Introduction
The complement system comprises several soluble and
membrane-bound proteins that, when activated, provide a congenital
defense against microbial infection. In addition to its defensive
role, complement activation elicits numerous biological effects,
including leukocyte recruitment, smooth muscle contraction and
increased vascular permeability (1–4).
Although the activation of the complement system is strictly
regulated by various regulators and inhibitors in the body fluids
and plasma (5,6), inappropriate or uncontrolled
activation of the complement system can lead to local or systemic
inflammation, tissue damage and disease (7,8). A
number of studies have provided novel insights into the structure
and function of complement proteins, which have improved our
understanding of how different components of the complement system
participate in the destruction or restoration of homeostasis in the
tumor microenvironment (TME). This increased knowledge has inspired
novel approaches for complement-targeted therapy (9,10).
Colorectal cancer (CRC) is one of the most
frequently occurring cancers worldwide, ranking third among all
cancers in incidence and second in terms of mortality (11). Despite certain improvements that
have been made to treatment regimens, including surgery,
radiotherapy and chemotherapy, the mortality rate of CRC remains
high (12). Notably, therapies
targeting the immune system have achieved significant advances in
CRC treatment, suggesting that the immune system plays a critical
role in the regulation of tumor progression. The tumor immune
microenvironment (TIME), which contains various immune components,
has received increasing attention due to its crucial role in
influencing the immune response in the TME. Indeed, the balance of
immunosuppressive cells, for example, myeloid-derived suppressor
cells (MDSCs) and regulatory T cells (Tregs), with antitumor immune
cells, for example, cytotoxic T cells and natural killer (NK)
cells, has been shown to determine the state of tumor progression
(13). Currently, tumor
immunotherapy is primarily focused on promoting the antitumor
effects of cytotoxic T lymphocytes and modulating inflammatory
responses (14). The complement
system, which is the main effector of innate immunity, is widely
present in the TIME, and plays a central role in regulating immune
responses in the TME. Complement activation was traditionally
considered to promote the killing of tumor cells via
complement-mediated cytotoxicity, with blocking of the complement
cascade recognized as a risk factor for tumor progression. For
example, inhibiting the activity of C3 and C5 convertases, which
serve as key enforcers of the complement cascade by upregulating
the expression of the complement regulatory protein (CRP) CD55, has
been found to be associated with a poor prognosis in patients with
CRC (15). However, complement
activation may fulfill complex roles in cancer progression. For
example, complement components from the TME, such as C1q, C5a and
C7, have been shown to facilitate tumor progression through the
modulation of angiogenesis, antitumor immunity and tumor growth
(16–18). Similarly, complement activation
products inside tumors, such as C3a, have been shown to exhibit
pro-tumor properties by inhibiting antitumor immunity (19).
In the present review, current knowledge of the role
of the complement system in the pathogenesis of CRC is summarized,
and the contribution, mechanism and functional modes of complement
components in CRC are also discussed. In particular, the review
highlights unconventional modes of complement activation in
extracellular and intracellular environments.
Complement system and its roles in CRC
It is widely acknowledged that the complement system
is a systemic, serum-based effector of innate immunity, with
activation typically comprising a cascade of enzymatic reactions
confined to the extracellular space, known as cascade-dependent
activation. However, it has also been shown that the complement
system can be activated by enzymes inside cells in a
cascade-independent manner (20).
Furthermore, several complement components, such as C1q, have been
shown to function without activation (21–23).
Recent experiments have established that complement components
themselves and their activation products, derived from either
cascade-dependent or -independent activation, are involved in
regulating the progression of CRC.
Complement cascade-dependent activation
pathway in CRC
Complement cascade-dependent
activation pathways
The complement system can be activated by three
different pathways, namely, the classical pathway (CP), the lectin
pathway (LP) and the alternative pathway (AP). These three pathways
are activated by different activators: The CP is activated by
antigen-antibody complexes, whereas the LP, also known as the
mannose-binding lectin (MBL) pathway, is triggered by the direct
recognition of sugar structures on the surface of pathogens by
MBLs, which are mainly present in the blood. The AP, also known as
the bypass pathway, is an antibody-independent pathway in which
microorganisms and xenobiotics initiate a cascade of enzymatic
reactions with the help of factors B, D and P (24). Activation of the CP and LP leads to
the cleavage of C4 and C2 and the formation of C3 convertase
(C4b2b). By contrast, the AP is activated by the spontaneous
hydrolysis of C3, leading to the formation of the alternative C3
convertase (C3bBb). C3 convertase cleaves C3 into an anaphylatoxin
(C3a) and a larger fragment (C3b). The deposition of C3b leads to
the formation of a new C3 convertase through a bypass pathway,
creating a positive feedback loop that results in the further
deposition of C3b (25).
The activation of C3 also leads to the formation of
C5 convertase (C4b2b3b or C3bBb3b). All three activation pathways
share a common terminal pathway: C5 convertase cleaves C5 into C5a
and C5b. Subsequently, C5b stably binds to C6 to form C5b6, which
spontaneously binds to C7, forming C5b67. The C7 component of the
complex is initially inserted into the lipid bilayer of the target
cell membrane, after which C8 binds with high affinity to C5b67
inserted into the membrane, thereby forming a stable C5b678 complex
that is deeply embedded in the cell membrane. This complex can bind
to multiple C9 molecules to form C5b6789n (C5b-9), which is termed
the ‘membrane attack complex’ (MAC) (26–28).
Notably, each step of the cascade-dependent
complement activation process is strictly regulated by a series of
CRPs. For example, C1 inhibitor (C1INH) is known to inactivate
active enzymes C1r, C1s of CP, and mannan-binding lectin serine
peptidase (MASP) of LP through by covalently binding to them
(29). Factor I is a serine
protease that degrades C3b and C4b in the presence of cofactors
(30). Factor H is a cofactor for
C3b degradation, whereas C4 binding protein is the primary cofactor
for C4b degradation. Other factors, such as complement receptor 1,
membrane cofactor protein and decay accelerating factor, limit the
formation and stability of C3/C5 convertases. Additionally,
clusterin, vitronectin and CD59 prevent the formation and membrane
insertion of the C5b-9 complex (31,32)
(Fig. 1).
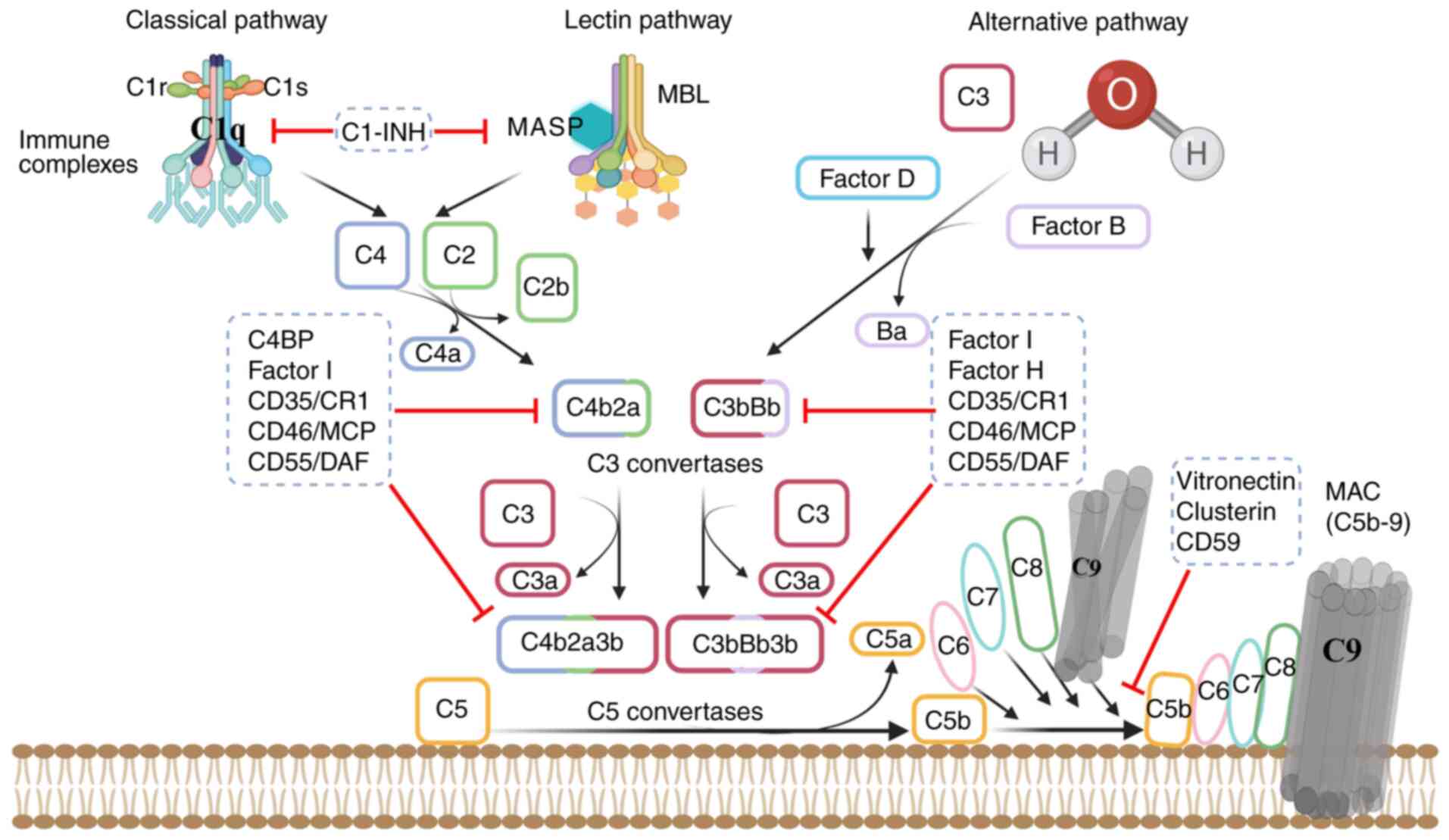 | Figure 1.Activation and regulation of
complement cascade-dependent processes. The complement system is
activated through three pathways: CP, LP and AP. The CP is
initiated when C1 (comprising C1q, C1r and C1s) binds to an
antibody; the LP is activated by the MBL-MASP complex on pathogens;
and the AP is triggered by spontaneous C3 hydrolysis or C3
tickover, which is amplified by regulators detecting unprotected
surfaces. All pathways lead to the formation of C3 convertases
(C3bBb or C4b2a), which cleave C3 into C3a and C3b. C3b is
deposited on target cells, where it promotes signaling via
complement receptors and forms a feedback loop in the AP, which
promotes C3b deposition and its conversion into C5 convertases. The
C5 convertases cleave C5 into C5a and C5b. The latter interacts
with C6, C7, C8 and C9 molecules to form the MAC. Regulation is
achieved by soluble factors, namely C1-INH, Factor I, C4BP, Factor
H, clusterin and vitronectin, and membrane proteins, namely
CD35/CR1, CD46/MCP, CD55/DAF and CD59. These regulators control
various activation stages: C1-INH influences C1r, C1s and MASP,
whereas C4BP, Factor H, Factor I, CD55 and CD35 regulate C3/C5
convertase formation and stability. Clusterin and vitronectin
inhibit the assembly or membrane insertion of the MAC by binding to
the C5b-7 complex, and CD59 inhibits the binding of C9 to C5b-8 by
competing for binding to a nascent epitope on C8, thereby
disrupting the association of C9 with C5b-8. CP, classical pathway;
LP, lectin pathway; AP, alternative pathway; MBL, mannose-binding
lectin; MASP, mannan-binding lectin serine protease; MAC, membrane
attack complex; C1-INH, C1 inhibitor; C4BP, C4 binding protein;
CR1, complement receptor 1; MCP, membrane cofactor protein; DAF,
decay accelerating factor. |
Upstream complement components in
CRC
Chronic inflammation serves as a significant driver
of CRC progression (33). In the
TIME, several inflammatory factors or chemokines produced during
chronic inflammation participate in the occurrence and development
of CRC (34,35). There is evidence to suggest that
upstream complement products from the cascade-dependent activation
pathway, such as C3a and C5a, are involved in influencing the TIME
(36,37). C3a levels have been found to be
significantly increased in the serum of patients with CRC, and C3a
upregulation has therefore been recommended as an important
biomarker for early diagnosis (36), suggesting that C3a may be an
important contributor to CRC progression. Mehrabani et al
(38) also found that C3a is
present at high levels in the serum of patients with CRC, and
clearly diminished after the patients received treatment, implying
that C3a plays a role in tumorigenesis. However, Krieg et al
(39) demonstrated that C3a
receptor (C3aR) deficiency promoted CRC development by the
induction of inflammatory responses characterized by pro-tumor
effects, suggesting that C3a/C3aR exerts antitumor effects on CRC
progression. Therefore, the role of C3a in CRC is not clear and
further research is necessary to elucidate it. Furthermore, the
complement cascade-dependent generation of C5a in the TIME has been
shown to promote CRC progression through multiple mechanisms:
Firstly, C5a induces the infiltration of immunosuppressive cells
into the TIME, which inhibits their antitumor activities. For
example, C5a recruits C5aR+ MDSCs into the colonic TIME
to suppress the antitumor activity of CD8+T cells
(40). Secondly, C5a promotes the
pro-tumor activities of tumor-associated macrophages (TAMs). For
example, C5a promotes colon cancer metastasis by activating
NF-κB-associated signaling pathways in C5aR+ TAMs and
stimulating macrophage differentiation toward an M2 phenotype with
tumor-promoting activity (41). In
addition, Piao et al (42)
used C5aR−/−mice to demonstrate that the
metastasis of CRC to the liver depends on monocyte chemoattractant
protein-1 (MCP-1) produced by macrophages, with high levels of
MCP-1 secreted by C5a, leading to activation of the PI3K-AKT
signaling pathway in macrophages. Thirdly, C5a prompts metastasis
in CRC by regulating CRC cell activities. For example, Xu et
al (43) demonstrated that, in
human CRC cell lines, the upregulation of C5a receptor 1 (C5aR1),
accelerates epithelial-mesenchymal transition (EMT) and activates
the Wnt/β-catenin signaling pathway, effects that are association
with CRC progression. Also, C5a has been identified as a potential
clinical biomarker for the early detection and prognosis of CRC,
based on the observation of elevated C5a levels in the plasma of
patients (44,45). Therefore, in addition to being
vital components of pro-tumor mechanisms in CRC, C3a and C5a are
also potentially valuable clinical biomarkers for this disease.
Overall, the anaphylatoxins C3a and C5a play
important roles during the progression of CRC by modulating the
inflammatory microenvironment within the TIME, or by directly
affecting tumor processes such as growth and metastasis. However,
their effects on CRC development are not consistent. Specifically,
C3a and its receptor C3aR display dual effects, while C5a and its
receptor C5aR display only pro-tumor effects. These differences may
be due to the involvement of distinct complement components and
different mechanisms. In addition, while signaling pathways such as
the NF-κB, PI3K-AKT and Wnt/β-catenin signaling pathways are being
considered as candidate mechanisms, further studies are necessary
to elucidate the molecular mechanisms underlying the regulation of
inflammatory cell function and CRC cell growth behaviors in greater
detail.
Although the complement-cascade activation products
C3a and C5a, derived from the cleavage of C3/C5 by C3/C5
convertase, are recognized as risk factors for CRC, the body
expresses several CRPs, including membrane proteins such as CD55
and CD46, and soluble proteins such as factor H and Factor I, that
can inhibit C3/C5 convertase activity, and thereby regulate CRC
progression. As noted by Talaat et al (46), CRC cells can evade
complement-dependent cytotoxicity by the overexpression of CD55,
particularly under hypoxic conditions, which prevents tumors from
being eradicated. Consistent with these findings, Lin et al
(47) found that the absence of
CD55 led to the dysregulation of complement-cascade activation,
which triggered the infiltration of inflammatory cells into the
intestinal mucosa, and eventually resulted in the excessive
production of pro-inflammatory cytokines, including IL-10, IL-12,
IL-6 and TNF-α, and aggravation of colonic inflammation. These
studies suggest that CD55 serves an antitumor role. However, other
studies have contradicted this by suggesting that CD55 performs a
pro-tumor role. CD55 expression has been shown to be upregulated in
CRC, particularly in the more advanced stages (48). Dho et al (49) developed a novel chimeric CD55
monoclonal antibody that activated the complement system, leading
to CRC cell proliferation, invasion and migration. Additionally,
this antibody exhibited a synergistic inhibitory effect with
5-fluorouracil (5-FU) on CRC cell growth, highlighting the
potential of CD55 as a therapeutic target for CRC. In multiple
clinical trials, the presence of CD55 has been considered a
potential hallmark of poor prognosis in patients with CRC (15,50,51).
The current perspective is that the conflicting roles of CD55 in
CRC may result from, for example, inconsistent modeling conditions,
modeling durations and tumor progression stages (46,52).
Mechanistically, CD55 may inhibit tumor growth by reducing the
levels of activation products C3a and C5a. By contrast, its
pro-tumor activities may result from its blockade of the complement
cascade-dependent activation pathway, leading to lower levels of
MAC formation and diminished MAC-induced antitumor effects. In
addition, CD46, along with CD55, has been found to be upregulated
in human colon cancer cells through the STAT3/STAT6/p38 MAPK
signaling pathway, and knocking out both CD46 and
CD55 induced tumor cell apoptosis and reduced colon cancer
growth in mice (52). This further
confirms the pro-tumor function of CRPs that inhibit C3/C5
convertases.
In addition to the aforementioned membrane CRPs,
soluble CRPs such as Factor I and Factor H have also been shown to
participate in the regulation of CRC tumorigenesis. In one study,
the expression of Factor I was demonstrated to be significantly
upregulated in colon cancer, and proposed as a diagnostic biomarker
for CRC (53). In addition, the
knockout of Factor I inhibited the proliferation, migration and
invasion of colon cancer cells, which was attributed to the
suppression of glycolysis as a consequence of blocking the
Wnt/β-catenin/c-Myc signaling pathway (53). In addition, Wilczek et al
(54) demonstrated that the levels
of Factor H and its split variant, Factor H-like protein, were
highly increased in patients with colon adenocarcinoma and
metastatic foci in the liver. The authors verified that the
upregulation of Factor H resulted in tumor growth and metastasis
through binding to C3b on the tumor cells and preventing the
subsequent formation of lytic components at the cell surface.
Moreover, after blocking the function of Factor H in the SW620 CRC
cell line, treatment of the cells with specific antibodies against
CD55 and CD59 rendered them more readily lysed (54). This demonstrates the strong
potential of either targeting Factor H or performing a combined
blockade of various CRPs, including CD55, CD59 and Factor H, in the
treatment of CRC.
Downstream complement components in
CRC
The MAC formed in the terminal stages of complement
activation has been suggested to exert cytotoxic effects on tumor
cells, thereby inhibiting tumor progression (55,56).
It has been reported that in patients with CRC, the expression of
CD59, which prevents MAC formation, significantly correlates with
tumor grade, suggesting that the high expression of CD59 in tumors
is a marker of poor prognosis (57). In addition, blocking CD59 with an
anti-CD59 monoclonal antibody was found to prevent MAC-mediated
cytolysis in HT29 CRC cells (58).
These data indicate that blocking the formation of MAC by CD59 in
CRC cells contributes to tumor growth. In a separate study, mice
with a deficiency of C6, an important component of the MAC,
developed more severe colitis symptoms than those of wild-type mice
following treatment with dextran sulfate sodium (59), suggesting that blocking MAC
formation may be a risk factor for colitis-associated cancer.
However, other studies have shown that MAC may
exacerbate tumor progression (55,60).
As summarized in a previous review, several studies have shown
that, in order to exert its pro-tumor effects, MAC must exist
mainly in its sublytic form (60).
The specific mechanisms that have been proposed for its pro-tumor
effects are as follows: i) Inducing the transcription of oncogenes
and the proliferation of tumor cells; ii) protecting tumor cells
from apoptosis; iii) promoting tumor angiogenesis; and iv)
regulating the expression of response gene to complement 32
(RGC-32), a gene that regulates the cell cycle via the
activation of Akt and cell division cycle protein 2 kinases
(60). Based on current
understanding, altering the transcriptional activity of CRC cells
to promote the expression of tumor-promoting molecules may be the
main mechanism by which sublytic MAC regulates CRC progression.
Epidermal growth factor (EGF) performs a vital role in tumor
development (61). Towner et
al (62) showed that sublytic
MAC induces the expression of the EGF receptor in CRC cells,
thereby driving CRC tumor progression. Additionally, other studies
have shown that strongly upregulating RGC-32 in colon cancer cells
promotes tumor growth via the regulation of cytoskeletal
reorganization or chromatin assembly (63,64).
Given that the activation of sub-lytic MAC significantly
upregulates the level of RGC-32, and considering the key role of
RGC-32 in the underlying mechanism of CRC, the role of the
sub-lytic MAC/RGC-32 signaling axis in the pathogenesis of CRC
merits further attention in the future. Although further studies of
CRC models are required to fully determine the pro-tumor effects of
sub-lytic MAC, the research that has already been completed to
elucidate the underlying pro-tumorigenic mechanisms has shed great
insights into the role of MAC in CRC.
These findings indicate that MAC exerts a dual role
in CRC, possessing both anti-cancer and cancer-promoting effects,
depending primarily on the pattern of MAC attack. In one scenario,
MAC initiates lethal attacks that directly induce cancer cell
death, which is the primary manifestation of its anticancer effect.
In the other scenario, sublytic MAC attacks, which are not
sufficiently robust to directly cause cell death, may stimulate
cancer cells to release growth factors or activate survival
signaling pathways, thereby promoting their survival, growth and
migration. This may be the primary mechanism underlying the
cancer-promoting effects of MAC. It should be emphasized, however,
that while the role of MAC in cancer is recognized, further
in-depth research and validation experiments are required in more
cancer models, such as CRC models, to fully elucidate the complex
association between MAC and cancer.
Complement cascade-independent activation
pathway in CRC
The complement system is a systemic, serum effector
in innate immunity, and complement activation is traditionally
viewed as a cascade of enzymatic reactions confined to the
extracellular space, driven by three cascade-dependent activation
pathways. Notably, the latest research has shown that the
complement system is also activated by enzymes inside cells in a
cascade-independent manner, and that this has an important role in
the pathogenesis of CRC. For example, C3 and C5, the core
complement components, have been found to be activated within cells
in CRC (65,66).
Intracellular C3 activation in CRC
Liszewski et al (65) initially discovered the
intracellular activation of C3 in human CD4+ T cells,
and confirmed that T-cell-intrinsic C3 can be cleaved into C3a and
C3b by the protease cathepsin L (CTSL). The CTSL-mediated
intracellular generation of C3a is essential for the survival of
resting T cells, as it sustains the tonic mammalian target of
rapamycin signaling by engaging C3aR expressed on the lysosomes. By
contrast, T cell receptor (TCR) activation induces the
translocation of intracellular C3a to the cell surface and induces
T helper 1 (Th1) immunity. In addition, the intracellular C3
product, C3b, can increase CD8+ T-cell immunity by
binding to the C3b receptor CD46 in an autocrine manner (67) (Fig.
2). Notably, in non-immune cells, such as Caco-2 cells,
pathogen-deposited C3 can be transported into the cells during
pathogen endocytosis, resulting in the activation of multiple
signaling pathways, including those regulated by NF-κB and
activator protein 1 (68).
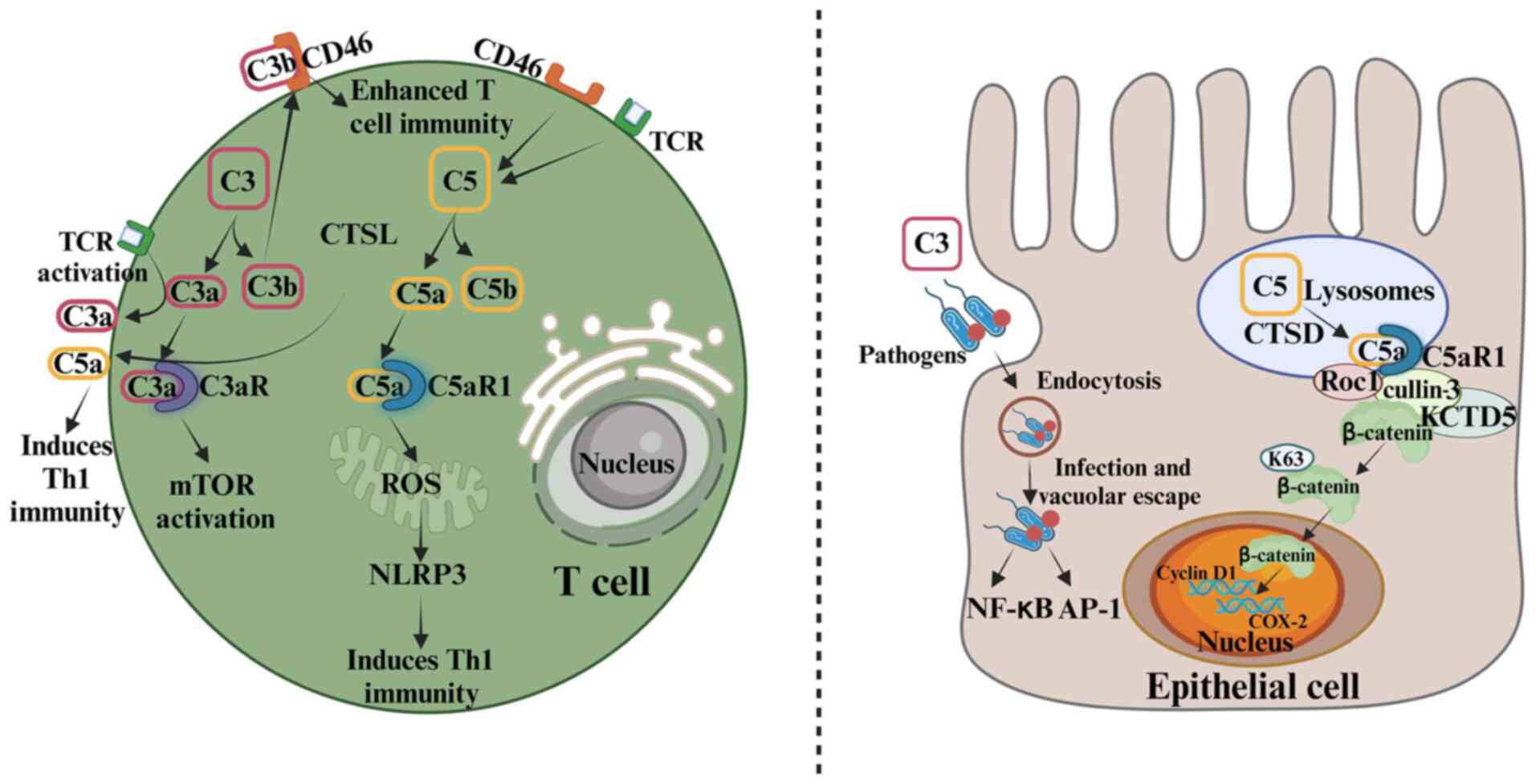 | Figure 2.Role of the complement system in the
regulation of immune cells and its impact on non-immune cells.
Intracellularly, complement primarily regulates immune cell
function whilst also influencing non-immune cells. The left panel
details the activation of C3 and C5 within a T-cell. CTSL cleaves
C3 into C3a and C3b. Intracellular C3a stabilizes mTOR signaling
via lysosomal C3aR, which is crucial for T-cell survival.
Intracellular C5a binds to C5aR1, inducing the production of ROS
and activating the NLRP3 pathway to promote the Th1 response. With
TCR activation, intracellular C3a translocates to the cell surface,
triggering Th1 immunity, while C3b binds to CD46 in an autocrine
manner, which enhances T-cell immunity. C5 in T cells can also be
activated by the co-stimulatory effect of TCR and CD46. The right
panel show the role of complement components in non-immune cells,
using epithelial cells as an example. Pathogen-deposited C3 is
endocytosed, activating pathways such as the NF-κB and AP-1
pathways. In addition, CTSD cleaves C5 in lysosomes and endosomes,
generating C5a. C5a binds to C5aR1, recruiting KCTD5, cullin-3 and
Roc1 to form a complex that stabilizes β-catenin through K63-linked
polyubiquitination. This stabilizes β-catenin in the nucleus,
thereby regulating gene transcription. Elevated β-catenin levels
and nuclear translocation are closely associated with poor
prognosis in colorectal cancer. CTSL, cathepsin L; C3aR, C3a
receptor; C5aR1, C5a receptor 1; mTOR, mammalian target of
rapamycin; ROS, reactive oxygen species; NLRP3, NOD-, LRR- and
pyrin domain-containing protein 3; Th1, T helper 1; TCR, T-cell
receptor; AP-1, activator protein-1; CTSD, cathepsin D; KCTD5,
K+ channel tetramerization domain 5; Roc1, regulator of
cullins-1; COX-2, cyclooxygenase-2. |
Recently, studies have indicated that intracellular
C3, particularly C3a, contributes to the promotion of CRC
progression. For example, Liu et al (69) showed that the proportions of M0 and
M1 macrophages and quiescent mast cells were increased in CRC
samples with high C3 expression, whereas the proportions of
activated dendritic cells (DCs), activated mast cells and memory
CD4+ T cells were decreased, implying that high levels
of C3 in CRC cells may promote CRC progression by altering the
TIME. In addition, the C3a produced by tumor cells has been shown
to reduce the infiltration of NK cells into solid tumors, such as
CRC, by promoting the interaction between C3aR and lymphocyte
function-associated antigen-1 in NK cells, which stimulates tumor
growth (70).
Studies have also shown that the intracellular
activation of C3, independent of the complement cascade, plays a
critical role in maintaining T-cell homeostasis and supporting
T-cell differentiation (65,71).
This pair of studies were in general agreement that,
intracellularly, C3a inhibits the differentiation of
CD4+ T cells into Tregs, leading to a reduction in the
level of the immunosuppressive cytokine IL-10, thereby inhibiting
antitumor activity (72). However,
no studies to date have explored whether C3a in T cells performs a
similar role in CRC progression. Given that T cells have an
important role as immune factors in the pathogenesis of CRC,
investigation of the impact of intracellular C3 on CRC progression
may emerge as an important area of future research.
Intracellular C5 activation in CRC
Within the lysosomes of colonic epithelial cells, C5
is cleaved by cathepsin D (CTSD) into C5a and C5b. Subsequently,
C5a binds to C5aR1, recruiting K+ channel
tetramerization domain 5, cullin-3 and regulator of cullins-1 to
form a complex that promotes the K63-linked polyubiquitination of
β-catenin, thereby stabilizing its expression in the nucleus. As
β-catenin is a pivotal component of the Wnt signaling pathway, it
stabilization regulates the transcription of downstream genes,
thereby influencing cell proliferation and differentiation
(66). As already mentioned, in
immune cells such as T cells, C3 can be activated by CTSL (65), and the intracellular C3 products
subsequently strengthen T-cell-mediated immune responses. In
addition, the C3 products can translocate into non-immune cells and
trigger multiple immune responses. Similarly to the activation of
intracellular C3 catalyzed by protease CTSL in human
CD4+ T cells, C5 can also undergo intracellular
activation upon TCR and CD46 co-stimulation in CD4+ T
cells (71). The intracellularly
generated C5a binds to C5aR1 and induces reactive oxygen species
production, which activates the NOD-, LRR-, and pyrin
domain-containing protein 3 signaling pathway, thereby promoting
Th1 responses (71) (Fig. 2).
A recent study reported that C5 inside colonic
cancer cells can be cleaved by CTSD to produce C5a, and the
subsequent C5a/C5aR1 signaling potentiates β-catenin stability and
promotes colorectal tumorigenesis (66). Therefore, C5a/C5aR1 signaling in
CRC cells has the potential to directly promote tumor growth. In
addition, the intracellular activation of urokinase-type
plasminogen activator-positive macrophages results in the release
of C5a, which has been demonstrated to regulate the tumorigenic
properties of C5aR1+ mast cells and macrophages, as well
as to inhibit the cytotoxicity of CD8+ T cells, thereby
promoting tumor growth (73).
Furthermore, intracellular complement activation products such as
C5a that are generated via the cascade-independent activation
pathway have been shown to have tumor-promoting effects (66).
Advances in complement system research have deepened
understanding of complement activation, expanding it from
extracellular cascade-dependent activation, as an extracellular
danger-sensing mechanism, to also include intracellular activation
as a cascade-independent intracellular effector system. Moreover,
this intracellular activation of complement proteins, such as C3a
and C5a, appears to have a key pro-tumor role in CRC
progression.
In summary, C3a and C5a, generated by complement
cascade activation and intracellular activation, function as
inflammatory mediators, capable of activating downstream signaling
pathways and triggering a series of biological effects. However,
these two activation pathways exhibit distinct differences in their
generative mechanisms, modes of action and resulting effects.
Complement cascade activation follows strict regulatory mechanisms,
producing C3a and C5a as immunomodulatory factors. Upon excessive
complement activation, these may induce inflammatory responses and
regulate CRC cell behavior, thereby promoting or inhibiting tumor
growth. By contrast, the generation of C3a and C5a by intracellular
activation may involve non-canonical complement activation or
direct cleavage via intracellular proteases. This process allows
them to directly influence the function of immune cells, for
example, T cells, DCs and MDSCs, as well as CRC cells. Also, the
extracellular release of C3a and C5a enables them to exert complex
effects on the TIME.
C1q protein in CRC beyond the borders of
activation
C1q consists of three distinct polypeptides,
designated C1qa, C1qb and C1qc, and functions as an initiating
component of the classical complement cascade (74). It is a type-II transmembrane
protein that is anchored to the membrane via the C1qa chain, prior
to being cleaved into its soluble form (74,75).
A number of studies have suggested that C1q functions beyond the
borders of complement activation; that is, C1q directly
participates in tumor progression in either its secreted or
membrane-anchored form (21–23,76,77).
Distinct from the majority of complement proteins, which are
derived from the liver, C1q is mainly secreted by macrophages, and
intestinal macrophages have been identified as the primary source
of C1q (78). C1q+
macrophages have been observed in both healthy and tumor tissues,
such as CRC and lung and liver cancers (79). And C1q produced by TAMs is
considered to facilitate tumor progression by inducing
immunosuppression, promoting T-cell exhaustion or facilitating
neoangiogenesis through interactions with endothelial cells
(76,77,80).
Similarly, C1q+ macrophages have been detected in both
healthy and tumor tissues in the colon, and single-cell analyses
have suggested a pro-inflammatory function of C1qc+
TAMs, which preferentially express phagocytosis- and antigen
presentation-associated genes (21). Another study suggested that in
patients with colon cancer, a high expression level of C1qc in the
cancerous tissue is associated with a worse prognosis (22), suggesting that C1q may serve as a
promoting factor of CRC progression. However, a subsequent study
revealed that, in addition to its action in the soluble form, the
membrane-anchored form of C1q plays a protective role in
inflammatory bowel diseases by enhancing the phagocytic capability
of macrophages against bacteria and inflammatory cells in the colon
(23). This study confirmed the
protective role of the monoterpenoid glycoside paeoniflorin, which
is mediated by the targeting of C1qa to increase the population of
membrane-anchored C1q, while reducing C1q secretion. Therefore, the
function of C1q depends on whether it is secretory or
membrane-bound. Secretory C1q exhibits a pathogenic role in colon
inflammation and cancer, even without activation of the complement
cascade, whereas membrane-anchored C1q has a protective role in
chronic colitis.
In summary, the complicated effects on CRC exerted
by complement activation products, such as C3a and C5a, and
complement proteins that function without undergoing activation,
such as C1q, have deepened our understanding of the role of
complement components in CRC. Firstly, the roles of activation
products, including C3a, C5a, and MAC, generated by the complement
cascade-dependent activation pathway are not always consistent. In
detail, the roles of C5a and C3a can be summarized as follows: C5a
promotes CRC progression by i) regulating antitumor immunity in the
TIME by recruiting immune cells, including MDSCs and TAMs, and
activating the NF-κB and PI3K-AKT signaling pathways; and/or ii)
directly promoting tumor behaviors, including tumor growth,
proliferation and invasion behavior, by activating the
Wnt/β-catenin signaling pathway. However, the role of C3a in CRC is
more complex, as the high plasma levels of C3a in patients with CRC
can significantly decrease after treatment, indicating that C3a may
have pro-tumor role in CRC, while C3a has also been indicated to
play an antitumor role via the suppression of inflammatory
responses typically associated with pro-tumor effects. Furthermore,
MAC can directly form on the surface of tumor cells, where it can
influence cell behavior. When MAC is formed in sufficient amounts,
it exerts cytotoxic effects that inhibit tumor progression.
However, when MAC is deposited at sublytic levels, it triggers
inflammatory signals in cells, which induce CRC progression. It is
worthy of note that the roles of C3a, C5a and MAC in CRC are also
regulated by various CRPs, including Factor I, Factor H, CD55 and
CD59. Secondly, some C3a and C5a is generated in cells, for
example, in tumor cells and macrophages, through
cascade-independent activation pathways in the TIME. These cells
can directly activate intracellular complement components by
cathepsins and secrete activation products such as C3a and C5a into
the TIME, where they exert their pro-tumor effects. Thirdly,
secretory C1q promotes tumorigenesis, whereas membrane-anchored C1q
is protective against chronic colitis. This progression is beyond
the borders of activation. The various associations between the
complement system and colon cancer are summarized in Table I.
 | Table I.Associations between complement
components and CRC. |
Table I.
Associations between complement
components and CRC.
| A, Complement
cascade-dependent pathway |
|---|
|
|---|
| Complement
components | Associations with
CRC | (Refs.) |
|---|
| C3a | C3a serves as a
potential clinical biomarker for the early detection and prognosis
assessment of CRC | (36) |
|
| High expression of
C3a in CRC and its significant reduction after treatment suggest
C3a plays a role in tumorigenesis (pro-tumor effects) | (38) |
|
| C3aR
deficiency promotes CRC development by inducing an inflammatory
response with pro-tumor effects, indicating that C3a also exerts
antitumor effects on CRC (antitumor effects) | (39) |
| C5a | C5a recruits
C5aR+ MDSCs, indirectly inhibiting CD8+ T
cells and promoting a tumor-friendly environment (pro-tumor
effects) | (40) |
|
| By activating
NF-κB-related signaling pathways in C5aR+ TAMs and
stimulating the differentiation of TAMs towards the M2 phenotype,
C5a exhibits tumor-promoting activity (pro-tumor effects) | (41) |
|
| By promoting the
secretion of macrophage-derived MCP-1 and activating the PI3K-AKT
signaling pathway in macrophages, C5a promotes the liver metastasis
of CRC (pro-tumor effects) | (42) |
|
| C5a promotes CRC
development by inducing EMT in CRC cells and activating
Wnt/β-catenin signaling pathways (pro-tumor effects) | (43) |
|
| C5a is a potential
clinical biomarker for early detection and prognosis of CRC | (44,45) |
| MAC | Blocking CD59 with
an anti-CD59 monoclonal antibody prevents HT29 CRC cells from
undergoing MAC-mediated cytolysis, thereby contributing to tumor
growth (antitumor effects) | (58) |
|
| Sublytic MAC
induces the expression of EGF receptors in CRC cells, thereby
driving the progression of CRC (pro-tumor effects) | (62) |
|
| Upregulated RGC-32
in colon cancer cells promotes tumor growth through the regulation
of cytoskeletal reorganization or chromatin assembly. It is
speculated that, given that sublytic MAC triggers the upregulation
of RGC-32 in tumor cells, sublytic MAC/RGC-32 may mediate pro-tumor
mechanisms in CRC | (63,64) |
| CD46 | CD46 upregulation
in colon cancer cells via STAT3/STAT6/p38 MAPK reduces apoptosis
and induces mouse cancer growth (pro-tumor effects) | (52) |
| CD55 | CRC tumors are
protected by the overexpression of CD55, especially during hypoxia
(pro-tumor effects) | (46) |
|
| CD55
deficiency abnormally activates the complement system, causing
intestinal inflammation, excess cytokine release, and worsening
colitis (antitumor effects) | (47) |
|
| Novel CD55
monoclonal antibody activates complement and inhibits CRC
proliferation, invasion and migration (pro-tumor effects) | (49) |
|
| CD55 is highly
upregulated in CRC, especially in the more advanced stages | (15,48,50,51) |
| CD59 | Expression of CD59
significantly correlates with tumor grade, and is a marker of poor
prognosis in patients with CRC |
|
|
| Blocking CD59 with
anti-CD59 monoclonal antibody prevents CRC cells from undergoing
MAC-mediated cytolysis, contributing to tumor growth (pro-tumor
effects) | (58) |
| Factor I | Factor I is
significantly increased in colon cancer cells, and considered to be
a diagnostic biomarker for CRC | (53) |
|
| Knocking out Factor
I inhibits colon cancer cell proliferation, migration and invasion
by blocking the Wnt/β-catenin/c-Myc pathway and suppressing
glycolysis (pro-tumor effects) | (53) |
| Factor H | Levels of factor H
and its split variant, factor H-like protein, are highly increased
in patients with colon adenocarcinoma and metastatic foci in the
liver | (54) |
|
| Upregulation of
factor H results in tumor growth and metastasis through binding to
C3b on tumor cells and preventing subsequent lytic component
formation at the cell surface (pro-tumor effects) | (54) |
|
| B, Complement
cascade-independent pathway |
|
| Complement
components | Associations
with CRC | (Refs.) |
|
| C3a | High C3 in CRC
increases M0/M1 macrophages and quiescent mast cells, but decreases
activated DCs, activated mast cells and memory CD4+ T
cells, suggesting C3 promotes CRC progression by altering the TIME
(pro-tumor effects) | (69) |
|
| C3a produced by
tumor cells reduces the infiltration of NK cells into solid tumors,
such as CRC, by promoting the interaction between C3aR and
lymphocyte function associated antigen 1 in NK cells, which
stimulates tumor growth (pro-tumor effects) | (70) |
|
| C3a inhibits
CD4+ T cell differentiation into Tregs and reduces IL-10
levels, suppressing antitumor activity. It is speculated that since
T cells play an important role as immune factors in the
pathogenesis of CRC, the impact of intracellular C3 may exert
pro-tumor effects on CRC by this mechanism (pro-tumor effects) | (72) |
| C5a | C5a/C5aR1 signaling
stabilizes β-catenin, promoting CRC cell proliferation and
differentiation (pro-tumor effects) | (66) |
|
| uPA+
macrophages release C5a, which regulates the carcinogenic
properties of | (73) |
|
| C5aR1+
mast cells and macrophages, and inhibits CD8+ T cell
toxicity, thereby promoting tumor growth (pro-tumor effects) |
|
|
| C, Beyond the
borders of activation |
|
| Complement
components | Associations
with CRC | (Refs.) |
|
| C1q | In the colon,
C1q+ TAMs are present in both healthy and tumor tissue,
and single-cell analyses suggest a pro-inflammatory function of
C1qc+ TAMs (pro-tumor effects) | (21) |
|
| Patients with colon
cancer who express a high level of C1qc in the colon cancer tissue
exhibit a worse prognosis (pro-tumor effects) | (22) |
|
| C1q plays a
protective role in inflammatory bowel diseases by enhancing the
phagocytic capability of macrophages against bacteria and
inflammatory cells in the colon (antitumor effects) | (23) |
Complexity of complement system in CRC
progression
As discussed above, complement components exhibit
complicated roles in CRC, with both pro-tumor and antitumor
effects. Differences in the roles of various complement components
may be responsible for this. For example, C5a produced by the
complement cascade-dependent activation pathway exerts pro-tumor
effects on CRC, whereas C3a produced analogously has antitumor
effects. Also, the distinct complement activation pathways appear
to contribute to these conflicting effects; while C3a produced by
the complement cascade-dependent activation pathway plays an
antitumor role in CRC, C3a produced by the complement
cascade-independent activation pathway promotes tumorigenesis. In
addition, the varying effects the complement system has on CRC may
be attributed to different CRC subtypes. According to the Consensus
Molecular Subtypes (CMS) classification system and gene expression
analysis, CRC may be divided into four subtypes: CMS1
(microsatellite instability immune), which exhibits clear immune
infiltration and activation resulting in poor survival after
relapse; CMS2 (canonical), which mainly exhibits epithelial
differentiation, although it is associated with superior survival
rates following relapse; CMS3 (metabolic), which is associated with
the occurrence of KRAS-activating mutations, and exhibits
prominent metabolic adaptation; and CMS4 (mesenchymal), which is
associated with poorer relapse-free and overall survival rates, and
displays high levels of complement-mediated inflammation, EMT and
activation of transforming growth factor-β signaling pathways
(81,82). Different CRC subtypes may exhibit
significant heterogeneity of the complement system, with distinct
patterns of complement expression potentially contributing to the
differences in tumor aggressiveness that have been observed among
CRC subtypes. For example, CMS4, characterized by
complement-mediated inflammation, has poorer relapse-free and
overall survival rates compared with those of other subtypes. These
heterogeneous features not only highlight the diverse effects of
the complement system on CRC, but also provide crucial insights
that may assist the development of personalized treatment
strategies. Understanding this variability is essential to
facilitate the development targeted therapies that can regulate
complement activity in CRC.
Clinical significance of complement system
in CRC
Several studies have investigated the possibility of
targeting the complement system to block tumor progression in CRC.
For example, Downs-Canner et al (83) demonstrated that cobra venom factor,
which depletes C3, and Staphylococcus aureus
superantigen-like protein 7, which inhibits C5, both effectively
suppressed tumor growth in a CRC mouse model. These antitumor
effects were mediated via the enhancement of immune cell
infiltration, specifically that of CD8+ T cells, and the
expression of chemokines, including C-C motif chemokine ligand 5,
and C-X-C motif chemokine ligands 10 and 11. Due to the important
role of the C5a/C5aR axis in immune cell infiltration, Ding et
al (84) discovered that
blocking this pathway, either by knocking out complement C5
or C5aR1, or by using the C5aR inhibitor PMX205, resulted in
the near-complete blockade of CRC progression. This was achieved
through the inhibition of MDSC infiltration and an increase in the
proportion of CD8+ T cells. Notably, the study also
demonstrated that the knockout of C5 and C5aR1
resulted in a smaller tumor size and fewer tumors, along with a
greater reduction in the number of infiltrating MDSCs and a more
pronounced increase in the proportion of CD8+ T cells
compared with that achieved with C3 knockout. This suggests
that targeting C5 is likely to more effective than targeting C3 in
complement-based therapies for CRC. However, although no clinical
treatment method has yet been devised to target the C5a/C5aR axis
for the treatment of CRC, the STELLAR-001 phase I trial has
explored the possibility of combining the anti-C5aR antibody
IPH5401 with the programmed cell death ligand 1 inhibitor
durvalumab for the treatment of advanced solid tumors (46).
Targeting CRPs has also been shown to delay the
progression of CRC, in addition to targeting the C5a/C5aR axis. The
expression of CD55 is upregulated in CRC cells under hypoxic
conditions, which protects the tumor from lysis (46). In this regard, Dho et al
(49) developed a novel CD55
chimeric monoclonal antibody that inhibits the proliferation,
invasion and migration of CRC cells by activation of the complement
system. In addition, a combination of the anti-CD55 antibody with
5-FU was found to provide synergistically improved therapeutic
effects on CRC (49).
In summary, while clinical applications are not yet
available, preclinical studies in which the complement system is
targeted have revealed promising therapeutic effects on CRC.
However, based on current research findings, several key areas
require further investigation to optimize the targeting of the
complement system for CRC treatment. For example, the complement
system is an important component of innate immunity, and
continuously blocking its activation may impair opsonization and
bacteriolytic activity, thereby increasing the risk of infection
(46). To overcome this challenge,
novel drug formulations, such as a next-generation ‘recycling’ form
of eculizumab, are being developed for potential use (85,86).
In addition, targeting C5aR instead of directly blocking C3 or C5
enables opsonization, which should protect cancer patients from the
risk of bacterial infection (46).
Therefore, it is necessary to carefully consider the complexity of
the function of a drug when studying the role of the complement
system in tumor progression, as this will provide a solid
theoretical foundation for the development of targeted therapeutic
strategies. Moreover, most current research efforts have focused on
in vitro experiments and animal models, which may not fully
represent the complex situation in the human body (87–89).
Therefore, further validation and adjustment of these strategies
are required prior to clinical application.
Conclusions and prospects
CRC remains one of the most commonly occurring
tumors worldwide, and the prognosis of patients with CRC is
unsatisfactory following treatment with traditional methods. As a
key component of the TIME, complement activation products derived
from both complement cascade-dependent activation in body fluids
and intracellular, cascade-independent activation play various
roles in CRC. These include the regulation of inflammatory
responses during tumorigenesis, the modulation of antitumor
immunity in the TME, and the control of tumor growth. At times,
complement proteins can affect tumor progression without being
activated. However, the effects of these components of the
complement system are not always consistent, and in some cases have
been shown to be contradictory. Further research is necessary to
explore the main factors of the complement system that contribute
to CRC progression. In addition, some early studies focused on the
changes of complement components in the plasma or feces rather than
the tumor tissues and did not explore the underlying mechanisms.
Comprehensive research elucidating the signaling pathways involved
in the interactions of complement components with other immune
cells or tumor cells within the TIME, particularly at various
developmental stages of CRC, are lacking. A deeper exploration of
these aspects may not only reveal novel therapeutic targets for
CRC, but also provide a more comprehensive analysis and
understanding of the conflicting roles of the complement system in
CRC.
Additionally, while several studies have suggested
that complement components play an antitumor role in CRC
development, current research is primarily focused on the pro-tumor
effects of these complement components. The inhibition of C3, C5
and CD55 has been shown to alleviate CRC development. The
therapeutic effects achieved by therapies targeting the complement
system indicate that the pro-tumor effects of the system play a
more dominant role than the antitumor effects in CRC progression.
In conclusion, given the important role of the complement system in
the pathogenesis of CRC, further exploration of therapies targeting
the complement system is likely to be invaluable.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant nos., 82000525 and 81873883) and Science
and Technology Support Plan for Youth Innovation of Colleges and
Universities of Shandong Province of China (grant no. 2021KJ106).
In addition, it was funded by Youth Innovation Team Project for
Talent Introduction and Cultivation in Universities of Shandong
Province and the domestic visiting project of Shandong Second
Medical University.
Availability of data and materials
Not applicable.
Authors' contributions
YX, ML and SL designed and edited the manuscript.
JZ, YW, JS and XF contributed to the search and analysis of
literature and the design of the manuscript. Data authentication is
not applicable. All authors read and approved the final version of
the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wiese AV, Duhn J, Korkmaz R, Quell KM,
Osman I, Ender F, Schröder T, Lewkowich I, Hogan S, Huber-Lang M,
et al: C5aR1 activation in mice controls inflammatory eosinophil
recruitment and functions in allergic asthma. Allergy.
78:1893–1908. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Khan MA, Nicolls MR, Surguladze B and
Saadoun I: Complement components as potential therapeutic targets
for asthma treatment. Respir Med. 108:543–549. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ali H and Panettieri RA Jr: Anaphylatoxin
C3a receptors in asthma. Respir Res. 6:192005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Khan MA, Maasch C, Vater A, Klussmann S,
Morser J, Leung LL, Atkinson C, Tomlinson S, Heeger PS and Nicolls
MR: Targeting complement component 5a promotes vascular integrity
and limits airway remodeling. Proc Natl Acad Sci USA.
110:6061–6066. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Trambas IA, Coughlan MT and Tan SM:
Therapeutic potential of targeting complement C5a receptors in
diabetic kidney disease. Int J Mol Sci. 24:87582023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Georg P, Astaburuaga-García R, Bonaguro L,
Brumhard S, Michalick L, Lippert LJ, Kostevc T, Gäbel C, Schneider
M, Streitz M, et al: Complement activation induces excessive T cell
cytotoxicity in severe COVID-19. Cell. 185:493–512.e25. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ricklin D, Reis ES and Lambris JD:
Complement in disease: A defence system turning offensive. Nat Rev
Nephrol. 12:383–401. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Morgan BP and Harris CL: Complement, a
target for therapy in inflammatory and degenerative diseases. Nat
Rev Drug Discov. 14:857–877. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang Y, Song Y, Wang X, Shi M, Lin Y, Tao
D and Han S: An NFAT1-C3a-C3aR positive feedback loop in
tumor-associated macrophages promotes a glioma stem cell malignant
phenotype. Cancer Immunol Res. 12:363–376. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Luan X, Lei T, Fang J, Liu X, Fu H, Li Y,
Chu W, Jiang P, Tong C, Qi H and Fu Y: Blockade of C5a receptor
unleashes tumor-associated macrophage antitumor response and
enhances CXCL9-dependent CD8+ T cell activity. Mol Ther.
32:469–489. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Miller KD, Nogueira L, Devasia T, Mariotto
AB, Yabroff KR, Jemal A, Kramer J and Siegel RL: Cancer treatment
and survivorship statistics, 2022. CA Cancer J Clin. 72:409–436.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yenyuwadee S, Aliazis K, Wang Q,
Christofides A, Shah R, Patsoukis N and Boussiotis VA: Immune
cellular components and signaling pathways in the tumor
microenvironment. Semin Cancer Biol. 86:187–201. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hu C, Qiao W, Li X, Ning ZK, Liu J,
Dalangood S, Li H, Yu X, Zong Z, Wen Z and Gui J: Tumor-secreted
FGF21 acts as an immune suppressor by rewiring cholesterol
metabolism of CD8+T cells. Cell Metab. 36:630–647.e8. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Durrant LG, Chapman MA, Buckley DJ,
Spendlove I, Robins RA and Armitage NC: Enhanced expression of the
complement regulatory protein CD55 predicts a poor prognosis in
colorectal cancer patients. Cancer Immunol Immunother. 52:638–642.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bulla R, Tripodo C, Rami D, Ling GS,
Agostinis C, Guarnotta C, Zorzet S, Durigutto P, Botto M and
Tedesco F: C1q acts in the tumour microenvironment as a
cancer-promoting factor independently of complement activation. Nat
Commun. 7:103462016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Corrales L, Ajona D, Rafail S, Lasarte JJ,
Riezu-Boj JI, Lambris JD, Rouzaut A, Pajares MJ, Montuenga LM and
Pio R: Anaphylatoxin C5a creates a favorable microenvironment for
lung cancer progression. J Immunol. 189:4674–4683. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Seol HS, Lee SE, Song JS, Rhee JK, Singh
SR, Chang S and Jang SJ: Complement proteins C7 and CFH control the
stemness of liver cancer cells via LSF-1. Cancer Lett. 372:24–35.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zha H, Wang X, Zhu Y, Chen D, Han X, Yang
F, Gao J, Hu C, Shu C, Feng Y, et al: Intracellular activation of
complement C3 leads to PD-L1 antibody treatment resistance by
modulating tumor-associated macrophages. Cancer Immunol Res.
7:193–207. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jackson SP, Darbousset R and Schoenwaelder
SM: Thromboinflammation: Challenges of therapeutically targeting
coagulation and other host defense mechanisms. Blood. 133:906–918.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang L, Li Z, Skrzypczynska KM, Fang Q,
Zhang W, O'Brien SA, He Y, Wang L, Zhang Q, Kim A, et al:
Single-cell analyses inform mechanisms of Myeloid-targeted
therapies in colon cancer. Cell. 181:442–459.e29. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Deng H, Chen Y, Liu Y, Liu L and Xu R:
Complement C1QC as a potential prognostic marker and therapeutic
target in colon carcinoma based on single-cell RNA sequencing and
immunohistochemical analysis. Bosn J Basic Med Sci. 22:912–922.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang Y, You K, You Y, Li Q, Feng G, Ni J,
Cao X, Zhang X, Wang Y, Bao W, et al: Paeoniflorin prevents
aberrant proliferation and differentiation of intestinal stem cells
by controlling C1q release from macrophages in chronic colitis.
Pharmacol Res. 182:1063092022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pouw RB and Ricklin D: Tipping the
balance: Intricate roles of the complement system in disease and
therapy. Semin Immunopathol. 43:757–771. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Afshar-Kharghan V: The role of the
complement system in cancer. J Clin Invest. 127:780–789. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Merle NS, Church SE, Fremeaux-Bacchi V and
Roumenina LT: Complement system part I-Molecular mechanisms of
activation and regulation. Front Immunol. 6:2622015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ling M and Murali M: Analysis of the
complement system in the clinical immunology laboratory. Clin Lab
Med. 39:579–590. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nesargikar PN, Spiller B and Chavez R: The
complement system: History, pathways, cascade and inhibitors. Eur J
Microbiol Immunol. 2:103–111. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hurler L, Toonen EJM, Kajdácsi E, van Bree
B, Brandwijk RJMGE, de Bruin W, Lyons PA, Bergamaschi L; Cambridge
Institute of Therapeutic Immunology and Infectious Disease-National
Institute of Health Research (CITIID-NIHR) COVID BioResource
Collaboration, ; Sinkovits G, et al: Distinction of early
complement classical and lectin pathway activation via
quantification of C1s/C1-INH and MASP-1/C1-INH complexes using
novel ELISAs. Front Immunol. 13:10397652022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hallam TM, Sharp SJ, Andreadi A and
Kavanagh D: Complement factor I: Regulatory nexus, driver of
immunopathology, and therapeutic. Immunobiology. 228:1524102023.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Song WC: Complement regulatory proteins
and autoimmunity. Autoimmunity. 39:403–410. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ghosh P, Sahoo R, Vaidya A, Chorev M and
Halperin JA: Role of complement and complement regulatory proteins
in the complications of diabetes. Endocr Rev. 6:272–288. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shah SC and Itzkowitz SH: Colorectal
cancer in inflammatory bowel disease: Mechanisms and management.
Gastroenterology. 162:715–730.e3. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang X, Wang J, Zhao J, Wang H, Chen J and
Wu J: HMGA2 facilitates colorectal cancer progression via
STAT3-mediated tumor-associated macrophage recruitment.
Theranostics. 12:963–975. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xu M, Wang S, Qi Y, Chen L, Frank JA, Yang
XH, Zhang Z, Shi X and Luo J: Role of MCP-1 in alcohol-induced
aggressiveness of colorectal cancer cells. Mol Carcinog.
55:1002–1011. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Habermann JK, Roblick UJ, Luke BT, Prieto
DA, Finlay WJ, Podust VN, Roman JM, Oevermann E, Schiedeck T,
Homann N, et al: Increased serum levels of complement C3a
anaphylatoxin indicate the presence of colorectal tumors.
Gastroenterology. 131:1020–1029. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nitta H, Wada Y, Kawano Y, Murakami Y,
Irie A, Taniguchi K, Kikuchi K, Yamada G, Suzuki K, Honda J, et al:
Enhancement of human cancer cell motility and invasiveness by
anaphylatoxin C5a via aberrantly expressed C5a receptor (CD88).
Clin Cancer Res. 19:2004–2013. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mehrabani D, Shamsdin SA, Dehghan A and
Safarpour A: Clinical significance of serum vascular endothelial
growth factor and complement 3a levels in patients with colorectal
cancer in southern Iran. Asian Pac J Cancer Prev. 15:9713–9717.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Krieg C, Weber LM, Fosso B, Marzano M,
Hardiman G, Olcina MM, Domingo E, El Aidy S, Mallah K, Robinson MD
and Guglietta S: Complement downregulation promotes an inflammatory
signature that renders colorectal cancer susceptible to
immunotherapy. J Immunother Cancer. 10:e0047172022. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Markiewski MM, DeAngelis RA, Benencia F,
Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, Coukos G and
Lambris JD: Modulation of the antitumor immune response by
complement. Nat Immunol. 9:1225–1235. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Piao C, Zhang WM, Li TT, Zhang CC, Qiu S,
Liu Y, Liu S, Jin M, Jia LX, Song WC and Du J: Complement 5a
stimulates macrophage polarization and contributes to tumor
metastases of colon cancer. Exp Cell Res. 366:127–138. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Piao C, Cai L, Qiu S, Jia L, Song W and Du
J: Complement 5a enhances hepatic metastases of colon cancer via
monocyte chemoattractant protein-1-mediated inflammatory cell
infiltration. J Biol Chem. 290:10667–10676. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xu D, Li M, Ran L, Li X, Sun X and Yin T:
C5aR1 promotes the progression of colorectal cancer by EMT and
activating Wnt/β-catenin pathway. Clin Transl Oncol. 25:440–446.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhu XL, Zhang L and Qi SX: Association of
complement components with risk of colorectal cancer: A systematic
review and meta-analysis. World J Gastrointest Oncol. 16:2168–2180.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Urbiola-Salvador V, Jabłońska A,
Miroszewska D, Kamysz W, Duzowska K, Drężek-Chyła K, Baber R,
Thieme R, Gockel I, Zdrenka M, et al: Mass spectrometry proteomics
characterization of plasma biomarkers for colorectal cancer
associated with inflammation. Biomark Insights.
19:117727192412577392024. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Talaat IM, Elemam NM and Saber-Ayad M:
Complement system: An immunotherapy target in colorectal cancer.
Front Immunol. 13:8109932022. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lin F, Spencer D, Hatala DA, Levine AD and
Medof ME: Decay-accelerating factor deficiency increases
susceptibility to dextran sulfate sodium-induced colitis: Role for
complement in inflammatory bowel disease. J Immunol. 172:3836–3841.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liu J, Fu N, Yang Z, Li A, Wu H, Jin Y,
Song Q, Ji S, Xu H, Zhang Z and Zhang X: The genetic and epigenetic
regulation of CD55 and its pathway analysis in colon cancer. Front
Immunol. 13:9471362022. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Dho SH, Cho EH, Lee JY, Lee SY, Jung SH,
Kim LK and Lim JC: A novel therapeutic anti-CD55 monoclonal
antibody inhibits the proliferation and metastasis of colorectal
cancer cells. Oncol Rep. 42:2686–2693. 2019.PubMed/NCBI
|
|
50
|
Nakagawa M, Mizuno M, Kawada M, Uesu T,
Nasu J, Takeuchi K, Okada H, Endo Y, Fujita T and Tsuji T:
Polymorphic expression of decay-accelerating factor in human
colorectal cancer. J Gastroenterol Hepatol. 16:184–189. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bao D, Zhang C, Li L, Wang H, Li Q, Ni L,
Lin Y, Huang R, Yang Z, Zhang Y and Hu Y: Integrative analysis of
complement system to prognosis and immune infiltrating in colon
cancer and gastric cancer. Front Oncol. 10:5532972020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tang G, Pan L, Wang Z, Zhu H, Yang Y, Wang
Z, Yue H, Shi Y, Wu D, Jiang Z and Jiang D: Knockdown of
membrane-bound complement regulatory proteins suppresses colon
cancer growth in mice through inducing tumor cell apoptosis. Int
Immunopharmacol. 114:1094502023. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Du YJ, Jiang Y, Hou YM and Shi YB:
Complement factor I knockdown inhibits colon cancer development by
affecting Wnt/β-catenin/c-Myc signaling pathway and glycolysis.
World J Gastrointest Oncol. 16:2646–2662. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wilczek E, Rzepko R, Nowis D, Legat M,
Golab J, Glab M, Gorlewicz A, Konopacki F, Mazurkiewicz M,
Sladowski D, et al: The possible role of factor H in colon cancer
resistance to complement attack. Int J Cancer. 122:2030–2037. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Fishelson Z and Kirschfink M: Complement
C5b-9 and cancer: Mechanisms of cell damage, cancer counteractions,
and approaches for intervention. Front Immunol. 10:7522019.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Reis ES, Mastellos DC, Ricklin D,
Mantovani A and Lambris JD: Complement in cancer: Untangling an
intricate relationship. Nat Rev Immunol. 18:5–18. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Watson NF, Durrant LG, Madjd Z, Ellis IO,
Scholefield JH and Spendlove I: Expression of the membrane
complement regulatory protein CD59 (protectin) is associated with
reduced survival in colorectal cancer patients. Cancer Immunol
Immunother. 55:973–980. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Bjørge L, Vedeler CA, Ulvestad E and Matre
R: Expression and function of CD59 on colonic adenocarcinoma cells.
Eur J Immunol. 24:1597–1603. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ding P, Li L, Huang T, Yang C, Xu E, Wang
N, Zhang L, Gu H, Yao X, Zhou X and Hu W: Complement component 6
deficiency increases susceptibility to dextran sulfate
sodium-induced murine colitis. Immunobiology. 221:1293–1303. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Vlaicu SI, Tatomir A, Rus V and Rus H:
Role of C5b-9 and RGC-32 in cancer. Front Immunol. 10:10542019.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Stefani C, Miricescu D, Stanescu-Spinu II,
Nica RI, Greabu M, Totan AR and Jinga M: Growth factors,
PI3K/AKT/mTOR and MAPK signaling pathways in colorectal cancer
pathogenesis: Where are we now? Int J Mol Sci. 22:102602021.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Towner LD, Wheat RA, Hughes TR and Morgan
BP: Complement membrane attack and tumorigenesis: A systems biology
approach. J Biol Chem. 291:14927–14938. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Vlaicu SI, Tegla CA, Cudrici CD, Fosbrink
M, Nguyen V, Azimzadeh P, Rus V, Chen H, Mircea PA, Shamsuddin A
and Rus H: Epigenetic modifications induced by RGC-32 in colon
cancer. Exp Mol Pathol. 88:67–76. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Tian J, Xu C, Yang MH and Li ZG:
Overexpression of response gene to complement-32 promotes
cytoskeleton reorganization in SW480 cell line. Nan Fang Yi Ke Da
Xue Xue Bao. 31:1179–1182. 2011.(In Chinese). PubMed/NCBI
|
|
65
|
Liszewski MK, Kolev M, Le Friec G, Leung
M, Bertram PG, Fara AF, Subias M, Pickering MC, Drouet C, Meri S,
et al: Intracellular complement activation sustains T cell
homeostasis and mediates effector differentiation. Immunity.
39:1143–1157. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Ding P, Xu Y, Li L, Lv X, Li L, Chen J,
Zhou D, Wang X, Wang Q, Zhang W, et al: Intracellular complement
C5a/C5aR1 stabilizes β-catenin to promote colorectal tumorigenesis.
Cell Rep. 39:1108512022. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Arbore G, West EE, Rahman J, Le Friec G,
Niyonzima N, Pirooznia M, Tunc I, Pavlidis P, Powell N, Li Y, et
al: Complement receptor CD46 co-stimulates optimal human CD8+ T
cell effector function via fatty acid metabolism. Nat Commun.
9:41862018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Tam JC, Bidgood SR, McEwan WA and James
LC: Intracellular sensing of complement C3 activates cell
autonomous immunity. Science. 345:12560702014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Liu Y and Wang X: Tumor
microenvironment-associated gene C3 can predict the prognosis of
colorectal adenocarcinoma: A study based on TCGA. Clin Transl
Oncol. 23:1923–1933. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Nandagopal S, Li CG, Xu Y, Sodji QH,
Graves EE and Giaccia AJ: C3aR signaling inhibits NK-cell
infiltration into the tumor microenvironment in mouse models.
Cancer Immunol Res. 10:245–258. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Arbore G, West EE, Spolski R, Robertson
AAB, Klos A, Rheinheimer C, Dutow P, Woodruff TM, Yu ZX, O'Neill
LA, et al: T helper 1 immunity requires complement-driven NLRP3
inflammasome activity in CD4+ T cells. Science.
352:aad12102016. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Wang Y, Zhang H and He YW: The complement
receptors C3aR and C5aR are a new class of immune checkpoint
receptor in cancer immunotherapy. Front Immunol. 10:15742019.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Medler TR, Murugan D, Horton W, Kumar S,
Cotechini T, Forsyth AM, Leyshock P, Leitenberger JJ, Kulesz-Martin
M, Margolin AA, et al: Complement C5a fosters squamous
carcinogenesis and limits T cell response to chemotherapy. Cancer
Cell. 34:561–578.e6. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Ghebrehiwet B, Hosszu KH and Peerschke EI:
C1q as an autocrine and paracrine regulator of cellular functions.
Mol Immunol. 84:26–33. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Ghebrehiwet B, Kandov E, Kishore U and
Peerschke EIB: Is the A-chain the engine that drives the diversity
of C1q functions? Revisiting its unique structure. Front Immunol.
9:1622018. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Bossi F, Tripodo C, Rizzi L, Bulla R,
Agostinis C, Guarnotta C, Munaut C, Baldassarre G, Papa G, Zorzet
S, et al: C1q as a unique player in angiogenesis with therapeutic
implication in wound healing. Proc Natl Acad Sci USA.
111:4209–4214. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Chen LH, Liu JF, Lu Y, He XY, Zhang C and
Zhou HH: Complement C1q (C1qA, C1qB, and C1qC) may be a potential
prognostic factor and an index of tumor microenvironment remodeling
in osteosarcoma. Front Oncol. 11:6421442021. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Earley AM, Graves CL and Shiau CE:
Critical role for a subset of intestinal macrophages in shaping gut
microbiota in adult zebrafish. Cell Rep. 25:424–436. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Revel M, Sautès-Fridman C, Fridman WH and
Roumenina LT: C1q+ macrophages: Passengers or drivers of cancer
progression. Trends Cancer. 8:517–526. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Roumenina LT, Daugan MV, Noé R, Petitprez
F, Vano YA, Sanchez-Salas R, Becht E, Meilleroux J, Clec'h BL,
Giraldo NA, et al: Tumor cells hijack Macrophage-produced
complement C1q to promote tumor growth. Cancer Immunol Res.
7:1091–1105. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Guinney J, Dienstmann R, Wang X, de
Reyniès A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda
G, Angelino P, et al: The consensus molecular subtypes of
colorectal cancer. Nat Med. 21:1350–1356. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Dienstmann R, Vermeulen L, Guinney J,
Kopetz S, Tejpar S and Tabernero J: Consensus molecular subtypes
and the evolution of precision medicine in colorectal cancer. Nat
Rev Cancer. 17:79–92. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Downs-Canner S, Magge D, Ravindranathan R,
O'Malley ME, Francis L, Liu Z, Sheng Guo Z, Obermajer N and
Bartlett DL: Complement inhibition: A novel form of immunotherapy
for colon cancer. Ann Surg Oncol. 23:655–662. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Ding P, Li L, Li L, Lv X, Zhou D, Wang Q,
Chen J, Yang C, Xu E, Dai W, et al: C5aR1 is a master regulator in
colorectal tumorigenesis via immune modulation. Theranostics.
10:8619–8632. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Zelek WM, Xie L, Morgan BP and Harris CL:
Compendium of current complement therapeutics. Mol Immunol.
114:341–35. 20192 View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Sheridan D, Yu ZX, Zhang Y, Patel R, Sun
F, Lasaro MA, Bouchard K, Andrien B, Marozsan A, Wang Y and
Tamburini P: Design and preclinical characterization of ALXN1210: A
novel anti-C5 antibody with extended duration of action. PLoS One.
13:e01959092018. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
van der Worp HB, Howells DW, Sena ES,
Porritt MJ, Rewell S, O'Collins V and Macleod MR: Can animal models
of disease reliably inform human studies? PLoS Med. 7:e10002452010.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Horvath P, Aulner N, Bickle M, Davies AM,
Nery ED, Ebner D, Montoya MC, Östling P, Pietiäinen V, Price LS, et
al: Screening out irrelevant cell-based models of disease. Nat Rev
Drug Discov. 15:751–769. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Gengenbacher N, Singhal M and Augustin HG:
Preclinical mouse solid tumour models: Status quo, challenges and
perspectives. Nat Rev Cancer. 17:751–765. 2017. View Article : Google Scholar : PubMed/NCBI
|














