respectively.
Cell transfection
The overexpression plasmids of SIRT1 [Ov-SIRT1;
vector, pEX-3(pGCMV/MCS/Neo)], TRAF6 [(Ov-TRAF6; vector,
pEX-3(pGCMV/MCS/Neo)] and negative control (NC) plasmid
pEX-3(pGCMV/MCS/Neo) were purchased from Genepharm Biotech Corp.
The cells were inoculated in a 6-well plate at a density of
1×105 cells/well. Following 24 h culture and upon
reaching 75% confluency, the cells were transfected using 2 µg
overexpression plasmid and Lipofectamine 2000®
(Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C for 48 h.
After 48 h, western blot analysis was performed to detect
transfection efficiency.
Cell Counting Kit (CCK)-8 assay
CCK-8 (BIOSS, cat. no. BA00208) was employed to
assess cell viability. NR8383 cells were inoculated into 96-well
plates at a density of 3×104/l. Following stimulation
with CSE, 10 µl CCK8 solution was added to each well and incubated
at 37°C for 2 h. The optical density at 450 nm was determined by a
microplate reader (Bio-Rad Laboratories, Inc.).
ELISA
The concentrations of TNFα (cat. no. JL13202-96T),
IL-1β (cat. no. JL20884-96T) and IL-6 (cat. no. JL20896-96T; all
JONLNBIO) in the cell supernatant were measured using ELISA kits
according to the manufacturer's instructions.
Western blot analysis
NR8383 cells were lysed in RIPA buffer with protease
inhibitor cocktail (both Beyotime Institute of Biotechnology).
After measuring the protein concentration by BCA assay, 20 µg/lane
protein was electrophoresed on 10% SDS-PAGE and transferred onto
PVDF membranes (Amersham Biosciences). The membrane was blocked in
5% BSA (Biofroxx; neoFroxx) at room temperature for 2 h and
incubated with anti-SIRT1 (1:1,000; cat. no. ab110304, Abcam),
anti-inducible nitric oxide synthase (iNOS; 1:1,500; cat. no.
AF0199, Affinity Biosciences), anti-CD86 (1:1,500; cat. no. DF6332,
Affinity Biosciences), anti-arginase 1 (Arg-1; 1:1,000; cat. no.
DF6657, Affinity Biosciences), anti-CD206 (1:1,500; cat. no.
DF4149, Affinity Biosciences), anti-TRAF6 (1:1,000; cat. no.
ab40675, Abcam), anti-NLRP3 (1:1,500; cat. no. DF7438, Affinity
Biosciences), anti-cleaved caspase-1 (1:1,000; cat. no. AF4022,
Affinity Biosciences), anti-pro-caspase-1 (1:1,000; cat. no.
DF6148, Affinity Biosciences) and anti-GAPDH (1:2,500; cat. no.
ab9485, Abcam) at 4°C overnight. The membrane was incubated with
secondary antibody (goat anti-rabbit IgG-HRP; 1:5,000, cat. no.
S0001, Affinity Biosciences) at 37°C for 2 h. The protein signals
were exposed using ECL reagent (Beyotime Institute of
Biotechnology) and analyzed using ImageJ software version 1.50
(National Institutes of Health).
Reverse transcription-quantitative
(RT-q)PCR
Total RNA was extracted from the NR8383 cells using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.). RT
was performed using the RevertAid RT kit (cat. no. MR101-02, Vazyme
Biotech Co., Ltd.) according to the manufacturer's instructions.
qPCR was performed at 95°C for 120 sec for initial denaturation,
followed by denaturation at 95°C for 15 sec and annealing and
extension at 60°C for 30 sec (40 cycles). Gene expression was
normalized to GAPDH and relative mRNA expression levels were
determined using the 2−ΔΔCq method (22). The primer sequences were obtained
from PrimerBank (pga.mgh.harvard.edu/primerbank) (Table I).
 | Table I.Primer sequences used for reverse
transcription-quantitative PCR. |
Table I.
Primer sequences used for reverse
transcription-quantitative PCR.
| Gene | Primer | Sequence,
5′→3′ |
|---|
| GAPDH | Forward |
GGCAAGTTCAACGGCACAGTC |
|
| Reverse |
TCGCTCCTGGAAGATGGTGATG |
| TNFα | Forward |
GCCCAGACCCTCACACTCAG |
|
| Reverse |
CCGCTTGGTGGTTTGCTACG |
| IL-1β | Forward |
GACTTCACCATGGAACCCGT |
|
| Reverse |
GGAGACTGCCCATTCTCGAC |
| IL-6 | Forward |
CACTTCACAAGTCGGAGGCT |
|
| Reverse |
TCTGACAGTGCATCATCGCT |
| TRAF6 | Forward |
AATCACTTGGCACGGCACTTG |
|
| Reverse |
GGAGAGGAGGCATCGCATGG |
Immunofluorescence assay
Transfected NR8383 cells were inoculated into
six-well plates and when the cell fusion reached 80%, the cells
were fixed with 4% neutral formaldehyde for 30 min at 4°C. Sealing
solution was added to each well followed by incubation at 37°C for
30 min. The primary antibodies [iNOS (1:200; cat. no. AF0199,
Affinity Biosciences) and Arg-1 (1:200; cat. no. DF6332, Affinity
Biosciences)] were incubated overnight at 4°C. The diluted
secondary antibody Goat Anti-Rabbit IgG (H+L) Fluor488-conjugated
(1:200; cat. no. S0018, Affinity Biosciences) was incubated at room
temperature without light for 1 h. After washing the cells with
PBS, nuclei of the cells were re-stained with DAPI solution
(Beyotime Institute of Biotechnology) at room temperature for 10
min and observed under a fluorescence microscope (200×) (Nikon
Corporation).
Co-immunoprecipitation (Co-IP)
assay
NR8383 cell lysates were prepared by IP lysate
(Beyotech Institute of Biotechnology; cat. no. P0013) (150 µl/well)
and incubated with anti-TRAF6 (1 µg) (1:100; cat. no. ab137452,
Abcam) overnight at 4°C followed by addition of 30 µl protein G
Agarose beads (Cytiva) at 4°C for 4 h. After being washed three
times with cold wash and once with lysis buffer (both New Cell and
Molecular Biotech Co., Ltd.), the complexes were isolated by
centrifuging at 1,000 × g at 4°C for 3 min. The immunoprecipitate
was resuspended in 30 µl loading buffer. The expression of NLRP3
was detected using western blot analysis as aforementioned.
Statistical analysis
All analyses were performed using GraphPad Prism 9
(Dotmatics) and data are expressed as the mean ± SD of at least
three independent experiments. All data were analyzed for normality
distribution using the Shapiro-Wilk test. Data were analyzed by
one-way ANOVA followed by Tukey's multiple comparisons post hoc
tests. P<0.05 was considered to indicate a statistically
significant difference.
Results
SIRT1 expression is decreased in
CSE-induced AMs
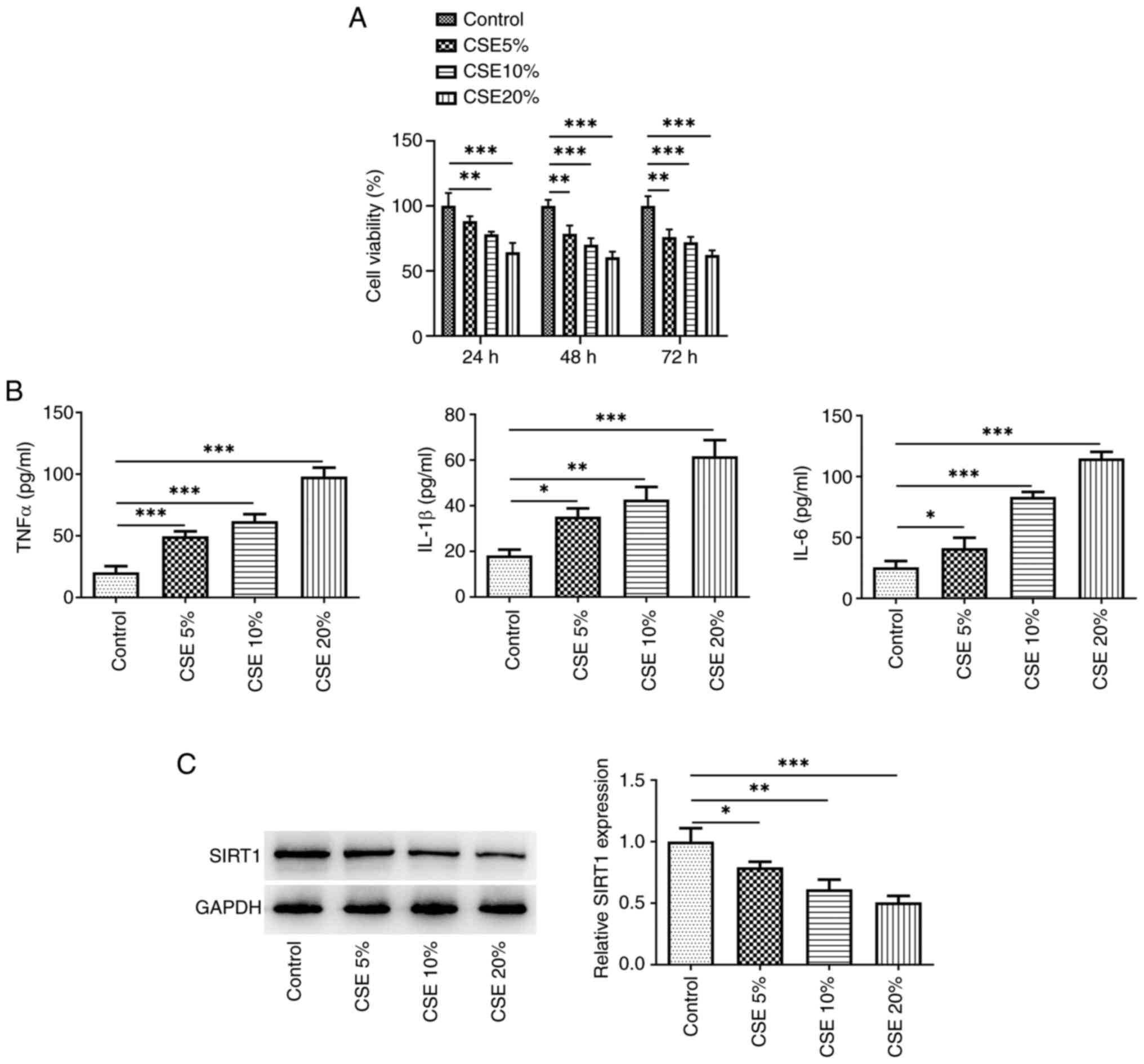
With increasing concentrations of CSE, cell
viability gradually decreased at 24 and 48 h, but there was no
significant difference between 48 and 72 h (Fig. 1A). Therefore, CSE was used to
induce cells for 48 h for subsequent experiments. The secretion of
inflammatory cytokines (TNFα, IL-β, and IL-6) was elevated
(Fig. 1B), whereas SIRT1
expression exhibited a dose-dependent decrease (Fig. 1C). The greatest effects were
observed at 20% CSE, therefore 20% CSE was used in further
experiments.
SIRT1 overexpression inhibits
CSE-induced AM M1-type polarization and inflammatory factor
release
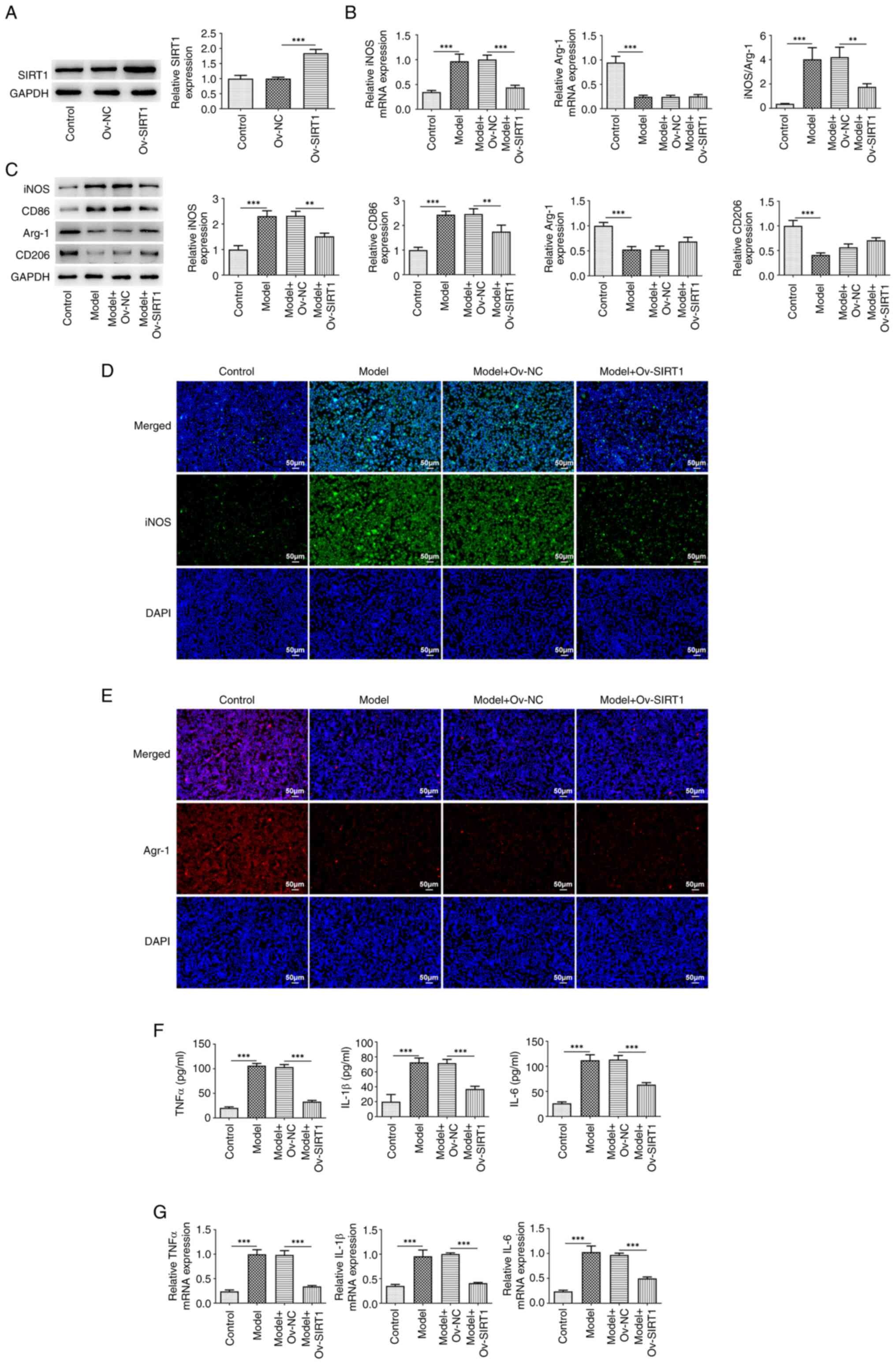
There was a significant increase in SIRT1 expression
in cells transfected with Ov-SIRT1 compared with both the control
and Ov-NC groups (Fig. 2A). The
detection of macrophage polarization markers indicated a
significant elevation in both mRNA (Fig. 2B) and protein expression levels
(Fig. 2C) of the M1 marker iNOS
following exposure to 20% CSE, along with an enhanced fluorescence
intensity of iNOS (Fig. 2D) and
augmented expression of CD86 (Fig.
2C). Following exposure to 20% CSE, there was a significant
decrease in both mRNA (Fig. 2B)
and protein expression levels (Fig.
2C) of the M2 marker Arg-1 accompanied by a reduced
fluorescence intensity of Arg-1 (Fig.
2E) and a diminished expression of CD206 (Fig. 2C). The iNOS/Arg-1 ratio exhibited a
significant decrease (Fig. 2B).
Moreover, exposure to 20% CSE increased expression of TNFα, IL-β
and IL-6 in the cells (Fig. 2F and
G). SIRT1 overexpression significantly reversed these changes
induced by CSE.
SIRT1 overexpression inhibits
TRAF6/NLRP3 signaling activation in CSE-induced macrophages
The present study assessed the TRAF6/NLRP3 signaling
pathway in macrophages stimulated by CSE. There was a significant
increase in the levels of TRAF6, NLRP3 and cleaved caspase-1
proteins in NR8383 cells following exposure to 20% CSE.
Overexpression of SIRT1 significantly reversed this trend (Fig. 3A). IP demonstrated the ability of
TRAF6 to interact with NLRP3 (Fig.
3B). Western blot analysis (Fig.
3C) and RT-qPCR (Fig. 3D)
revealed a significant increase in both the protein and mRNA levels
of TRAF6 in NR8383 cells following transfection with Ov-TRAF6.
Moreover, TRAF6 overexpression attenuated the effects of Ov-SIRT1
on expression of TRAF6, NLRP3 and cleaved caspase-1 proteins in
CSE-induced macrophages (Fig.
3E).
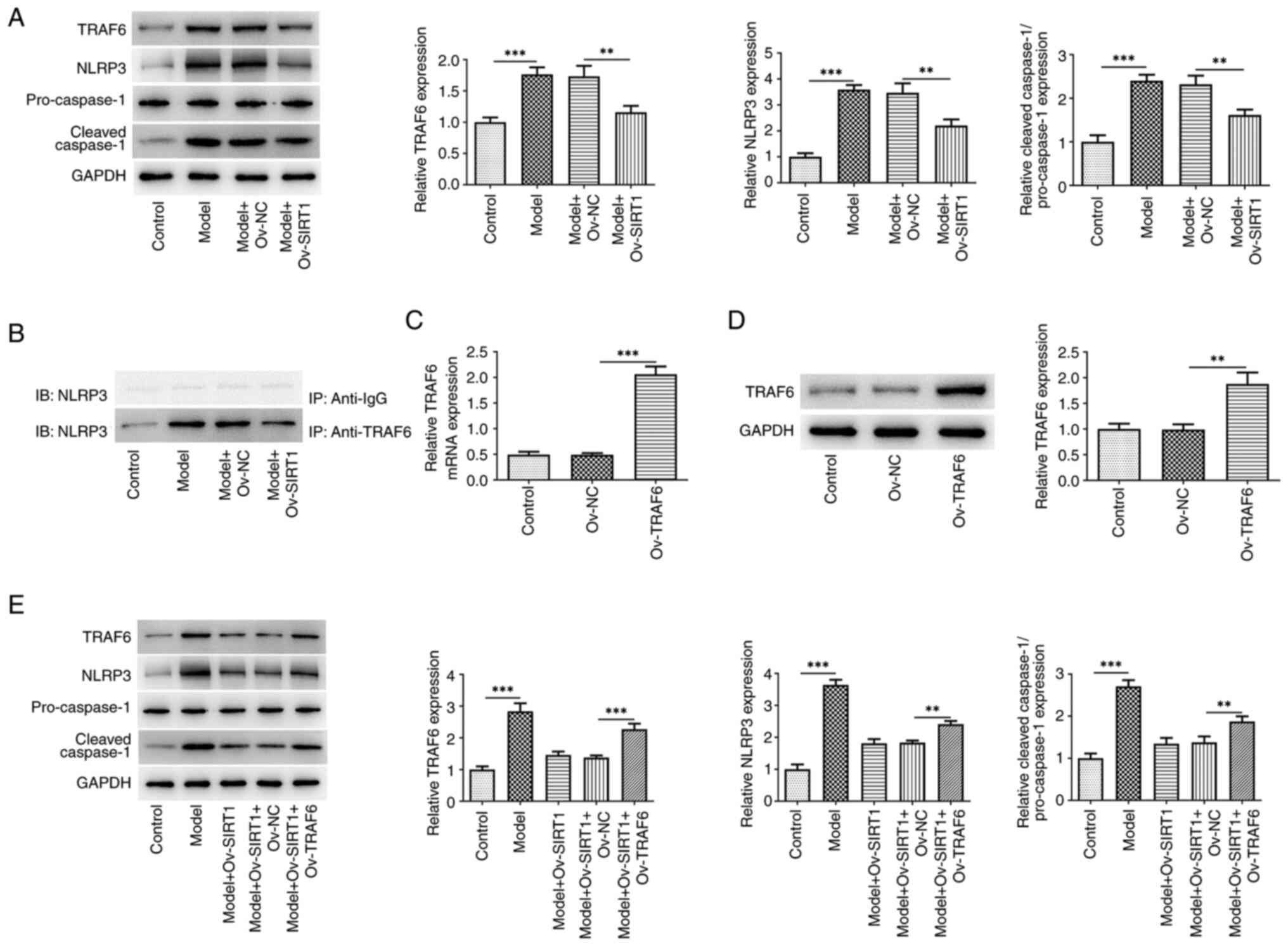 | Figure 3.Ov-SIRT1 overexpression inhibits
TRAF6/NLRP3 signaling activation in CSE-induced macrophages. (A)
TRAF6, NLRP3 and cleaved caspase-1 protein expression in NR8383
cells. (B) Validation of TRAF6 binding ability to NLRP3. TRAF6 (C)
protein and (D) mRNA and (E) TRAF6, NLRP3 and cleaved caspase-1
protein expression in NR8383 cells following TRAF6 overexpression.
**P<0.01 and ***P<0.001; n=3. SIRT1, silent information
regulator 1; CSE, cigarette smoke extract; TRAF6, TNF
receptor-associated factor 6; NLRP3, NOD-like receptor thermal
protein domain associated protein 3; Ov, overexpression; NC,
negative control; IB, immunoblotting; IP, immunoprecipitation. |
Overexpression of SIRT1 inhibits
CSE-induced AM M1-type polarization and inflammatory factor release
by suppressing TRAF6/NLRP3 signaling
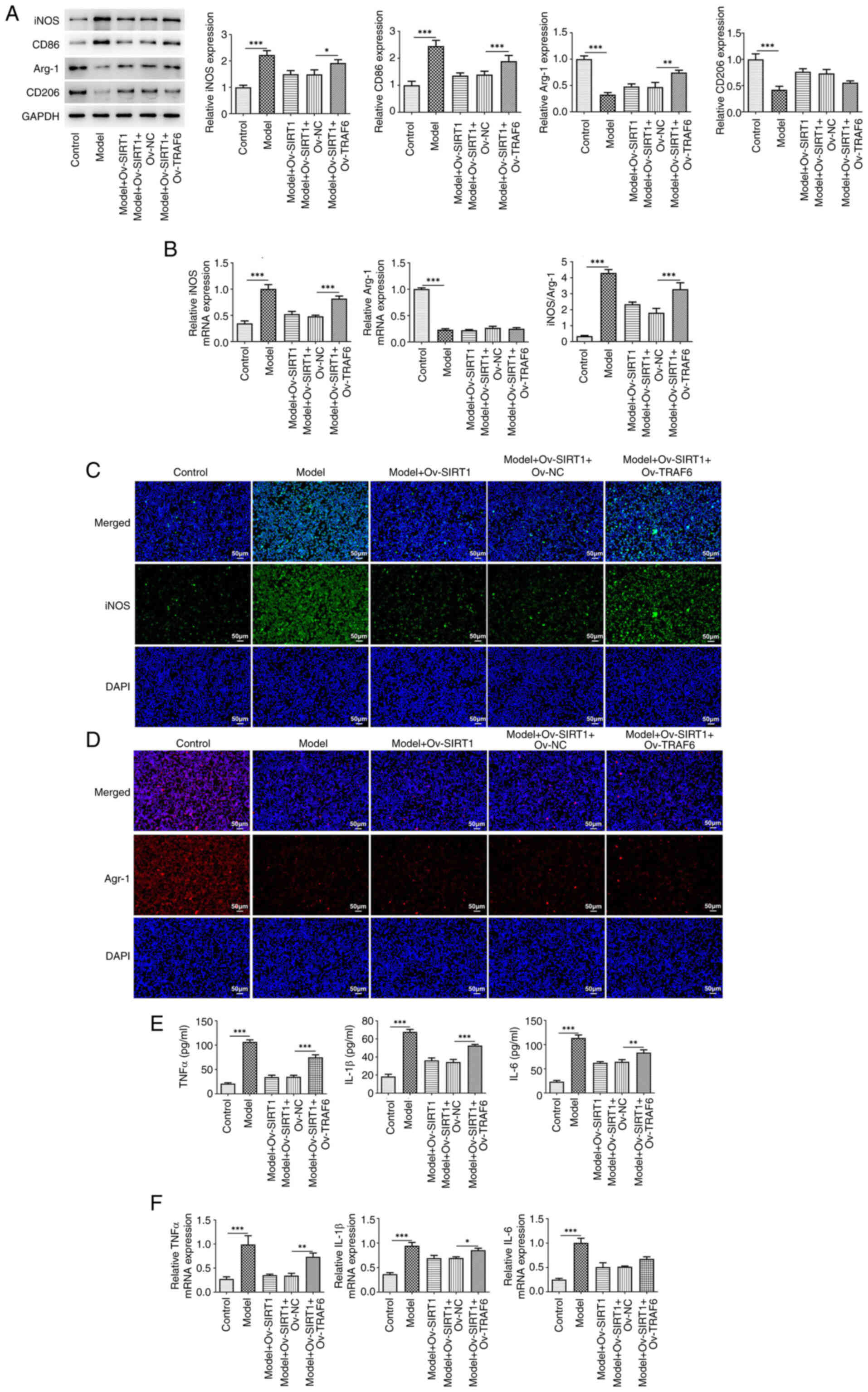
Finally, the present study used the TRAF6
overexpression vector to examine whether TRAF6/NLRP3 signaling
mediates the role of SIRT1. The results revealed marked suppression
of the effects of Ov-SIRT1 on CSE-induced M1 polarization and
inflammatory release in NR8383 cells, supported by the upregulated
protein expression of the M1 markers iNOS and CD86 and the
downregulated protein expression of the M2 markers Arg-1 and CD206
following TRAF6 overexpression (Fig.
4A). Following TRAF6 overexpression, there was an increase in
iNOS mRNA levels (Fig. 4B) and
fluorescence intensity (Fig. 4C),
whereas Arg-1 mRNA levels (Fig.
4B) and fluorescence intensity decreased (Fig. 4D). The release and mRNA levels of
inflammatory cytokines (TNFα, IL-β, and IL-6) also exhibited an
increase (Fig. 4E and F).
Discussion
The present study investigated the involvement of
SIRT1 in AMs induced by CSE. The results revealed a decrease in
SIRT1 expression in NR8383 cells induced by CSE. SIRT1
overexpression suppressed M1 polarization, inflammatory factor
release and TRAF6/NLRP3 signaling activation in NR8383 cells
exposed to CSE. However, enhancing TRAF6 expression markedly
diminished the protective effects of SIRT1 overexpression on NR8383
cells.
Inflammation is a key pathological feature of COPD
(23). Smoking is a contributing
factor for COPD. CS stimulates release of inflammatory mediators
and activates pathways associated with inflammation, leading to
pulmonary inflammation (24). As
the concentration of CSE increased, TNFα, IL-β, and IL-6 release in
NR8383 cells increased, accompanied by a gradual decrease in
cellular viability.
SIRT1 is involved in oxidative stress and chronic
inflammatory responses, known for its anti-aging properties and
associated with the development and progression of COPD (25). Previous research has revealed a
decrease in SIRT1 levels in pulmonary macrophages of patients with
COPD (26), suggesting its
potential involvement in pathogenesis of COPD through the
modulation of these macrophages. AMs are key components of innate
immune responses (27). Depending
on environmental stimuli, they are activated classically (also
known as macrophage M1 polarization) or alternatively (also known
as macrophage M2 polarization). The two polarization directions of
AMs suggest two different inflammatory states. M1-type macrophages
primarily release inflammatory factors (TNFα, IL-β and IL-6) and
promote development of inflammation, and their markers mainly
include iNOS and CD86. M2-type macrophages primarily release
anti-inflammatory factors (TGF-β and IL-10) and inhibit progression
of inflammation, and their markers mainly include CD206 and Arg-1
(28). Dysregulated macrophage
function exacerbates pulmonary inflammation in COPD by affecting
initiation of inflammation, disrupting alveolar architecture and
remodeling the airways (29).
Modulating the balance of M1/M2 macrophage polarization is
effective in alleviating COPD (28). Macrophage polarization is one of
the pathogenic mechanisms of COPD. Feng and Zheng (10) found that M1 polarization of AMs is
inhibited in smokers and patients with COPD and that CS may inhibit
LPS-induced M1 polarization of AMs by suppressing NLRP3. Both
macrophages from individuals with COPD and (30), and CSE-induced macrophages tend to
be M1-type (29). Mu et al
(31) showed that CSE promotes
macrophage polarization towards the M1 type and high-mobility group
box-1 is involved in regulation of macrophage polarization. Li
et al (32) demonstrated
that Fritillaria cirrhosa D. Don inhibits CSE
treatment-induced macrophage M1 polarization, thereby attenuating
inflammatory responses. It has been reported that salidroside
serves a protective role in COPD by inhibiting JNK/c-Jun to reduce
the M1 polarization of AMs induced by CS (7). BML-111 (lipoxin receptor agonist)
treatment decreases iNOS levels and increases Arg-1 expression,
which has the potential to convert macrophages from a
pro-inflammatory M1 to an anti-inflammatory M2 phenotype, thereby
preventing COPD (33).
Rosiglitazone, an exogenous ligand of PPARγ, has been shown to
inhibit CS-induced M1 macrophage polarization and decrease M1/M2
ratio, thereby attenuating emphysema induced by CS exposure and
inflammatory responses (34). The
present study demonstrated a significant decrease in inflammatory
factor release in NR8383 cells following the overexpression of
SIRT1. This reduction was accompanied by a decrease in expression
of M1 polarization markers, such as iNOS and CD86, induced by CSE
in NR8383 cells, along with an increase in expression of M2
markers, including Arg-1 and CD206. Numerous studies have
investigated the role of SIRT1 in COPD: For example, SIRT1 improves
COPD by regulating CS-induced autophagy (35), endoplasmic reticulum stress
(36), airway remodeling and
epithelial-mesenchymal transition (37). To the best of our knowledge, the
present study is the first to reveal that SIRT1 has the capability
to inhibit the M1 polarization of AMs induced by CSE. This provides
novel insight into the role of SIRT1 in COPD.
SIRT1 exerts a protective effect against COPD by
influencing downstream signaling molecules. For example, SIRT1
activates the proliferator-activated receptor-γ
coactivator-1α/NF-κB signaling axis, mitigating oxidative stress
induced by CS in mice with COPD (38). SIRT1 has also been shown to inhibit
inflammation in vivo via the Nrf2/p65 NF-κB pathway, thereby
reversing oxidative stress and inflammation induced by CS (39). Therefore, the present study
explored the mechanisms underlying SIRT1-mediated regulation of AM
polarization. SIRT1 mediates the toll-like receptor 4 (TLR4)/NF-κB
signaling pathway to regulate the polarization of microglial cells
(40). Tetramethylpyrazine
improves acute lung injury by inhibiting the
TLR4/TRAF6/NF-κB/NLRP3/caspase-1 signaling pathway (41). This suggests a potential
association between SIRT1 and TLR4/TRAF6 and their downstream
signaling. Previous research has suggested that TRAF6 can interact
with NLRP3, thereby activating the NLRP3 signaling pathway
(19). IP experiments demonstrated
the ability of TRAF6 to interact with NLRP3. The present study
revealed overexpression of SIRT1 significantly reversed the
increased expression of TRAF6, NLRP3 and cleaved caspase-1 induced
by exposure to 20% CSE treatment. Conversely, overexpression of
TRAF6 attenuated the effects of SIRT1 overexpression on NR8383
cells, indicating that the regulatory role of SIRT1 in CSE-induced
polarization and inflammation of NR8383 cells was mediated by the
TRAF6/NLRP3 signaling pathway. However, further experimental
validation is required to determine whether this process involves
other up- and downstream components of the TRAF6/NLRP3 pathway.
Additionally, the present study primarily examined the role of
SIRT1 in the polarization of AMs induced by CSE at the cellular
level. However, in vitro experiments may not completely
replicate the complexity of the in vivo environment. Thus,
further studies are warranted to validate the findings of the
present study by establishing an animal model of COPD.
In conclusion, the present study conducted a
preliminary exploration of the role of SIRT1 in AMs. The
overexpression of SIRT1 reduced CSE-induced M1 polarization and
inflammatory release in NR8383 cells by inhibiting the TRAF6/NLRP3
signaling pathway.
Acknowledgements
Not applicable.
Funding
The present study was supported by National Natural Science
Foundation Committee, Regional Science Fund Project (grant no.
82160002) and Guangxi Natural Science Foundation, Youth Science
Project (grant no. 2021GXNSFBA220071).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
FY conceptualized the study and wrote the
manuscript. HQ was responsible for conceptualization. CQ and BH
designed and performed experiments. FG, YL, YT, YM and QY analyzed
data and constructed figures. CW designed experiments and edited
the manuscript. All authors have read and approved the final
manuscript. FY and CW confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Celli B, Fabbri L, Criner G, Martinez FJ,
Mannino D, Vogelmeier C, Montes de Oca M, Papi A, Sin DD, Han MK
and Agusti A: Definition and nomenclature of chronic obstructive
pulmonary disease: Time for its revision. Am J Respir Crit Care
Med. 206:1317–1325. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cunha AS, Raposo B, Dias F, Henriques S,
Martinho H and Pedro AR: Management of chronic obstructive
pulmonary disease: Constraints in patient pathway and mitigation
strategies. Port J Public Health. 42:93–100. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gai X, Allwood B and Sun Y:
Post-tuberculosis lung disease and chronic obstructive pulmonary
disease. Chin Med J (Engl). 136:1923–1928. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dorababu A and Maraswami M: Recent
advances (2015–2020) in drug discovery for attenuation of pulmonary
fibrosis and COPD. Molecules. 28:36742023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Han MK: From conundrum to cures,
pioneering breakthroughs in chronic obstructive pulmonary disease
research: Introduction to an AJRCCM special issue. Am J Respir Crit
Care Med. 208:339–340. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Barnes PJ: Inflammatory mechanisms in
patients with chronic obstructive pulmonary disease. J Allergy Clin
Immunol. 138:16–27. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Feng H, Zhang D, Yin Y, Kang J and Zheng
R: Salidroside ameliorated the pulmonary inflammation induced by
cigarette smoke via mitigating M1 macrophage polarization by
JNK/c-Jun. Phytother Res. 37:4251–4264. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Baßler K, Fujii W, Kapellos TS, Dudkin E,
Reusch N, Horne A, Reiz B, Luecken MD, Osei-Sarpong C,
Warnat-Herresthal S, et al: Alveolar macrophages in early stage
COPD show functional deviations with properties of impaired immune
activation. Front Immunol. 13:9172322022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Akata K and van Eeden SF: Lung macrophage
functional properties in chronic obstructive pulmonary disease. Int
J Mol Sci. 21:8532020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brake SJ, Lu W, Chia C, Haug G, Larby J,
Hardikar A, Singhera GK, Hackett TL, Eapen MS and Sohal SS:
Transforming growth factor-β1 and SMAD signalling pathway in the
small airways of smokers and patients with COPD: Potential role in
driving fibrotic type-2 epithelial mesenchymal transition. Front
Immunol. 14:12165062023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Feng H and Zheng R: Cigarette smoke
prevents M1 polarization of alveolar macrophages by suppressing
NLRP3. Life Sci. 327:1218542023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kato R, Mizuno S, Kadowaki M, Shiozaki K,
Akai M, Nakagawa K, Oikawa T, Iguchi M, Osanai K, Ishizaki T, et
al: Sirt1 expression is associated with CD31 expression in blood
cells from patients with chronic obstructive pulmonary disease.
Respir Res. 17:1392016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li S, Huang Q and He B: SIRT1 as a
potential therapeutic target for chronic obstructive pulmonary
disease. Lung. 201:201–215. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
He B, Zhang W, Qiao J, Peng Z and Chai X:
Melatonin protects against COPD by attenuating apoptosis and
endoplasmic reticulum stress via upregulating SIRT1 expression in
rats. Can J Physiol Pharmacol. 97:386–391. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li BS, Zhu RZ, Lim SH, Seo JH and Choi BM:
Apigenin alleviates oxidative stress-induced cellular senescence
via modulation of the SIRT1-NAD[Formula: See text]-CD38 Axis. Am J
Chin Med. 49:1235–1250. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sul OJ, Choi HW, Oh J and Ra SW: GSPE
attenuates CSE-induced lung inflammation and emphysema by
regulating autophagy via the reactive oxygen species/TFEB signaling
pathway. Food Chem Toxicol. 177:1137952023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rajendrasozhan S, Yang SR, Kinnula VL and
Rahman I: SIRT1, an antiinflammatory and antiaging protein, is
decreased in lungs of patients with chronic obstructive pulmonary
disease. Am J Respir Crit Care Med. 177:861–870. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ren W, Xi G, Li X, Zhao L, Yang K, Fan X,
Gao L, Xu H and Guo J: Long non-coding RNA HCG18 promotes M1
macrophage polarization through regulating the miR-146a/TRAF6 axis,
facilitating the progression of diabetic peripheral neuropathy. Mol
Cell Biochem. 476:471–482. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu X, Zhang Y, Zhang Y, Xia L, Yang Y,
Wang P, Xu Y, Ren Z and Liu H: MST4 attenuates NLRP3
inflammasome-mediated neuroinflammation and affects the prognosis
after intracerebral hemorrhage in mice. Brain Res Bull. 177:31–38.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wan SY, Li GS, Tu C, Chen WL, Wang XW,
Wang YN, Peng LB and Tan F: MicroNAR-194-5p hinders the activation
of NLRP3 inflammasomes and alleviates neuroinflammation during
intracerebral hemorrhage by blocking the interaction between TRAF6
and NLRP3. Brain Res. 1752:1472282021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu SW, Zhang YJ, Liu WM, Zhang XF, Wang Y,
Xiang SY, Su JC and Liu ZB: Cigarette smoke extract-induced
inflammatory response via inhibition of the TFEB-mediated autophagy
in NR8383 cells. Exp Lung Res. 49:39–48. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Uwagboe I, Adcock IM, Lo Bello F, Caramori
G and Mumby S: New drugs under development for COPD. Minerva Med.
113:471–496. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cheng Y, Wang D, Wang B, Li H, Xiong J, Xu
S, Chen Q, Tao K, Yang X, Zhu Y and He S: HMGB1 translocation and
release mediate cigarette smoke-induced pulmonary inflammation in
mice through a TLR4/MyD88-dependent signaling pathway. Mol Biol
Cell. 28:201–209. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Conti V, Corbi G, Manzo V, Pelaia G,
Filippelli A and Vatrella A: Sirtuin 1 and aging theory for chronic
obstructive pulmonary disease. Anal Cell Pathol (Amst).
2015:8973272015.PubMed/NCBI
|
|
26
|
Yanagisawa S, Papaioannou AI,
Papaporfyriou A, Baker JR, Vuppusetty C, Loukides S, Barnes PJ and
Ito K: Decreased Serum Sirtuin-1 in COPD. Chest. 152:343–352. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cruz T, López-Giraldo A, Noell G,
Casas-Recasens S, Garcia T, Molins L, Juan M, Fernandez MA, Agustí
A and Faner R: Multi-level immune response network in mild-moderate
chronic obstructive pulmonary disease (COPD). Respir Res.
20:1522019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Deng YS, Fan YP, Zheng YQ, Xi LH, Li WY,
Liang TW, Huang H and Lin J: Traditional Chinese medicine and
active ingredients regulate M1/M2 macrophage polarization balance
to treat chronic obstructive pulmonary disease: A review. Zhongguo
Zhong Yao Za Zhi. 49:4298–4312. 2024.(In Chinese). PubMed/NCBI
|
|
29
|
Sun X, Liu Y, Feng X, Li C, Li S and Zhao
Z: The key role of macrophage depolarization in the treatment of
COPD with ergosterol both in vitro and in vivo. Int
Immunopharmacol. 79:1060862020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Le Y, Cao W, Zhou L, Fan X, Liu Q, Liu F,
Gai X, Chang C, Xiong J, Rao Y, et al: Infection of Mycobacterium
tuberculosis Promotes Both M1/M2 Polarization and MMP production in
cigarette smoke-exposed macrophages. Front Immunol. 11:19022020.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mu Q, Wang Q, Yang Y, Wei G, Wang H, Liao
J, Yang X and Wang F: HMGB1 promotes M1 polarization of macrophages
and induces COPD inflammation. Cell Biol Int. Oct 4–2024.(Epub
ahead of print). doi: 10.1002/cbin.12252. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li L, Sun J, Li Q, Sun K and Jiang J:
Fritillaria cirrhosa D. Don Alleviates Inflammatory Progression and
Suppresses M1 Polarization of Macrophages in Chronic Obstructive
Pulmonary Disease. Int Arch Allergy Immunol. 16:1–9. 2024.
View Article : Google Scholar
|
|
33
|
Cao E, Xu J, Gong Y, Yuan J, Chen A, Liu
J, Fan Y, Fan X and Kuang X: Effect of the Lipoxin Receptor Agonist
BML-111 on cigarette smoke extract-induced macrophage polarization
and inflammation in RAW264.7 Cells. Int J Chron Obstruct Pulmon
Dis. 18:919–932. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Feng H, Yin Y, Zheng R and Kang J:
Rosiglitazone ameliorated airway inflammation induced by cigarette
smoke via inhibiting the M1 macrophage polarization by activating
PPARγ and RXRα. Int Immunopharmacol. 97:1078092021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Iqbal IK, Bajeli S, Sahu S, Bhat SA and
Kumar A: Hydrogen sulfide-induced GAPDH sulfhydration disrupts the
CCAR2-SIRT1 interaction to initiate autophagy. Autophagy.
17:3511–3529. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tang F and Ling C: Curcumin ameliorates
chronic obstructive pulmonary disease by modulating autophagy and
endoplasmic reticulum stress through regulation of SIRT1 in a rat
model. J Int Med Res. 47:4764–4774. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Guan R, Wang J, Cai Z, Li Z, Wang L, Li Y,
Xu J, Li D, Yao H, Liu W, et al: Hydrogen sulfide attenuates
cigarette smoke-induced airway remodeling by upregulating SIRT1
signaling pathway. Redox Biol. 28:1013562020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang S, He N, Xing H, Sun Y, Ding J and
Liu L: Function of hesperidin alleviating inflammation and
oxidative stress responses in COPD mice might be related to
SIRT1/PGC-1α/NF-κB signaling axis. J Recept Signal Transduct Res.
40:388–394. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Deng M, Tong R, Bian Y and Hou G:
Astaxanthin attenuates cigarette smoking-induced oxidative stress
and inflammation in a sirtuin 1-dependent manner. Biomed
Pharmacother. 159:1142302023. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wu J, Hao Z, Wang Y, Yan D, Meng J and Ma
H: Melatonin alleviates BDE-209-induced cognitive impairment and
hippocampal neuroinflammation by modulating microglia polarization
via SIRT1-mediated HMGB1/TLR4/NF-κB pathway. Food Chem Toxicol.
172:1135612023. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jiang R, Xu J, Zhang Y, Zhu X, Liu J and
Tan Y: Ligustrazine Alleviate acute lung injury through suppressing
pyroptosis and apoptosis of alveolar macrophages. Front Pharmacol.
12:6805122021. View Article : Google Scholar : PubMed/NCBI
|