Introduction
Acute lymphoblastic leukemia (ALL) is a
hematological malignancy characterized by an abundance of immature
lymphocytes, composed of 80–85% B cells and 20–25% T cells. This
condition leads to the arrest of differentiation and abnormal
proliferation of lymphocytes (1).
ALL is the most prevalent form of cancer in children, accounting
for ~30% of all childhood malignancies. Among acute leukemias, the
incidence of ALL is five-times higher than that of acute myeloid
leukemia (2). ALL is a significant
global health concern, with an average incidence rate ranging from
0.4 to 2 per 100,000 individuals. The disease primarily affects
children <15 years old, with the highest rates observed in
high-income countries due to advanced diagnostic capabilities. Male
individuals are more frequently affected than female individuals,
with an incidence rate of 2.66 per 100,000 compared with 1.92 for
female individuals. While high-income regions have successfully
reduced mortality rates through improved treatment protocols,
low-income countries continue to face higher death rates due to
limited healthcare resources and late diagnoses. Addressing this
disparity remains critical in the global fight against ALL
(3). Notably, ALL is the most
common type of cancer that occurs in childhood, with a peak
occurrence between the ages of 2 and 5 years, as well as after the
age of 50 years, with ~60% of cases occurring in individuals <20
years old (4). However, there is
no significant sex difference in ALL incidence (5,6).
Although its 5-year survival rate is ~90%, 20% of children with ALL
experience relapse with a poor prognosis (7); in adults, the relapse rate is higher,
reaching 40–50% (8,9). Therefore, continuous efforts are
required to improve ALL treatment. Furthermore, children with ALL
exhibit poorer social, physical and emotional health compared with
their age-matched peers and siblings (10); these children may experience
depression, anxiety and attention problems (11,12).
Current ALL treatments primarily involve combination chemotherapy
using high-dose methotrexate (MTX), mercaptopurine (6-MP) and other
drugs, followed by the regular oral or injectable administration of
anticancer drugs, such as dexamethasone (DEX), vincristine,
cytarabine, Endoxan, 6-MP and MTX (13–16).
Recurrence can occur due to the presence of residual cancer cells
that are difficult to detect in the body after treatment. When
these residual cancer cells proliferate, the disease re-emerges;
thus, there remains an unmet medical need for treatment. In
addition, unresolved medical issues, which include the high risk of
relapse after the first remission and refractory disease after
relapse (17), necessitate further
investigation.
Previous studies have detected the sustained
activation of the JAK2/STAT3 signaling pathway in various types of
human cancer, including blood cancer, and its association with poor
prognosis (18,19). The small molecule compound Stattic
is a potent STAT3 inhibitor, which selectively blocks the SH2
domain, regardless of its phosphorylation status (20,21).
This signal transduction is selectively inhibited by Stattic,
together with the activation, dimerization and nuclear
translocation of STAT3. The consequence of this inhibition is a
greater apoptotic rate of STAT3-dependent cancer cells (22). Based on previous studies, the
present study aimed to determine whether Stattic can achieve
anticancer effects by regulating the role of STAT3 in ALL. The aim
was to reveal the cellular and molecular mechanisms underlying the
anticancer effects of Stattic. Understanding the related role and
mechanisms of Stattic in ALL may have extensive clinical and
immunological significance, and the results of the present study
may aid in the development of effective treatments for patients
with ALL and provide novel insights into the basic treatment of
ALL.
Materials and methods
Cell culture
The human T-cell ALL (T-ALL) cell lines CCRF-CEM and
Jurkat (American Type Culture Collection) were maintained in RPMI
1640 medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented
with 10% fetal bovine serum (HyClone; Cytiva), 1 mM sodium pyruvate
(HyClone; Cytiva), 100 U/ml penicillin, and 100 µg/ml streptomycin
(Gibco; Thermo Fisher Scientific, Inc.) in a humidified atmosphere
containing 5% CO2 at 37°C.
Reagents
Stattic (cat. no. HY-13818; MedChemExpress), a
selective inhibitor of STAT3, and dimethyl sulfoxide (DMSO), which
was used as a vehicle control, were procured from Sigma-Aldrich;
Merck KGaA. For cell treatments, Stattic was dissolved in DMSO for
stock preparation. In the in vitro experiments, the vehicle
control group received a final DMSO concentration of 0.05%, while
the 5, 2.5 and 1.25 µM Stattic groups received DMSO concentrations
of 0.05, 0.025 and 0.0125%, respectively.
Cell viability assay
The cytotoxic effects of Stattic on CCRF-CEM and
Jurkat cells were assessed using the Cell Counting Kit-8 (CCK-8;
cat. no. 96992; Sigma-Aldrich; Merck KGaA) according to the
manufacturer's instructions. Briefly, a total of 1×105
cells/well were seeded in 200 µl culture medium in a 96-well cell
culture plate, and were then treated at 37°C for 24 h with various
concentrations of Stattic (0.625, 1.25, 2.5, 5 and 10 µM) or an
equivalent volume of the vehicle control. Subsequently, CCK-8
solution was added, and plates were incubated for another 2–3 h.
Absorbance at 450 nm was then measured using a microplate reader
(Enspire 2300–0000; PerkinElmer, Inc.).
Western blot analysis
For western blot analysis, cells were treated prior
to collection. In the dose-dependent experiment, cells were treated
with DMSO or different concentrations of Stattic (1.25, 2.5 and 5
µM) for 24 h. In the time-course experiment, cells were treated
with DMSO or 5 µM Stattic for 8, 16 and 24 h. After treatment, the
cells were first washed with PBS prior to collection, and were then
lysed in ice-cold Tris buffer (50 mM, pH 7.5) containing the
following: 5 mM EDTA, 300 mM NaCl, 0.1% Igepal, 0.5 mM NaF, 0.5 mM
Na3VO4, 0.5 mM PMSF and antiprotease mixture
(Roche Molecular Diagnostics), and centrifuged at 13,000 × g for 10
min at 4°C. Protein concentrations were determined according to the
Bradford procedure. Equal amounts of protein (25 µg) were then
separated by SDS-PAGE on 10 and 12% gels, and were transferred to
PVDF membranes. After blocking with 5% non-fat milk dissolved in
PBS at room temperature for 1 h, the membranes were incubated with
primary antibodies against p-STAT3 (1:1,000; cat. no. 9145S; Cell
Signaling Technology, Inc.), STAT3 (1:1,000; cat. no. 30835S; Cell
Signaling Technology, Inc.), pro-caspase-3 (1:1,000; cat. no.
9662S; Cell Signaling Technology, Inc.), cleaved caspase-3
(1:1,000; cat. no. 9662S; Cell Signaling Technology, Inc.), LC3B
(1:1,000; cat. no. 83506S; Cell Signaling Technology, Inc.), p62
(1:1,000; cat. no. 5114; Cell Signaling Technology, Inc.), Bcl-2
(1:1,000; cat. no. 26593-1-AP; Proteintech Group, Inc.), PARP-1
(1:1,000; cat. no. sc-74470; Santa Cruz Biotechnology, Inc.), ATG5
(1:1,000; cat. no. sc-133158; Santa Cruz Biotechnology, Inc.),
BECN1 (1:500; cat. no. sc-11427; Santa Cruz Biotechnology, Inc.)
and β-actin (1:5,000; cat. no. 3700S; Cell Signaling Technology,
Inc.), followed by incubation with an antimouse IgG, HRP-linked
secondary antibody (1:7,000; cat. no. 7076P2; Cell Signaling
Technology, Inc.) or an anti-rabbit IgG, HRP-linked secondary
antibody (1:7,000; cat. no. 7074P2; Cell Signaling Technology,
Inc.). The ATG5 antibody used in the present study can detect the
ATG5-ATG12 conjugate with a molecular weight of ~50 kDa. Bands were
visualized using an Enhanced Chemiluminescence Detection Kit
(MilliporeSigma) and the Alliance Q9 (Uvitec Ltd.). The protein
expression levels were normalized to β-actin. The band intensity
was measured using ImageJ software (version 1.50i; National
Institutes of Health).
Flow cytometric analysis
Apoptosis was quantified based on Annexin V-FITC and
propidium iodide (PI) double staining. Briefly, 4×105
CCRF-CEM cells and 3×105 Jurkat cells were treated with
5 µM Stattic or DMSO in a 24-well plate for 24 h. After treatment,
the cells were collected and stained using the Annexin V-FITC/PI
apoptosis kit according to the manufacturer's protocol (Elabscience
Bionovation Inc.). Stained cells were analyzed by flow cytometry
within 1 h of staining. Fluorescence intensities were measured
using a flow cytometer (FACSCanto II; BD Biosciences) with BD
FACSDiva software (version 8.0.1; BD Biosciences). Each experiment
was performed in duplicate, at least three times independently.
Xenograft T-ALL mouse model
Female NOD/SCID mice (age, 9 weeks; weight, 18–22 g)
were procured from the National Laboratory Animal Center (Taipei,
Taiwan). The mice were maintained in an individually ventilated
caging system, adhering to a 12-h light/dark cycle, at a
temperature of 22 ± 2°C and a humidity of 50–70%, with ad
libitum access to food and water. All experimental protocols
were approved by the Animal Care and Use Committee of the Taichung
Veterans General Hospital (IACUC no. La-1132052; Taichung, Taiwan).
To create tumors, 1×106 CCRF-CEM cells suspended in a
1:1 mixture of BD Matrigel™ (cat. no. 356231; Corning, Inc.) and
RPMI-1640 medium were subcutaneously injected into the flanks of
each mouse. On day 1, T-ALL cells were subcutaneously injected into
mice under gas anesthesia using isoflurane (induction, 4–5%
isoflurane for 3 min; maintenance, 1–3% isoflurane for an
additional 1 min). After the procedure, the mice were placed in a
warm, dry environment and were continuously monitored for vital
signs during recovery. The mice were returned to their original
housing only after fully recovering from anesthesia. Post-surgery,
the mice were examined at least once daily, monitoring the
injection site for wound condition, including any signs of
secretion, as well as assessing body weight, eating, urination and
defecation. Analgesics were not used, as they could impact the
experimental results; instead, physical pain management strategies,
such as environmental enrichment items (e.g., toys), were provided.
Mice received wooden sticks, paper houses and similar enrichment
items. Mice were subsequently randomized into the following five
groups (n=6 mice/group): Vehicle control group and four treatment
groups, each receiving three different doses of Stattic (7.5, 15
and 30 mg/kg) or DEX (1 mg/kg; Taiwan Biotech Co., Ltd.). Starting
from day 6 after tumor cell injection, Stattic and DEX were
administered by intraperitoneal injection three times a week; the
volume of intraperitoneal injection per mouse was 300 µl. Stattic
was dissolved in DMSO for stock preparation. The final
concentrations of DMSO within the administered Stattic doses (7.5,
15, and 30 mg/kg) were 0.75, 1.5 and 3%, respectively. For the
vehicle control group, the injection consisted of normal saline
containing 3% DMSO. Tumor sizes were measured using a digital
caliper thrice weekly until the day of sacrifice and tumor volume
was calculated using the following formula: Tumor volume=length ×
width2. All mice were euthanized on day 23 for
subsequent analyses. Mice were placed into a euthanasia chamber,
which was gradually filled with CO2 at a flow rate of
30% volume/min. After the gas infusion, the mice were observed for
3 min to ensure proper euthanasia; the signs confirming death
included the cessation of heartbeat, lack of respiratory activity
and pupil dilation.
Statistical analysis
Data are presented as the mean ± SEM of at least
three independent experiments. To compare differences between the
treatment and control groups, statistical significance was assessed
using one-way ANOVA followed by Tukey's multiple comparisons test.
Data were analyzed using GraphPad Prism software (version 6;
Dotmatics). P<0.05 was considered to indicate a statistically
significant difference.
Results
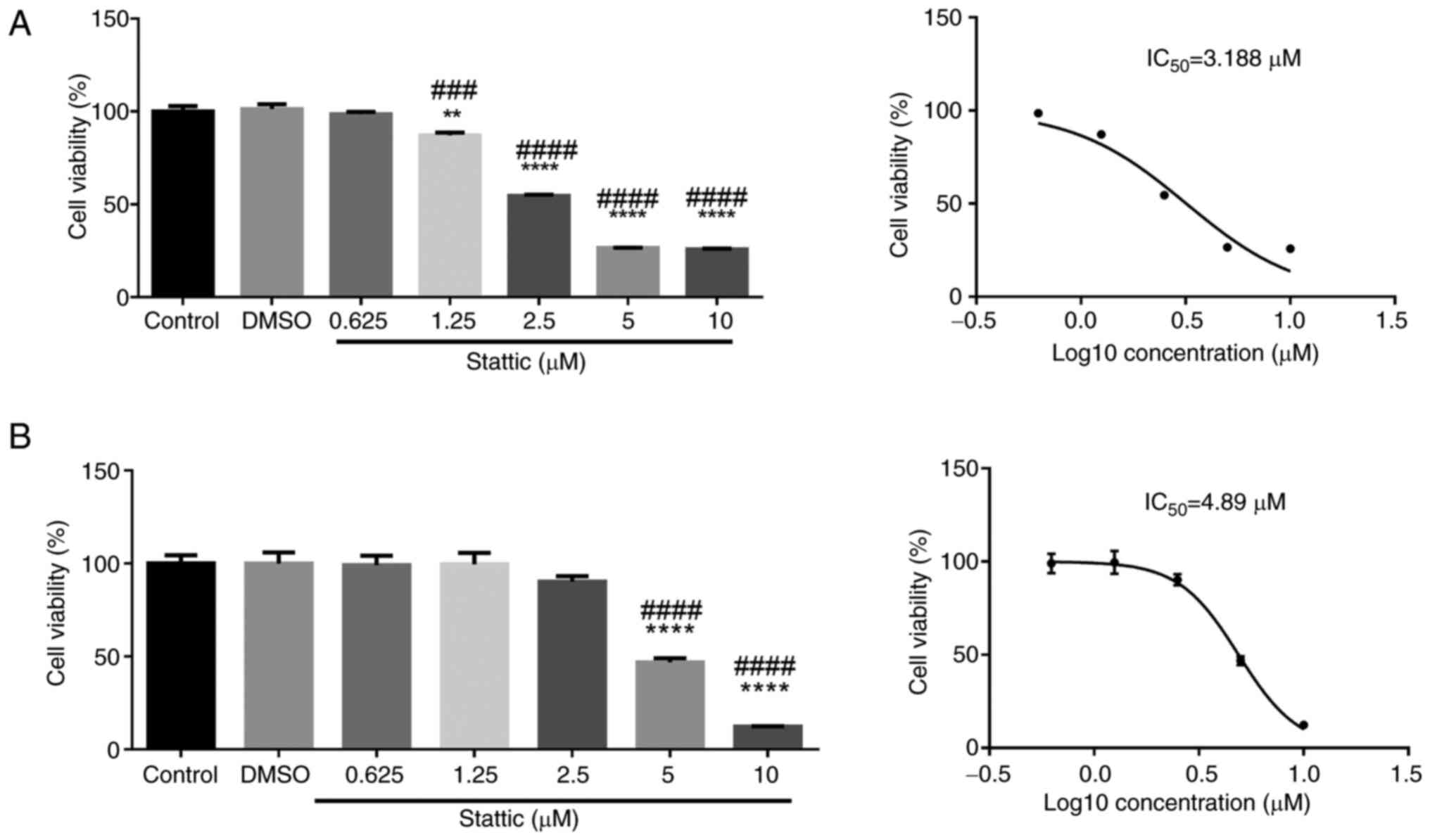
Stattic inhibits the viability of
CCRF-CEM and Jurkat cells in a dose-dependent manner
To assess the cytotoxic effects of Stattic on T-ALL
cells, CCRF-CEM (Fig. 1A) and
Jurkat cells (Fig. 1B) were
treated with increasing concentrations of Stattic (0.625, 1.25,
2.5, 5 and 10 µM) or vehicle control (DMSO) for 24 h. Cell
viability was measured using the CCK-8 assay. In CCRF-CEM cells,
Stattic treatment resulted in a significant, dose-dependent
reduction in cell viability. A statistically significant reduction
was observed at 1.25 µM, and cell viability was further decreased
at 2.5 µM and higher concentrations. The half maximal inhibitory
concentration (IC50) value for CCRF-CEM cells was
determined to be 3.188 µM, indicating that these cells were
sensitive to Stattic-induced inhibition of viability (Fig. 1A). In Jurkat cells, a similar
dose-dependent reduction in viability was observed; however, the
inhibitory effect was less pronounced compared with in CCRF-CEM
cells. Significant reductions in viability were detected at 5 and
10 µM concentrations. The IC50 value for Jurkat cells
was 4.89 µM, suggesting that Jurkat cells are slightly more
resistant to Stattic than CCRF-CEM cells (Fig. 1B). These results indicated that
Stattic may effectively reduce the viability of CCRF-CEM and Jurkat
cells in a dose-dependent manner, with CCRF-CEM cells showing
greater sensitivity. The observed differential sensitivity between
the two cell lines highlights the potential for Stattic as a
targeted therapeutic agent in T-ALL.
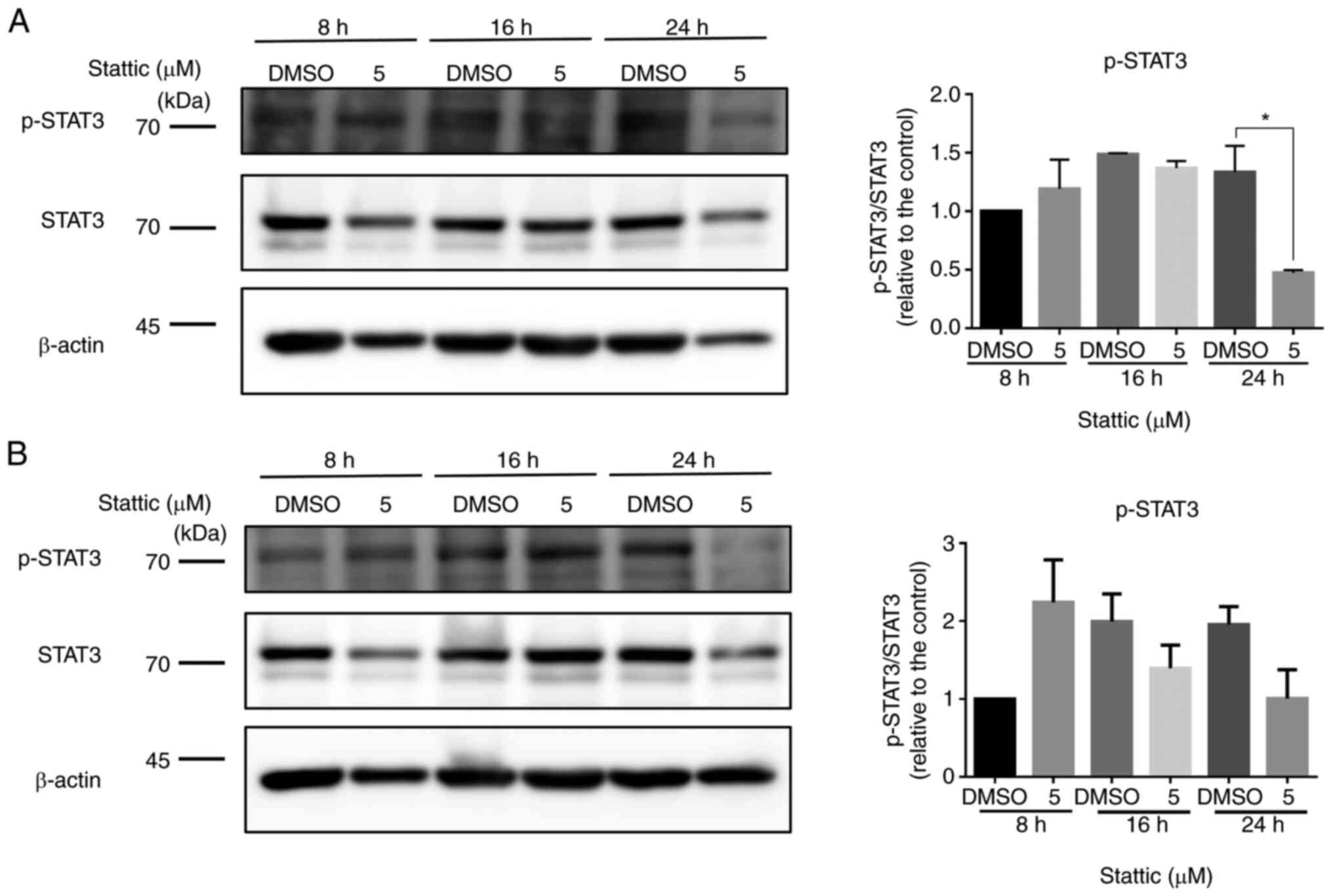
Stattic suppresses p-STAT3 levels in
CCRF-CEM and Jurkat cells
To investigate the effect of Stattic on STAT3
signaling, the expression levels of p-STAT3 were we examined in
CCRF-CEM (Fig. 2A) and Jurkat
cells (Fig. 2B) at 8, 16 and 24 h
following treatment with 5 µM Stattic or DMSO. Total STAT3 and
β-actin were used as loading controls. In CCRF-CEM cells, Stattic
treatment resulted in a reduction in p-STAT3 levels over time.
Notably, a significant decrease in p-STAT3 was observed at 24 h,
indicating that Stattic effectively suppressed STAT3
phosphorylation with prolonged exposure. However, total STAT3
levels remained stable across all time points, suggesting that
Stattic may specifically inhibit STAT3 activation without affecting
its overall expression (Fig. 2A).
In Jurkat cells, although p-STAT3 levels fluctuated, no
statistically significant differences were observed at any of the
time points compared with the DMSO-treated controls. Total STAT3
expression remained constant, similar to in CCRF-CEM cells
(Fig. 2B). These findings
indicated that Stattic may inhibit STAT3 activation more
effectively in CCRF-CEM cells than in Jurkat cells, reflecting a
differential response between the two T-ALL cell lines. The
suppression of STAT3 phosphorylation suggested that Stattic may
exert its anti-proliferative effects, at least in part, through the
inhibition of STAT3-mediated signaling in CCRF-CEM cells.
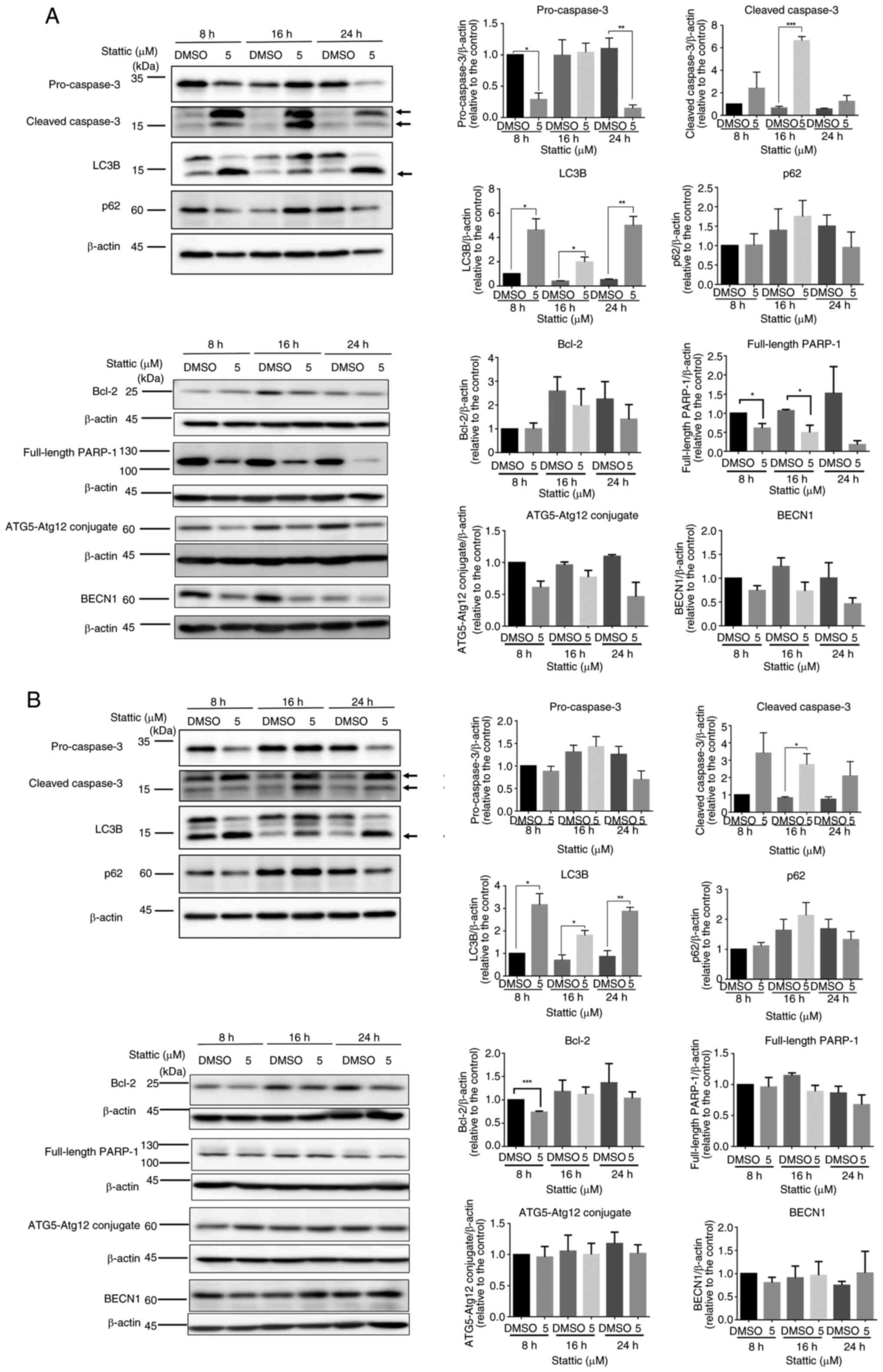
Stattic induces apoptosis and
autophagy-related changes in T-ALL cells
Western blot analysis was conducted to investigate
the time-dependent effects of Stattic (5 µM) on apoptosis and
autophagy markers in two T-ALL cell lines: CCRF-CEM (Fig. 3A) and Jurkat (Fig. 3B). Both cell types were treated for
8, 16 and 24 h with Stattic or with DMSO as a control. In CCRF-CEM
cells, an inhibition of pro-caspase-3 expression was observed at 8
and 24 h after Stattic treatment. Furthermore, a significant
increase was detected in cleaved caspase-3 expression 16 h after
Stattic treatment, indicating the activated apoptotic cascade.
Stattic also induced a time-dependent upregulation of LC3B
expression at 8, 16 and 24 h, suggesting enhanced autophagic
activity. Meanwhile, p62 protein levels showed a decreasing trend
at 24 h, although this change was not statistically significant,
further supporting autophagy activation. Bcl-2 expression remained
relatively stable, and full-length PARP-1 displayed a significant
reduction at 8 and 16 h, reflecting apoptotic progression. Markers
related to autophagy initiation, such as ATG5-ATG12 conjugate and
BECN1, did not exhibit substantial changes during the observation
period (Fig. 3A). In Jurkat cells,
the apoptotic response to Stattic was less pronounced. While
pro-caspase-3 levels remained relatively stable, cleaved caspase-3
exhibited a slight increase at 16 h. LC3B levels also demonstrated
a significant increase at 8,16 and 24 h. Similarly, p62 levels
showed a decreasing trend at 24 h, indicating autophagy activation,
though less prominent compared with in CCRF-CEM cells. Bcl-2
expression was significantly reduced at 8 h but remained unchanged
thereafter. Full-length PARP-1 and autophagy-related proteins,
including ATG5-ATG12 conjugate and BECN1, showed no significant
changes across all time points (Fig.
3B). Together, these results indicated that Stattic could
induce both apoptotic and autophagic processes in CCRF-CEM and
Jurkat cells, with more robust effects observed in CCRF-CEM cells.
The differential responses between the two cell lines highlight the
potential variability in the sensitivity of T-ALL subtypes to
Stattic treatment.
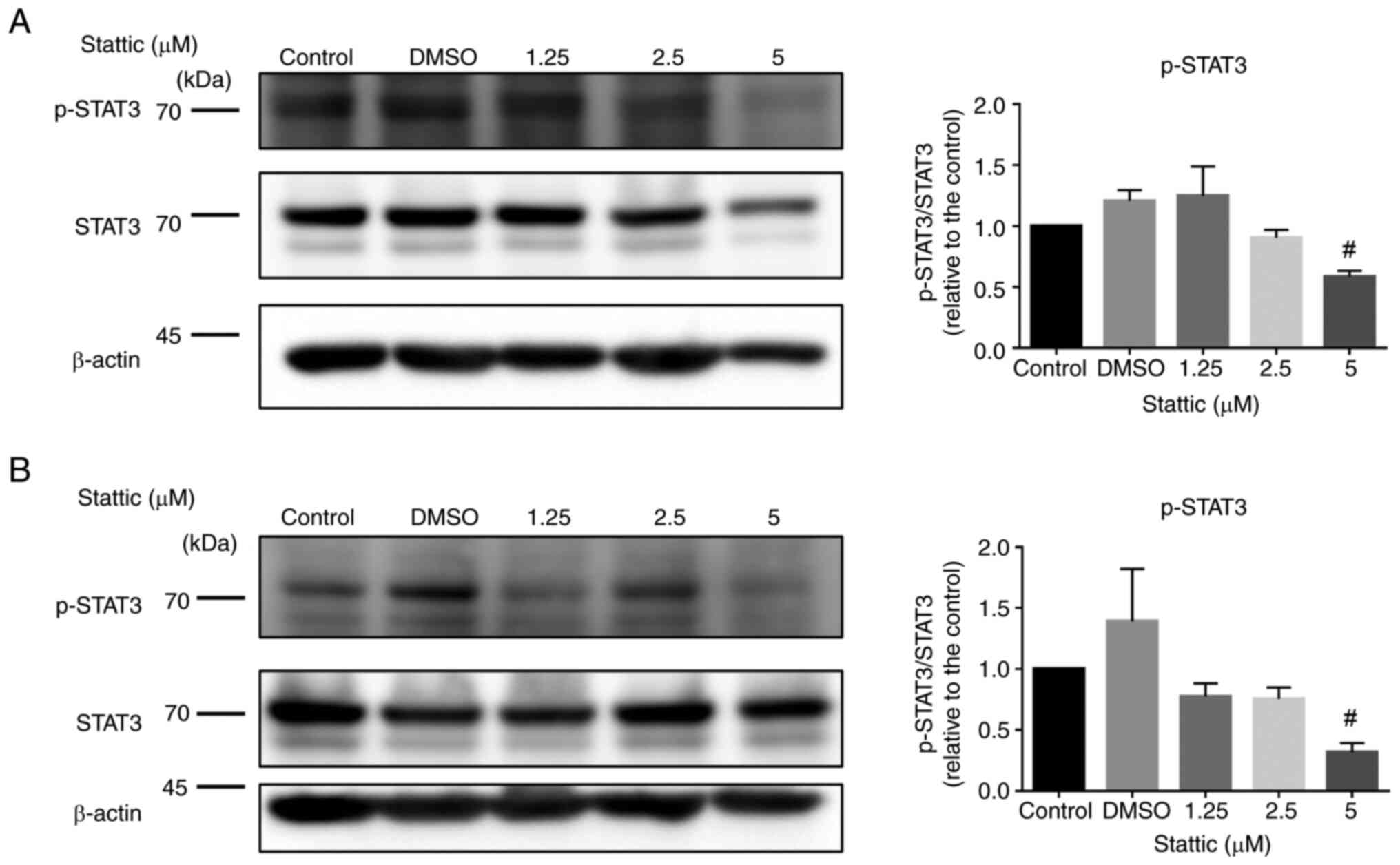
Stattic dose-dependently inhibits
p-STAT3 expression in CCRF-CEM and Jurkat cells
To further explore the impact of Stattic on STAT3
signaling, CCRF-CEM (Fig. 4A) and
Jurkat cells (Fig. 4B) were
treated with increasing concentrations of Stattic (1.25, 2.5 and 5
µM) or vehicle control (DMSO) for 24 h. The expression levels of
p-STAT3 and total STAT3 were measured by western blotting, with
β-actin serving as the loading control. In CCRF-CEM cells, Stattic
reduced p-STAT3 levels in a dose-dependent manner. A marked
reduction in p-STAT3 was observed at the 5 µM concentration, with
statistical significance indicated. However, total STAT3 protein
expression remained unchanged across all treatment groups,
suggesting that Stattic specifically inhibited STAT3
phosphorylation without affecting total STAT3 levels (Fig. 4A). Similarly, in Jurkat cells,
p-STAT3 levels were reduced with increasing Stattic concentrations.
A significant decrease was evident in response to the 5 µM
concentration, while total STAT3 levels showed no major changes,
indicating selective inhibition of phosphorylation by Stattic
(Fig. 4B). These results indicated
that Stattic may effectively inhibit STAT3 activation in a
dose-dependent manner in both CCRF-CEM and Jurkat cells. The
suppression of p-STAT3 without affecting total STAT3 levels further
supports the role of Stattic as a selective inhibitor of STAT3
signaling, which could underlie its therapeutic potential in
T-ALL.
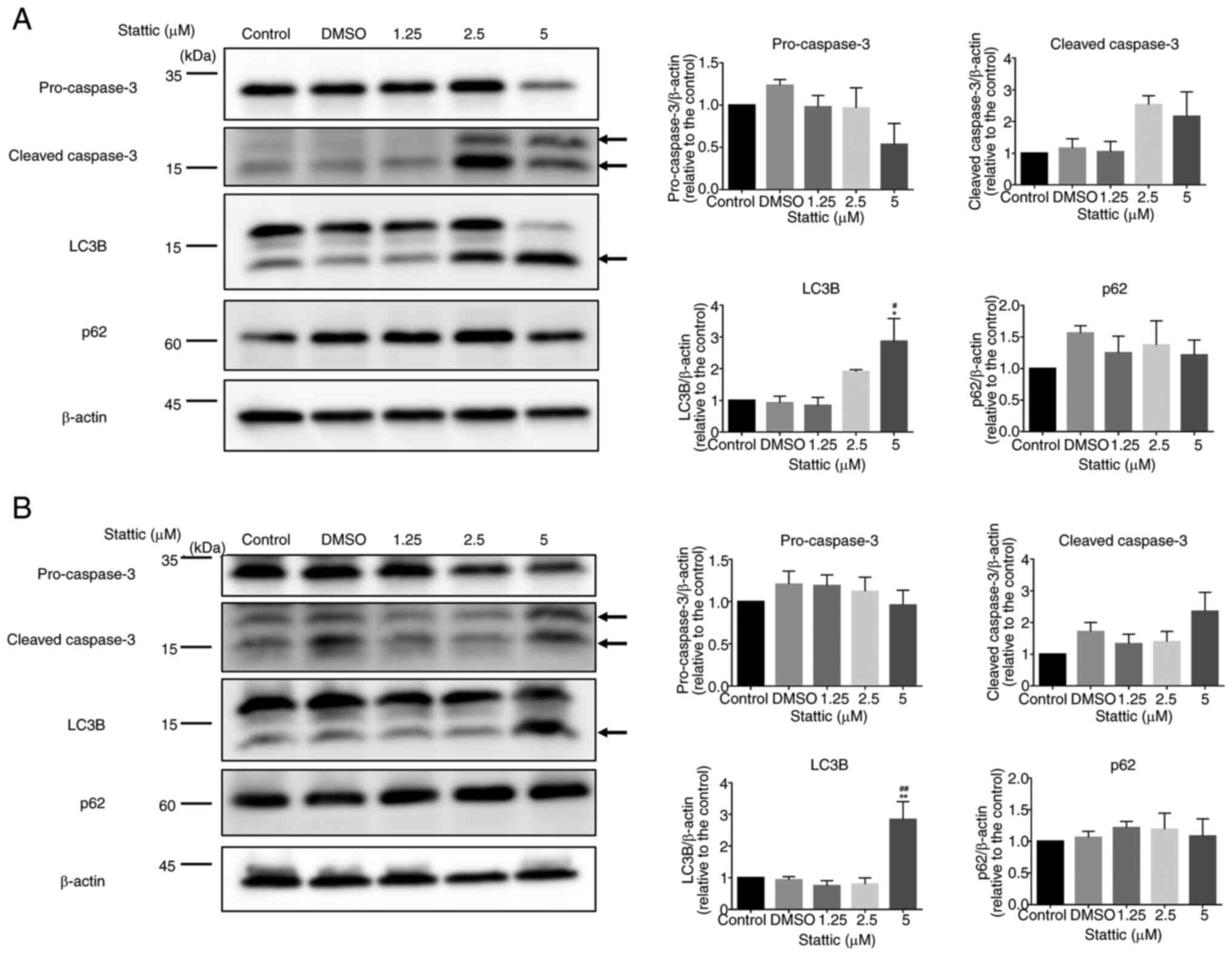
Stattic modulates apoptosis and
autophagy markers in CCRF-CEM and Jurkat cells in a dose-dependent
manner
To investigate the effects of Stattic on apoptosis
and autophagy, CCRF-CEM (Fig. 5A)
and Jurkat cells (Fig. 5B) were
treated with increasing concentrations of Stattic (1.25, 2.5 and 5
µM) or vehicle control (DMSO) for 24 h. Protein expression levels
of apoptotic markers (pro-caspase-3, cleaved caspase-3) and
autophagy markers (LC3B, p62) were assessed by western blotting,
with β-actin used as a loading control. In CCRF-CEM cells, cleaved
caspase-3 levels showed an increasing trend with higher
concentrations of Stattic, indicating enhanced apoptotic activity,
although the changes were not statistically significant. LC3B
levels were significantly increased at 5 µM concentrations,
suggesting induction of autophagy. By contrast, the expression
levels of p62, a marker of autophagic flux, remained relatively
unchanged across all treatment groups, indicating incomplete
autophagic flux. Pro-caspase-3 levels remained stable, further
supporting that apoptosis was primarily indicated by the cleaved
form (Fig. 5A). In Jurkat cells, a
similar trend was observed. Cleaved caspase-3 levels increased
slightly with higher Stattic concentrations, but the changes were
not statistically significant. LC3B expression was significantly
increased in response to 5 µM Stattic, indicating the activation of
autophagic processes. However, as in CCRF-CEM cells, p62 levels did
not show a significant reduction, suggesting a potential blockade
in autophagic flux. Pro-caspase-3 levels also remained constant
across the different Stattic concentrations (Fig. 5B). These findings demonstrated that
Stattic significantly increased autophagy markers in CCRF-CEM and
Jurkat cells, while also showing a trend toward increased apoptosis
in both cell lines. The differential expression patterns of LC3B
and p62 suggested that Stattic may trigger autophagy but not
complete autophagic degradation. The increased levels of cleaved
caspase-3 suggest a potential role of Stattic in promoting
apoptosis in these T-ALL cell lines, although the changes were not
statistically significant.
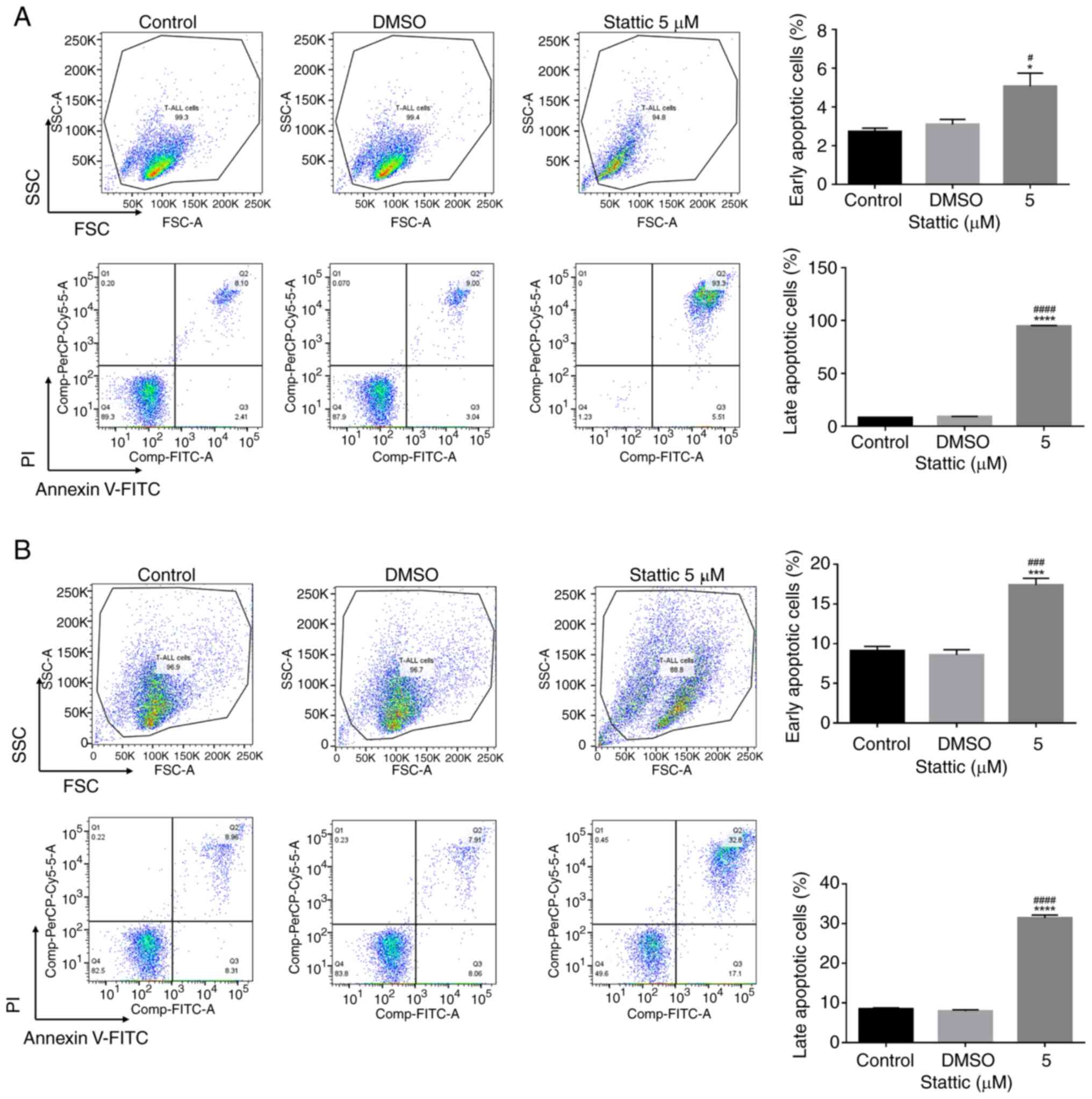
Stattic induces early and late
apoptosis in CCRF-CEM and Jurkat cells
To further confirm the pro-apoptotic effects of
Stattic, flow cytometry was performed to analyze apoptosis in
CCRF-CEM (Fig. 6A) and Jurkat
cells (Fig. 6B) after treatment
with 5 µM Stattic for 24 h. Cells were stained with Annexin V-FITC
and PI to differentiate between early and late apoptotic cells,
which were detected in quadrants 3 and 2, respectively. The forward
scatter (FSC)/side scatter (SSC) plots in Fig. 6A and B show the FSC and SSC
characteristics of the cells, which provide information about cell
size and granularity, respectively. From the results of the 5 µM
Stattic treatment, the FSC/SSC plots in CCRF-CEM (Fig. 6A) and Jurkat (Fig. 6B) cells showed an increase in the
proportion of cells with reduced size, indicating cell shrinkage
typically associated with apoptosis. These results were obtained
after Annexin V/PI staining, highlighting the effects of Stattic on
inducing apoptotic changes in both cell lines. In CCRF-CEM cells,
Stattic treatment significantly increased both early and late
apoptotic populations compared with the control and DMSO groups.
The percentage of early apoptotic cells increased significantly
upon treatment with 5 µM Stattic. Moreover, late apoptotic cells
showed a significant increase in the Stattic-treated group,
indicating that Stattic strongly induced the apoptosis of CCRF-CEM
cells (Fig. 6A). Similarly, in
Jurkat cells, Stattic treatment led to a significant increase in
apoptosis. Early apoptotic cells were significantly elevated, and
late apoptotic cells increased substantially following treatment
with 5 µM Stattic compared with in the control and DMSO-treated
groups (Fig. 6B). These results
indicated that Stattic effectively promoted both early and late
apoptosis in CCRF-CEM and Jurkat cells, with a particularly strong
effect on late apoptosis. This supports the role of Stattic as a
potent inducer of apoptosis in T-ALL cells, highlighting its
therapeutic potential for T-ALL treatment.
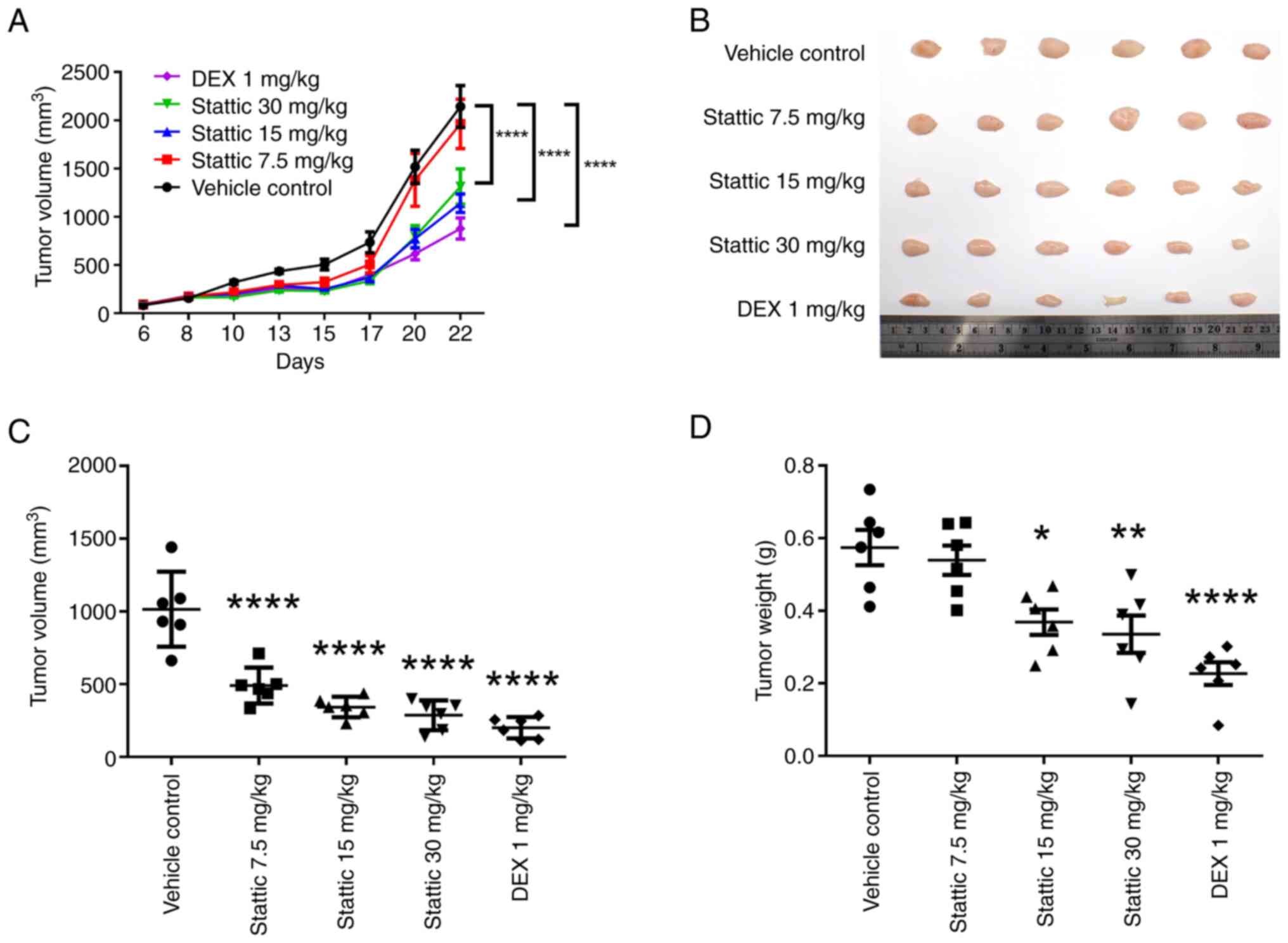
Stattic inhibits tumor growth in the
xenograft model of T-ALL
Using a xenograft mouse model of T-ALL,
CCRF-CEM-xenografted mice were intraperitoneally injected with
Stattic three times a week. The results revealed that while the
control group showed a progressive growth in tumor volume over a
22-day period, the Stattic groups (15 and 30 mg/kg) and the DEX
group (1 mg/kg; positive control), showed a significant reduction
in tumor growth; the antitumor effect of Stattic was
dose-dependent, with a peak effect observed at 30 mg/kg (Fig. 7A). Excised tumor volumes measured
at the end of treatment (day 23) confirmed such a dose-dependent
reduction in tumor size (Fig. 7B),
and the significant decrease in volume relative to the control
group in response to all doses of Stattic (Fig. 7C). In addition, a significant
reduction in tumor weight was detected in the 15 and 30 mg/kg
Stattic, and 1 mg/kg DEX treatment groups relative to the control
(Fig. 7D). These results suggested
that Stattic, along with DEX, effectively reduced tumor burden in a
dose-dependent manner, with 30 mg/kg Stattic being the most
effective dose.
Discussion
The present study on the effects of Stattic on T-ALL
provided compelling evidence for its therapeutic potential.
Notably, Stattic exhibited a dose-dependent inhibitory effect on
the viability of T-ALL cells, affirming its capacity to suppress
the survival of T-ALL cells. The findings indicated that Stattic
not only inhibited cell viability and p-STAT3 expression in a
dose-dependent manner, but also induced cell death through
apoptosis and autophagy. In addition, Stattic suppressed tumor
growth in a xenograft model of T-ALL, suggesting its potential as a
therapeutic agent for this malignancy.
The observed dose-dependent reduction in the
viability of CCRF-CEM and Jurkat cells underscores the potent
cytotoxic effects of Stattic against T-ALL cells. The findings
suggested that Stattic could be effective in curtailing T-ALL
progression by inhibiting cell proliferation and promoting cell
death. Moreover, the significant suppression of p-STAT3 expression
after Stattic treatment confirmed its action as a STAT3 inhibitor,
affirming its therapeutic potential in targeting abnormal STAT3
signaling pathways in T-ALL (23).
The results of the present study demonstrated that Stattic
treatment may lead to a reduction in p-STAT3 levels in both
CCRF-CEM cells and Jurkat cells, although the magnitude and timing
of inhibition differed between the two cell lines. Specifically,
CCRF-CEM cells, which have higher basal p-STAT3 levels, exhibited
significant inhibition only after 24 h, whereas Jurkat cells, with
inherently lower p-STAT3 expression, displayed a similar trend but
with less pronounced changes. These data suggested that the effect
of Stattic on p-STAT3 is influenced by the initial expression level
of p-STAT3 in the T-ALL cell line being studied. The data from both
CCRF-CEM and Jurkat cells strengthen the hypothesis regarding the
ability of Stattic to modulate p-STAT3-dependent pathways and
provide a solid foundation for future investigations involving
additional cell lines.
The present results showed a significant reduction
in p-STAT3 levels only after 24 h of Stattic treatment, whereas
shorter treatments (8 and 16 h) did not yield statistically
significant changes. This observation is distinct from findings in
some previous studies (24,25),
which reported more rapid inhibition of STAT3 phosphorylation.
Several factors could explain this discrepancy. First, the cell
type-specific response might serve a role, as the present
experiments were conducted in CCRF-CEM and Jurkat cells, which are
T-ALL cell lines. These cells may exhibit a more delayed response
to Stattic due to differences in the activation state of STAT3 or
varying levels of basal p-STAT3 expression compared with other cell
lines used in prior studies, such as solid tumor cells or other
hematological malignancies. Additionally, the stability of p-STAT3
and the rate of dephosphorylation may vary among different cell
lines. In some systems, STAT3 is rapidly turned over, while in
others, the phosphorylation status may be sustained for longer
periods. The delayed inhibition observed in the current study
suggested that Stattic might require sustained exposure to
accumulate sufficiently in the cells, or that a certain threshold
concentration must be reached to effectively inhibit upstream
kinases or disrupt STAT3 dimerization. Furthermore, experimental
conditions, such as cell density, medium composition and Stattic
concentration could influence the kinetics of p-STAT3 inhibition.
The present study used 5 µM Stattic, and it is possible that lower
concentrations or shorter time points in previous studies led to
different kinetic profiles. The delayed inhibition of p-STAT3 in
the current study could reflect the need for extended Stattic
exposure to overcome cellular compensatory mechanisms or gradual
inhibition of signaling pathways upstream of STAT3. This might
suggest that T-ALL cells are more resistant to immediate STAT3
inactivation but become vulnerable with prolonged Stattic exposure,
which could be therapeutically relevant. In summary, the longer
treatment duration required for significant STAT3 inhibition in the
present experiments highlights the context-dependent nature of
STAT3 signaling and suggests that prolonged Stattic exposure might
be necessary to achieve optimal therapeutic effects in T-ALL
models.
The present study also demonstrated the complex
interaction between apoptosis and autophagy induced by Stattic in
CCRF-CEM and Jurkat cells. Increased expression levels of both
cleaved caspase-3 and LC3B markers of apoptotic and autophagic cell
death suggested a dual mechanism regarding the promotion of cell
death by Stattic. The dose-dependent nature of these responses
further highlighted the ability of Stattic to effectively modulate
these key cell death pathways (26).
The translational significance of the present
results is supported by the in vivo efficacy of Stattic in
reducing tumor growth in a xenograft mouse model of T-ALL. Stattic
led to a dose-dependent decrease in tumor growth, with the highest
dose generating the greatest antitumor effect. Targeting STAT3 is
known to inhibit tumor growth in various types of cancer, such as
colorectal cancer (27), breast
cancer (28) and glioma (29). These findings corroborate the
present in vitro data, and highlight the potential of
Stattic as a targeted therapeutic for T-ALL. These in vivo
results further indicated the translational potential of Stattic,
predicting its move into clinical trial phase. Such an approach
provides a new option for T-ALL therapy that targets STAT3
signaling, a critical pathway in the pathogenesis of numerous
malignancies.
The primary limitations of the present study include
the reliance on the CCRF-CEM and Jurkat cell lines, and a xenograft
mouse model, which might not fully capture the biological
complexity and heterogeneity of human T-ALL. The short-term nature
of these experiments cannot reflect long-term outcomes or potential
resistance mechanisms to Stattic treatment. Further research is
needed to fully understand the therapeutic potential and
limitations of Stattic in treating T-ALL. Extending studies to
include diverse T-ALL subtypes, long-term treatment effects and
comprehensive safety profiles would provide more robust data to
support clinical applications, and strengthen the preliminary
findings of the current study. As part of our future studies, we
plan to use CRISPR-Cas9 or RNA interference approaches to knock out
or knock down STAT3 expression in CCRF-CEM cells and compare the
resulting effects with those of Stattic treatment. This additional
work will provide more direct evidence of STAT3 dependency.
In conclusion, the results of the present study
support the potential of Stattic as a therapeutic agent against
T-ALL by inhibiting STAT3 signaling, and inducing programmed cell
death through apoptosis and autophagy. The present findings may
pave the way for further clinical investigations into Stattic and
emphasize the importance of targeting dysregulated STAT3 signaling
in leukemia therapy.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the Tri-Service General
Hospital Penghu Branch, Taiwan (grant no. TSGH-PH_D_112002) and
Taichung Veterans General Hospital, Taiwan (grant nos.
TCVGH-1136505C, TCVGH-HK1138001 and TCVGH-YM1130108). This project
was also supported by grants from the National Science and
Technology Council, Taiwan (grant nos. NSTC 112-2914-I-075A-002-A1
and NSTC 111-2314-B-075A-009).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
CLL, HYC and FLH conceptualized the study. CLL, HYC,
JCY and SJY designed the methodology. CLL, TYC and SWY performed
the formal analysis. CLL, HYC, JCY and SJY conducted the
investigation. CLL prepared the original draft, while CLL, HYC and
FLH reviewed and edited the manuscript. CLL, HYC and FLH managed
the project. HYC, JCY, HYC and FLH secured funding. CLL, SJY and
FLH confirm the authenticity of all the raw data. All authors have
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
The experimental protocols were approved by the
Animal Care and Use Committee of the Taichung Veterans General
Hospital (IACUC no. La-1132052).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Inaba H, Greaves M and Mullighan CG: Acute
lymphoblastic leukaemia. Lancet. 381:1943–1955. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Puumala SE, Ross JA, Aplenc R and Spector
LG: Epidemiology of childhood acute myeloid leukemia. Pediatric
Blood Cancer. 60:728–733. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang N, Wen D, Wang T and Deng J:
Disparities in incidence and mortality of pediatric acute
lymphoblastic leukemia across countries with different incomes.
Leukemia. October 4–2024.(Epub ahead of print). View Article : Google Scholar
|
|
4
|
Lupo PJ and Spector LG: Cancer progress
and priorities: Childhood cancer. Cancer epidemiology, biomarkers
& prevention: A publication of the American Association for
Cancer Research, cosponsored by the American Society of Preventive
Oncology. 29:1081–1094. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Faderl S, O'Brien S, Pui CH, Stock W,
Wetzler M, Hoelzer D and Kantarjian HM: Adult acute lymphoblastic
leukemia: Concepts and strategies. Cancer. 116:1165–1176. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Redaelli A, Laskin BL, Stephens JM,
Botteman MF and Pashos CL: A systematic literature review of the
clinical and epidemiological burden of acute lymphoblastic
leukaemia (ALL). Eur J Cancer Care (Engl). 14:53–62. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tzoneva G, Perez-Garcia A, Carpenter Z,
Khiabanian H, Tosello V, Allegretta M, Paietta E, Racevskis J, Rowe
JM, Tallman MS, et al: Activating mutations in the NT5C2
nucleotidase gene drive chemotherapy resistance in relapsed ALL.
Nat Med. 19:368–371. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fielding AK, Richards SM, Chopra R,
Lazarus HM, Litzow MR, Buck G, Durrant IJ, Luger SM, Marks DI,
Franklin IM, et al: Outcome of 609 adults after relapse of acute
lymphoblastic leukemia (ALL); an MRC UKALL12/ECOG 2993 study.
Blood. 109:944–950. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sas V, Moisoiu V, Teodorescu P, Tranca S,
Pop L, Iluta S, Pasca S, Blag C, Man S, Roman A, et al: Approach to
the adult acute lymphoblastic leukemia patient. J Clin Med.
8:11752019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bansal M, Sharma KK, Bakhshi S and Vatsa
M: Perception of Indian parents on health-related quality of life
of children during maintenance therapy of acute lymphoblastic
leukemia: A comparison with siblings and healthy children. J
Pediatr Hematol Oncol. 36:30–36. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Reinfjell T, Lofstad GE, Nordahl HM, Vikan
A and Diseth TH: Children in remission from acute lymphoblastic
leukaemia: Mental health, psychosocial adjustment and parental
functioning. Eur J Cancer Care (Engl). 18:364–370. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Khalifa AS, Bishry Z, Tantawy AAG, Ghanem
MH, Effat SM, Shahawy HE and Ebeid FSE: Psychiatric morbidity in
Egyptian children with acute lymphoblastic leukemia and their care
providers. Hematol Oncol Stem Cell Ther. 7:76–84. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kantarjian H, Thomas D, O'Brien S, Cortes
J, Giles F, Jeha S, Bueso-Ramos CE, Pierce S, Shan J, Koller C, et
al: Long-term follow-up results of hyperfractionated
cyclophosphamide, vincristine, doxorubicin, and dexamethasone
(Hyper-CVAD), a dose-intensive regimen, in adult acute lymphocytic
leukemia. Cancer. 101:2788–2801. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mantadakis E, Cole PD and Kamen BA:
High-dose methotrexate in acute lymphoblastic leukemia: Where is
the evidence for its continued use? Pharmacotherapy. 25:748–755.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schmiegelow K, Nielsen SN, Frandsen TL and
Nersting J: Mercaptopurine/Methotrexate maintenance therapy of
childhood acute lymphoblastic leukemia: Clinical facts and fiction.
J Pediatr Hematol Oncol. 36:503–517. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Burnett AK, Milligan D, Prentice AG,
Goldstone AH, McMullin MF, Hills RK and Wheatley K: A comparison of
low-dose cytarabine and hydroxyurea with or without all-trans
retinoic acid for acute myeloid leukemia and high-risk
myelodysplastic syndrome in patients not considered fit for
intensive treatment. Cancer. 109:1114–1124. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huguet F and Tavitian S: Emerging
biological therapies to treat acute lymphoblastic leukemia. Expert
Opin Emerg Drugs. 22:107–121. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pan Y, Zhou F, Zhang R and Claret FX:
Stat3 inhibitor stattic exhibits potent antitumor activity and
induces chemo- and radio-sensitivity in nasopharyngeal carcinoma.
PLoS One. 8:e545652013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Buettner R, Mora LB and Jove R: Activated
STAT signaling in human tumors provides novel molecular targets for
therapeutic intervention. Clin Cancer Res. 8:945–954.
2002.PubMed/NCBI
|
|
20
|
Zhang Q, Zhang C, He J, Guo Q, Hu D, Yang
X, Wang J, Kang Y, She R, Wang Z, et al: STAT3 inhibitor stattic
enhances radiosensitivity in esophageal squamous cell carcinoma.
Tumour Biol. 36:2135–2142. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schust J, Sperl B, Hollis A, Mayer TU and
Berg T: Stattic: A small-molecule inhibitor of STAT3 activation and
dimerization. Chem Biol. 13:1235–1242. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lin L, Liu A, Peng Z, Lin HJ, Li PK, Li C
and Lin J: STAT3 is necessary for proliferation and survival in
colon cancer-initiating cells. Cancer Res. 71:7226–7237. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kanna R, Choudhary G, Ramachandra N,
Steidl U, Verma A and Shastri A: STAT3 inhibition as a therapeutic
strategy for leukemia. Leuk Lymphoma. 59:2068–2074. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li CH, Xu LL, Jian LL, Yu RH, Zhao JX, Sun
L, Du GH and Liu XY: Stattic inhibits RANKL-mediated
osteoclastogenesis by suppressing activation of STAT3 and NF-κB
pathways. Int immunopharmacol. 58:136–144. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mikyskova R, Sapega O, Psotka M, Novotny
O, Hodny Z, Balintova S, Malinak D, Svobodova J, Andrys R, Rysanek
D, et al: STAT3 inhibitor Stattic and its analogues inhibit STAT3
phosphorylation and modulate cytokine secretion in senescent tumour
cells. Mol Med Rep. 27:812023. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Debnath J, Gammoh N and Ryan KM: Autophagy
and autophagy-related pathways in cancer. Nat Rev Mol Cell Biol.
24:560–575. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang L, Zhao Y, Shan M, Wang S, Chen J,
Liu Z and Xu Q: Targeting crosstalk of STAT3 between
tumor-associated M2 macrophages and Tregs in colorectal cancer.
Cancer Biol Ther. 24:22264182023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yue P, Zhu Y, Brotherton-Pleiss C, Fu W,
Verma N, Chen J, Nakamura K, Chen W, Chen Y, Alonso-Valenteen F, et
al: Novel potent azetidine-based compounds irreversibly inhibit
Stat3 activation and induce antitumor response against human breast
tumor growth in vivo. Cancer Lett. 534:2156132022.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang L, Alizadeh D, Van Handel M,
Kortylewski M, Yu H and Badie B: Stat3 inhibition activates tumor
macrophages and abrogates glioma growth in mice. Glia.
57:1458–1467. 2009. View Article : Google Scholar : PubMed/NCBI
|