Introduction
Insulin receptor (IR) tyrosine kinase substrate
(IRTKS), also known as brain-specific angiogenesis inhibitor
1-associated protein 2 (BAIAP2)-like 1 (BAIAP2L1), was first
identified as a phosphorylation substrate for the IR (1,2). The
human IRTKS gene was initially cloned from endocrine organs
(GenBank accession number, AF119666.2) and is located on chromosome
7 at q21.3-q22.1 (gene ID, 55971). The full-length human IRTKS is
109,441 bp (NC_000007.14) and includes 14 exons. Its coding region
comprises 1,536 bp, which encodes a 511-amino acid protein
(GenBank, AAF17223.2) with a molecular weight of ~57 kDa (1,3–5).
Human IRTKS is widely distributed in various tissues, with broad
expression in the stomach, colon, duodenum, prostate, skin, thyroid
and lung, and trace amounts in the spleen, ovary, brain and bone
marrow (4).
I-BAR family
IRTKS is a member of the Bin-amphiphysin-Rvs (BAR)
superfamily. The members of this family share the defining element
of a BAR domain, which is a coiled structure that dimerizes into
modules with membrane-binding and curvature-sensing abilities
(6). The BAR domain protein was
initially identified in the mammalian proteins Bin1 and
amphiphysin, as well as the budding yeast Rvs167 protein (7). Based on crystal structures, the BAR
domain superfamily includes several classes, including the
classical BARs, the Fes/CIP4 homology-BARs (F-BARs) and the
inverse-BARs (I-BARs) (8). These
members differ in their effects on cell membrane remodeling
(9). The classical BAR and F-BAR
domains drive the formation of positive membrane curvature by
forming banana-shaped α-helical dimers, which facilitate the
formation of plasma membrane invaginations (8,10).
However, the I-BAR domain forms a zeppelin-shaped structure that
binds to phosphoinositide-rich membranes and generates negative
membrane curvature in a phosphatidylinositol
4,5-bisphosphate-dependent manner, thereby inducing plasma membrane
protrusions such as filopodia and lamellipodia (6,7).
There are five genes in the I-BAR family, namely
IRSp53 (BAIAP2), missing-in-metastasis (MIM), IRTKS, FLJ22582 (also
known as BAIAP2-like 2) and actin-binding protein ABBA, that encode
homologous N-terminal sequences and were identified via human
genome alignment (3). The
N-terminal helical domains of the I-BAR proteins IRSp53 and MIM
were the first shown to be evolutionarily conserved; hence, the
I-BAR domain was originally named the IRSp53/MIM homology domain
(IMD) (3,7).
In addition to the N-terminal IMD, the human I-BAR
family proteins each possess a Wiskott-Aldrich syndrome protein
(WASP)-homology 2 (WH2) domain in the C-terminus. The IRSp53, IRTKS
and FLJ22582 proteins are distinctive in that they contain a
canonical Src homology 3 (SH3) domain in the C-terminal half;
therefore, these proteins are classified into the IRSp53 subfamily,
primarily because IRSp53 was studied early and extensively. IRTKS
and IRSp53 each have a WW domain-binding motif, which is in
agreement with their close genetic relationship, with ~40.9%
similarity (1,7) (Fig.
1). Therefore, the comparative study of these two proteins is a
major topic in IRTKS research.
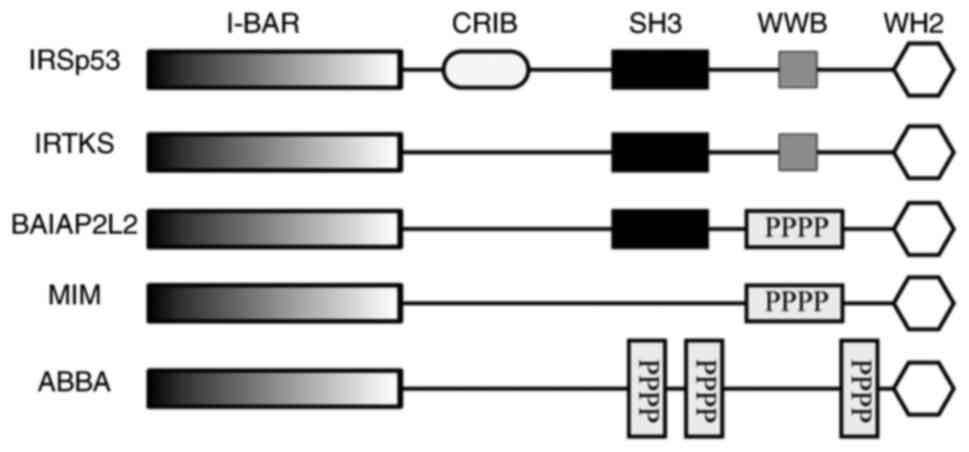 | Figure 1.Structures of I-BAR family members.
ABBA, actin-bundling protein with BAIAP2 homology; CRIB,
Cdc42⁄Rac-interactive-binding domain; FLJ22582 (BAIAP2L2),
brain-specific angiogenesis inhibitor 1-associated protein 2-like
2; I-BAR, inverse Bin-amphiphysin-Rvs domain; IRSp53, insulin
receptor tyrosine kinase substrate p53; IRTKS, insulin receptor
tyrosine kinase substrate; MIM, missing-in-metastasis; PPPP,
proline-rich region; SH3, Src homology 3 domain; WH2,
Wiskott-Aldrich syndrome protein homology 2 domain; WWB, WW binding
domain. |
IRTKS and cellular morphology
A number of cellular processes, including cell
migration, phagocytosis and axon pathfinding, are dependent on
membrane deformation and cytoskeletal rearrangement, which are
regulated by an array of signaling complexes that control actin
assembly. As already noted, I-BAR domains, as sensors of membrane
curvature, can induce membrane bending and anisotropic cytoskeletal
rearrangement (1,7,10).
Cytoskeletal rearrangements involve multiple cell
types and are regulated by various biological molecules.
Extracellular signals converge on small GTPases, which activate
downstream effectors such as the WASP family verprolin-homologous
protein (WAVE) complex, which stimulates the actin-related protein
2/3 (Arp2/3) complex. This complex nucleates new actin filaments,
leading to the branching and crosslinking of preexisting filaments
(11–13). Capping proteins bind to the barbed ends of filaments,
which terminates filament growth and stabilizes the filament
structure (14). By contrast,
enabled (Ena)/vasodilator-stimulated phosphoprotein (VASP) proteins
prevent filament capping, resulting in the elongation of actin
filaments (15,16), and diaphanous-related formin 2
promotes fibrous actin (F-actin) elongation (17). In addition, numerous actin-bundling
and crosslinking proteins are crucial for stabilizing actin
filaments and for facilitating the extension of membrane
projections at the cell periphery (18,19).
IRSp53, which shares 40.9% sequence homology with IRTKS, interacts
with Rho-GTPases Rac and cell division cycle 42 (Cdc42) (1). This interaction, in collaboration
with various proteins, such as mammalian enabled, an Ena/VASP
family member (20), the
actin-bundling/capping protein known as epidermal growth factor
receptor (EGFR) kinase substrate 8 (Eps8) (21), WAVE1/2 (22–24), mammalian
diaphanous isoforms 1/2 (23,25)
and dynamin 1 (26), promotes the
formation of filopodia (1,4,27,28).
IRTKS-overexpressing HT1080 cells have been
demonstrated to spread more than their parent cells, and to display
F-actin clusters and the formation of lamellipodia, which
demonstrates the role of IRTKS in cellular morphology (29). Studies have also shown that IRTKS
plays a role in cytoskeletal rearrangement, membrane tubulation and
the formation of protrusions, by I-BAR- and WH2 domain-dependent
mechanisms (1,2,27,30).
Rho, Rac and Cdc42 are small GTPases of the Rho
family, which play roles in the formation and rearrangement of
actin stress fibers (31,32). Millard et al (1) revealed that IRTKS interacts with Rac,
but not Cdc42, via its I-BAR domain, while its SH3 domain has an
autoregulatory function in the control of I-BAR activity. Moreover,
the ectopic expression of IRSp53 and IRTKS causes distinct changes
in actin cytoskeletal organization. In COS7 cells, IRSp53 induces
the formation of numerous long, wavy filopodia-like extensions,
while low levels of IRTKS expression result in small actin
microspikes at the cell periphery. By contrast, higher levels of
IRTKS expression lead to the formation of clusters of short actin
bundles around the cell periphery, rather than the development of
filopodia (1,33).
Sudhaharan et al (27) established that the Rho in filopodia
(Rif)-IRTKS-Eps8-WAVE2 pathway promotes the formation of dorsal
filopodia and membrane ruffles via a coexpression system comprising
IRTKS and its interacting partners in cancer cells. In this
pathway, the Rho GTPase Rif interacts with IRTKS via its I-BAR
domain in the GTP and GDP forms, and Eps8 and WAVE2 act as
downstream modulators. Specifically, Eps8 reduces the size and
increases the number of dorsal filopodia and membrane ruffles,
while WAVE2 modulates the activity of dorsal membrane ruffling
(Fig. 2) (27). In addition, the study reported that
IRTKS did not interact with Cdc42, RhoA or Rac (27), which differs from the study by
Millard et al (1), which
reported that IRTKS binds with Rac.
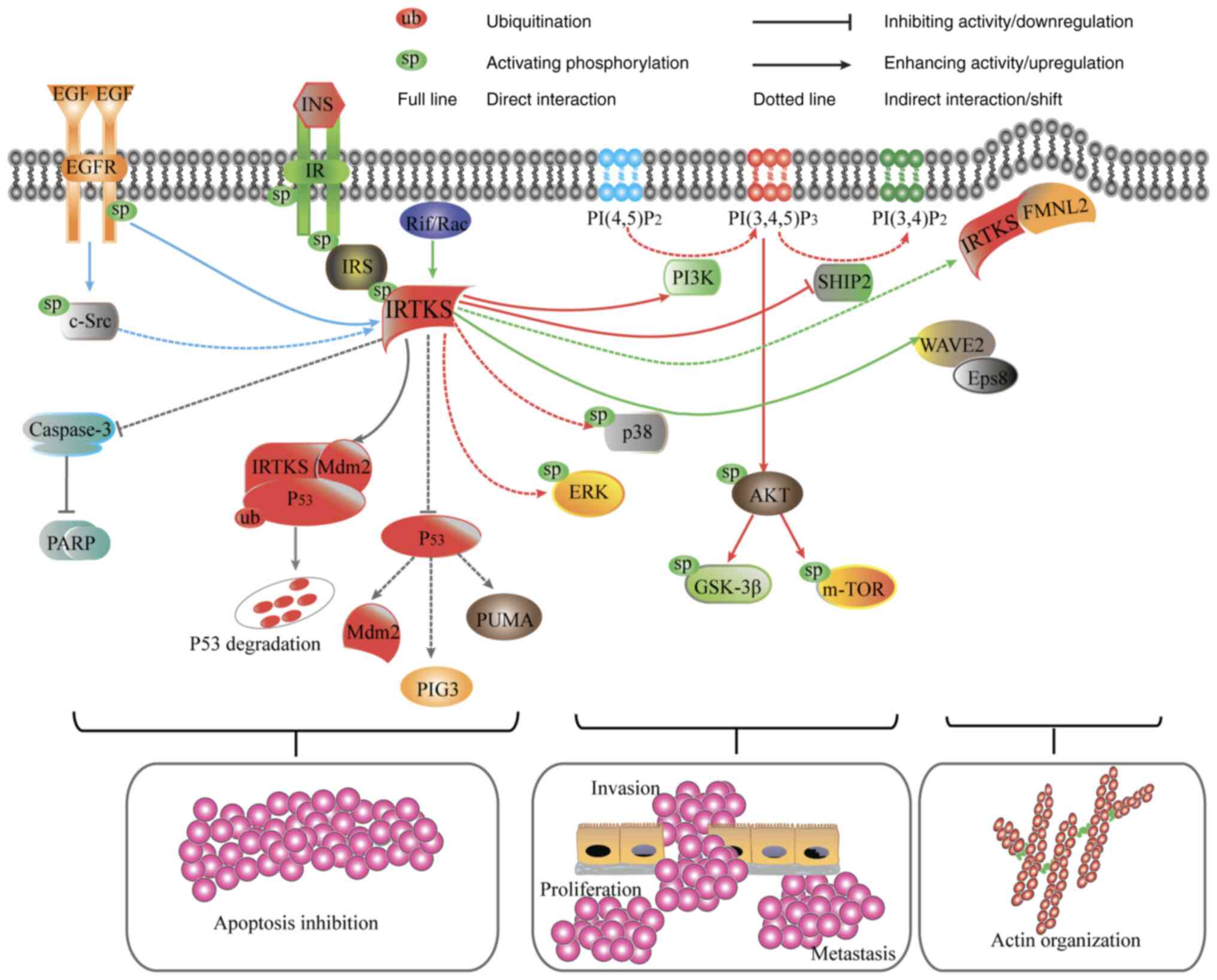 | Figure 2.IRTKS-associated signaling pathways.
IRTKS positively regulates IR-IRS-PI3K-AKT signaling.
Overexpression of IRTKS increases the phosphorylation levels of
ERK, p38, AKT, mTOR and GSK-3β but attenuates SHIP2 activity (red
lines). IRTKS inhibits the caspase-3-dependent apoptosis pathway
and restricts the activity of p53, promotes IRTKS-Mdm2-p53 complex
formation and thereby increases Mdm2-mediated p53 ubiquitination
and degradation in unstressed cells (gray lines). IRTKS also
initiates and maintains actin organization by interacting with Rif,
Eps8, WAVE2 and FMNL2 (green lines). IRTKS activity is modulated by
Src/EGFR (blue lines). EGF, epidermal growth factor; EGFR, EGF
receptor; Eps8, EGFR kinase substrate 8; FMNL2, formin-like 2;
GSK-3β, glycogen synthase kinase-3β; INS, insulin; IR, insulin
receptor; IRTKS, IR tyrosine kinase substrate; IRS, IR tyrosine
kinase substrate; Mdm2, mouse double minute 2 homolog; mTOR,
mammalian target of rapamycin; PARP, poly (ADP-ribose) polymerase;
PI3K, phosphatidylinositol 3-kinase; PI(3,4)/(4,5)P2,
phospatidylinositol 3,4/4,5-diphosphate; PI(3,4,5)P3,
phosphatidylinositol triphosphate; PIG3, p53-inducible gene 3;
PUMA, p53 upregulated modulator of apoptosis; Rif, Rho in
filopodia; SHIP2, SH2 containing inositol polyphosphate
5-phosphatase-2; WAVE2, Wiskott-Aldrich syndrome protein family
verprolin-homologous protein 2. |
Formins are multidomain proteins that act as actin
polymerization factors. Formin-like 2 (FMNL2) is associated with
filopodium formation in multiple cell types (34,35).
Fox et al (30) recently
identified IRTKS as a novel FMNL2-binding protein in melanoma
cells, and proposed an updated hierarchical model based on the
interdependence of IRTKS and FMNL2 in filopodia assembly. In this
model, FMNL2 first docks to the plasma membrane via
N-myristoylation and initiates membrane bending. IRTKS is then
recruited to these membrane-bending sites and remains anchored to
the membrane via its interaction with FMNL2. The two proteins
mutually facilitate actin polymerization, leading to the initiation
of filopodia assembly and growth (Fig.
2) (30).
Notably, the widely accepted notion that the
outwardly curved I-BAR domain induces protrusions has also been
challenged (36,37). Veltman et al (36) reported that in the amoeba
Dictyostelium, the I-BARa protein, which contains a single
I-BAR/SH3 domain similar to that of mammalian IRSp53 family
proteins, is involved in clathrin-mediated endocytosis rather than
the formation of actin-driven protrusions. This suggests that the
physiological function of the I-BAR domain is not limited to
protrusion formation, and further extends the functional areas of
I-BAR domain proteins such as IRTKS.
IRTKS and pathogen-driven actin
assembly
IRTKS and microvilli
Microvilli, also known as brush borders, are
evolutionarily ancient cell surface protrusions that fulfill
diverse functions in epithelial cells, enabling them to move and
sense the surrounding environment. The growth, elongation,
directional motility and collapse of microvilli are supported by
actin bundles, with IRTKS and its binding partner Eps8 playing
important roles in these processes (38–40). The two crucial
proteins are located at the epithelial cell surface, where they
mark future sites of microvillus growth (38). They together promote the elongation
of microvilli (38) and help to
maintain the directed motion of nascent microvilli during early
differentiation (39); moreover,
the sharp loss of Eps8 or IRTKS from the distal tip destabilizes
nascent microvilli, leading to their collapse (40). Postema et al (38) suggested two distinct mechanisms by
which IRTKS promotes microvillar elongation: Eps8-dependent and
Eps8-independent mechanisms. In the Eps8-dependent mechanism, IRTKS
uses its SH3 domain to promote Eps8 enrichment at the microvillar
tips to drive microvillar elongation, while in the Eps8-independent
mechanism, IRTKS promotes elongation via a direct mechanism
involving its actin-binding WH2 domain.
While a temporal molecular framework for
understanding the assembly and dynamics of new microvilli is
currently available, the mechanisms regulating the number of actin
filaments per core bundle and the initial recruitment of distal
tip-enriched factors to the plasma membrane remain unclear
(40).
IRTKS and intestinal infections
Intestinal microvilli are tiny, finger-like
projections on the apical end of enterocytes, that facilitate the
digestion and absorption of nutrients, while also providing a
barrier against luminal pathogens and toxins (38,41).
The destruction of microvilli by pathogenic microbes can lead to
nutrient malabsorption and osmotic imbalances and may even be
life-threatening (42).
Enterohemorrhagic E. coli (EHEC), particularly the O157:H7
serotype, is an important human pathogen causing diarrheal and
systemic diseases. A hallmark of EHEC infections is epithelial
attaching and effacing (A/E) lesions, which are characterized by
microvilli effacement, actin assembly and the formation of
‘pedestal’ structures beneath the host cell membrane at sites of
bacterial attachment (43).
A number of studies have shown that, by targeting
the A/E lesion machinery, the EHEC serotype O157:H7 triggers
intestinal colonization via two essential effector proteins, namely
translocated intimin receptor (Tir) and E. coli-secreted
protein F-like protein encoded on prophage U (EspFU) (33,44–46).
‘Type III’ secretion systems are complex bacterial structures that
provide pathogenic bacteria with a unique virulence mechanism
enabling them to inject bacterial effector proteins directly into
host cells. These effector molecules manipulate host cells and
contribute to a number of different infectious diseases (47,48).
Through the ‘type III’ secretion system, Tir is translocated into
host cells, localizes at sites of bacterial attachment, and
initiates a signaling cascade upon binding to intimin, which leads
to actin assembly and ‘pedestal’ formation.
IRSp53 and IRTKS play important roles in the
pathogenesis of EHEC O157:H7 (49). The N-terminal I-BAR of IRTKS binds
to Tir in a manner dependent on the Asn-Pro-Tyr 458 (NPY458)
sequence of the latter; in addition, the C-terminal SH3 domain of
IRTKS interacts with the C-terminal tandem PxxP motifs of EspFU,
which results in EspFU recruitment (50,51).
Therefore, IRTKS physically links the two EHEC effectors, Tir and
EspFU (49–51). In host cells, the N-terminal repeat sequence of
EspFU binds to the GTPase binding domain (GBD) of neuronal WASP
(N-WASP). This activates N-WASP by destabilizing its interaction
with an inhibitory protein. Subsequently, activated N-WASP
stimulates the Arp2/3 actin nucleator complex, which promotes actin
polymerization (Fig. 3).
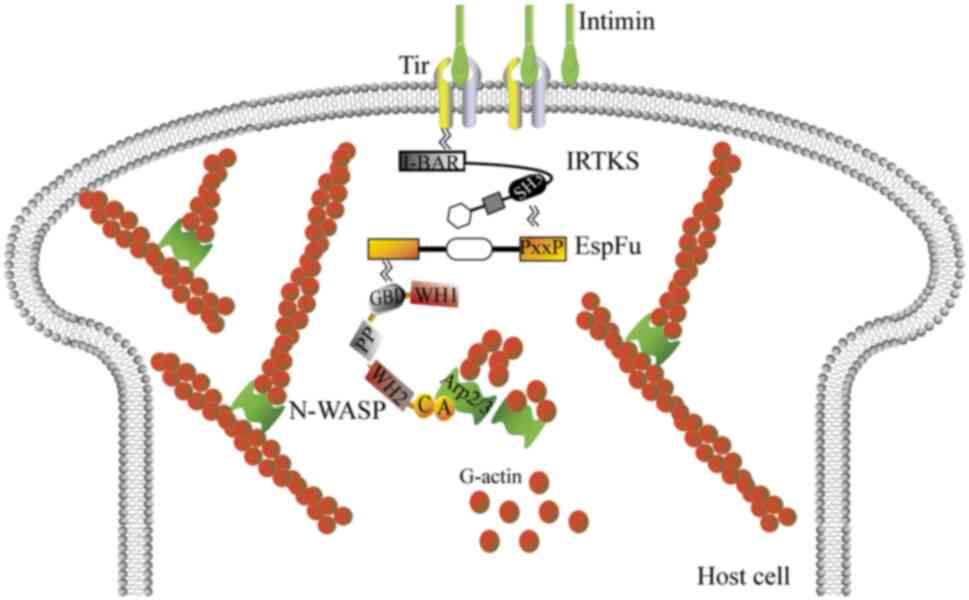 | Figure 3.IRTKS initiates and maintains actin
pedestal assembly in E. coli O157:H7. IRTKS links with the
actin assembly effectors of E. coli O157:H7, including Tir,
EspFU, WASP and Arp2/3. This leads to actin polymerization and
subsequently generates ‘pedestals’ beneath bound bacteria. A,
acidic peptide; ARP2/3, actin-related protein 2/3; C, connector
region; EspFU, E. coli-secreted protein F-like protein
encoded on prophage U; G-actin, globular actin; GBD, GTPase binding
domain; I-BAR, inverse Bin-amphiphysin-Rvs; IRTKS, insulin receptor
tyrosine kinase substrate; N-WASP, neuronal WASP; PP, proline-rich
domain; SH3, Src homology 3 domain; Tir, translocated intimin
receptor; WASP, Wiskott-Aldrich syndrome protein; WH1/2,
WASP-homology 1/2 domain; WH2CA, WH2 connector acidic region. |
In summary, IRTKS links Tir and the
EspFU:GBDWASP/N-WASP:Arp2/3 complex, which results in
recruitment of the ternary complex and thus actin polymerization,
subsequently generating ‘pedestals’ beneath bound bacteria
(50–52).
IRTKS and insulin signaling
Tyrosine phosphorylation of IRTKS in response to
insulin stimulation was observed in COS-7 cells cotransfected with
IRTKS and the IR β-subunit, which led to the initial identification
of IRTKS as a substrate for the IR by Millard et al
(1). These results suggest that
IRTKS is associated with insulin signaling. However, following the
identification of IRTKS in mammals, its role in regulating the
formation of membrane protrusions and triggering pathogen-driven
actin assembly attracted considerable attention, and less attention
was given to its role in insulin signaling.
A study by Huang et al (53) found that IRTKS-knockout mice
display characteristics of insulin resistance, including
hyperglycemia, hyperinsulinemia and glucose intolerance, which can
be reversed by ectopic IRTKS expression. Furthermore, the study
demonstrated that the phosphorylation of key molecules in the
insulin signaling pathway, including IR, AKT and glycogen synthase
kinase-3β (GSK-3β), is attenuated in response to IRTKS knockout or
knockdown in vivo and in vitro, and this defective
insulin signaling can be reversed by ectopic IRTKS. These findings
indicate that IRTKS, as an adaptor of the IR, positively regulates
insulin signaling (53). In
addition, the study revealed that the expression of IRTKS in the
livers of diabetic patients and mice is downregulated. Moreover,
DNA hypermethylation of the human IRTKS promoter, which is
associated with the downregulation of IRTKS expression in diabetes,
was detected via bisulfite-treatment DNA sequencing (53). An increase in the phosphorylation
level of extracellular regulated protein kinase (ERK) was also
reported to occur following IRTKS overexpression (53).
SH2 containing inositol polyphosphate
5-phosphatase-2 (SHIP2) is a potent negative regulator of
phosphatidylinositol 3-kinase (PI3K)-AKT signaling. In 2019, Wu
et al (54) revealed that
IRTKS interacts with SHIP2 in human liver cancer cell lines via the
SH3 domain of IRTKS and the inositol polyphosphate-5-phosphatase
catalytic domain of SHIP2. In addition, they demonstrated that
phosphorylated active IRTKS attenuates the activity of SHIP2, and
thereby suppresses the conversion of phosphatidylinositol
3,4,5-triphosphate (PIP3) to phosphatidylinositol
3,4-bisphosphate, resulting in PIP3 accumulation
(54). The study also found that
in addition to directly inhibiting SHIP2 activity, IRTKS
overexpression also evokes the phosphorylation of AKT, mammalian
target of rapamycin (mTOR) and GSK-3β, which partially relieves the
inhibitory effect of SHIP2 on insulin downstream molecules and
promotes cancer cell proliferation (54) (Fig.
2).
These findings indicate that IRTKS, by promoting IR
phosphorylation and inhibiting SHIP2 activity, modulates the
IR-IRS-PI3K-AKT signaling pathway, which may affect glucose and
lipid metabolism, and even cell growth and proliferation.
Consequently, the expression and phosphorylation of IRTKS, the
methylation status of the IRTKS gene, and IRTKS-interacting
proteins, may have therapeutic potential against diabetes, cancer
and related conditions.
IRTKS and tumors
IRTKS promotes the proliferation of
tumor cells
There is a considerable quantity of literature
reporting that IRTKS, as a scaffold protein, interacts with a
variety of cancer-related gene products, thereby controlling cancer
cell growth, differentiation, apoptosis and motility. The
expression of IRTKS is upregulated in numerous types of human
cancer tissues compared with that in adjacent normal tissues,
including hepatocellular carcinoma (HCC), ovarian cancer (OC),
colorectal cancer (CRC), gastric cancer (GC) and pancreatic cancer
(55–59). It has been shown that the knockdown of endogenous IRTKS
inhibits the proliferation of these cancer cells, whereas the
ectopic expression of IRTKS promotes their proliferation
(54–59).
An integrative database analysis of IRTKS revealed
that in OC tissues and cell lines, the level of IRTKS upregulation
does not differ among various cell types, stages or grades of each
histologic subtype (55). However,
in HCC the upregulation of IRTKS expression is significantly
associated with tumor volume and patient age, although not with
sex, hepatitis B surface antigen status, differentiation or
tumor-node-metastasis stage (58).
In addition, IRTKS can interact with EGFR and positively regulate
the EGFR-ERK signaling pathway in HCC cells, resulting in increased
proliferation, which is also associated with an increase in the
G1-to-S transition of the cell cycle (58). Another regulatory mechanism of
IRTKS in HCC cells involves its interaction with SHIP2, which
attenuates SHIP2 activity and leads to the accumulation of
PIP3, a substrate of SHIP2 (54). This process activates the
phosphorylation of downstream insulin signaling molecules,
including AKT, mTOR and GSK-3β (Fig.
2). Consequently, this process also promotes cell proliferation
(54).
In CRC cells, the overexpression of IRTKS promotes
basic fibroblast growth factor-induced cell proliferation via the
phosphorylation of AKT; however, no significant correlation was
detected between IRTKS and SHIP2 via database analysis (55). However, other research has shown
that IRTKS-overexpressing HCC cells do not exhibit changes in the
phosphorylated AKT/AKT ratio compared with that in control HCC
cells (58). Therefore, the
mechanism by which IRTKS triggers cell proliferation remains
unclear, and more studies with different cell types, stress factors
or stress durations are required to elucidate the dynamic
regulation of IRTKS-related signaling pathways.
IRTKS promotes tumor invasion and
metastasis
Membrane fusion between tumor cells and neighboring
tissues is a fundamental biological process of tumor cell invasion
(60). A study revealed that in
RAW264.7 macrophages, IRTKS is induced in response to stimulation
with receptor activator of NF-κB ligand, which contributes to
osteoclastogenesis. In addition, the study indicated that IRTKS
interacts with Talin domain-containing protein kinase substrate 5
(Tks5), a known regulator of invadopodia in cancer cells, via
different SH3 sites on IRTKS (61). Therefore, IRTKS may drive
osteoclast-osteoclast fusion as well as osteoclast-cancer cell
fusion in concert with Tks5 (61).
Actin polymerization, cytoskeletal remodeling and
membrane deformation are important for cell mobility (62). IRTKS is involved in actin dynamics
and the development of membrane protrusions, which implies that
IRTKS contributes to tumor invasion and metastasis. Notably, it has
been observed that in cases of OC, the level of IRTKS expression in
metastatic sites is higher than that in primary cancers (56). In HeLa cells, in addition to
increasing the phosphorylation of ERK1/2 and p38 and activation of
the small GTPases Rac1 and Cdc42, IRTKS also promotes the
chemotactic response to serum and increases cellular polarity, as
observed by an elongated cytoplasm and increased numbers of
lamellipodia at the leading edges of the cells (63). Importantly, the SH3 domain of IRTKS
is key to the process of IRTKS-mediated cell migration associated
with p38 phosphorylation and cytomembrane polarity (63).
Src functions as both a substrate and an upstream
activator of receptor tyrosine kinases, which regulate various
cellular functions, including adhesion, migration and invasion
(64,65). Notably, it has been shown that
IRTKS promotes the motility of HT1080 fibrosarcoma cells in a
Src-dependent manner, with the tyrosine phosphorylation of IRTKS by
c-Src or EGFR contributing to this process (Fig. 2) (65).
IRTKS suppresses the apoptosis of
tumor cells
IRTKS is considered to act as an inhibitor of
apoptosis during cancer progression. It prevents cell apoptosis
through two key mechanisms: Inhibition of the caspase-3-dependent
apoptosis pathway and suppression of p53 activity (56,57,66,67).
The knockdown of IRTKS in OC cells treated with ultraviolet
irradiation or cisplatin increases the protein levels of cleaved
caspase-3 and poly (ADP-ribose) polymerase (56), which are key indicators of the
execution phase of apoptosis (68). This suggests that IRTKS may protect
cells from apoptosis by blocking the caspase-3-dependent apoptosis
pathway (Fig. 2) (56).
p53 protects against genomic instability and
oncogene expression via the induction of cell cycle arrest and
apoptosis (69). Mouse double
minute 2 homolog (Mdm2) regulates p53 levels and nuclear export in
a concentration-dependent manner: High levels of Mdm2 promote p53
polyubiquitination and proteasomal degradation, whereas low levels
of Mdm2 mediate the monoubiquitination and cytoplasmic localization
of p53 (62). In unstressed cells
with low levels of Mdm2, ectopic IRTKS inhibits the transactivating
effect of p53 on its downstream genes, such as p53-inducible gene
3, p53 upregulated modulator of apoptosis and Mdm2. Simultaneously,
IRTKS promotes the interaction between p53 and Mdm2, leading to the
induction of p53 ubiquitination and cytoplasmic localization.
Ultimately, IRTKS mediates apoptosis resistance under low-Mdm2
conditions (Fig. 2) (67). Furthermore, studies have suggested
the existence of an IRTKS-Mdm2-p53 tertiary complex, and
demonstrated that IRTKS overexpression promotes Mdm2-mediated p53
ubiquitination and degradation, suggesting a new cancer-promoting
mechanism of IRTKS (57,67). However, when DNA damage occurs,
IRTKS undergoes serine/threonine phosphorylation by checkpoint
kinase 2 (57). This induces the
dissociation of IRTKS from p53, and promotes the IRTKS-Mdm2
interaction (67). Subsequently,
the binding of p53 to Mdm2 is blocked, which induces IRTKS
ubiquitination and degradation but attenuates p53 ubiquitination
and degradation, ultimately increasing p53-initiated DNA repair or
apoptosis (57).
Notably, IRTKS overexpression has been shown to have
no effect on the expression of p53, the phosphorylation and
acetylation of certain amino acid residues of p53, and
Mdm2-mediated p53 neddylation (67).
IRTKS fusion genes and tumors
Chromosomal translocations/rearrangements leading to
kinase activation are important contributors to tumorigenesis, and
IRTKS has been reported to undergo fusion to certain oncogenes.
Fibroblast growth factor receptor 3 (FGFR3)
activation by mutation or overexpression is frequently detected in
cases of bladder cancer, and another mechanism of activation
involves chromosomal rearrangements that generate constitutively
activated fusion genes, such as the FGFR3-IRTKS fusion gene, which
has been identified by several research groups (70–72). In 293T
cells, the overexpression of IRTKS, as a fusion partner in
FGFR3-IRTKS fusions, introduces dimerization motifs that activate
FGFR fusion kinases (72). In
addition, the stable expression of FGFR3-IRTKS in telomerase
reverse transcriptase-expressing human mammary epithelial cell
lines increases downstream ERK1/2 and STAT1 phosphorylation and
promotes proliferation (72).
Another study identified the FGFR3-IRTKS fusion gene in patients
with bladder cancer and lung cancer, and demonstrated that
FGFR3-IRTKS has potent tumorigenic activity. Specifically, it
revealed that FGFR3-IRTKS not only activates growth signals. such
as those generated by the mitogen-activated protein kinase pathway.
but also inhibits tumor-suppressive signals, including those
generated by the p53, retinoblastoma 1 and cyclin dependent kinase
inhibitor 2A pathways (70).
Williams et al (71)
demonstrated a chromosomal translocation t(4;7) with breakpoints at
FGFR3 on chromosome 4 and IRTKS on chromosome 7 in SW780 bladder
cancer cells. All the aforementioned studies indicate that
FGFR3-IRTKS-positive cells are sensitive to selective FGFR
inhibitors (70–72). Therefore, the presence of a fusion gene may
aid in the selection of patients for FGFR-targeted therapy.
Several other types of IRTKS gene fusion have been
identified; for example, BRAF and IRTKS fusion has been detected by
RNA sequencing in spitzoid melanoma (73), and an oncogenic mesenchymal MET and
IRTKS fusion has been identified in papillary renal carcinoma,
which results in constitutive activation of the MET kinase
(74,75). Although these IRTKS-containing
fusion genes have been shown to be oncogenic, the biology of gene
fusion has not been characterized extensively, and responses to
single-agent or combination IRTKS-directed targeted therapy are
underexplored.
IRTKS and epigenetic
reprogramming
In 2023, Cui et al (76) revealed for the first time that
IRTKS can induce the deubiquitination of the histone
methyltransferase SET domain bifurcated histone lysine
methyltransferase 1 (SETDB1), thereby blocking proteasome-mediated
SETDB1 degradation and allowing SETDB1 to accumulate through the
recruitment of OUT deubiquitinase 4 (OTUD4). The upregulation of
SETDB1 subsequently increases histone H3 lysine 9 trimethylation
(H3K9me3), which leads to a reduction in chromatin accessibility.
Accordingly, chromatin inaccessibility represses the transcription
of numerous genes, including that encoding E-cadherin, which
results in epithelial-mesenchymal transition and the metastasis of
malignant cells (76). In
addition, elevated IRTKS/SETDB1 levels in clinical tumor specimens
have been found to be negatively associated with survival time
(76). These results advance our
understanding of the epigenetic reprogramming caused by IRTKS, and
suggest that the IRTKS-OTUD4-SETDB1-H3K9me3 axis may constitute a
new tumor therapeutic focus in the future.
Further findings from the same research team
revealed that IRTKS localizes not only to the cell membrane and
cytoplasm but also the nucleus under certain conditions. In
addition, a noncanonical role of IRTKS as a nuclear protein was
identified, in which it regulates heterochromatin formation via
liquid-liquid phase separation (77). The research also showed that IRTKS
recruits ubiquitin-conjugating enzyme E2, small ubiquitin modifier
9 (Ubc9) to sumoylate heterochromatin protein 1-α (HP1α), a
critical factor for heterochromatin formation and maintenance,
thereby stabilizing HP1α. Also, IRTKS can integrate into and
promote the phase separation of HP1α, facilitating heterochromatin
formation (77). By contrast, the
absence of IRTKS leads to heterochromatin loss, increased global
chromatin accessibility and the reactivation of repetitive DNA
elements, which contributes to the enrichment of gene sets
associated with cellular senescence and aging. The aforementioned
molecular events accelerate cellular senescence and activate the
cyclic GMP-AMP synthase-stimulator of interferon (IFN) genes
pathway to trigger the senescence-associated secretory phenotype
response (77). These discoveries
indicate that IRTKS functions as an epigenetic regulator to
stabilize heterochromatin architecture, which emphasizes the link
between heterochromatin loss and cellular senescence. However, the
underlying mechanism and whether IRTKS alleviates aging require
further investigation.
Based on the findings of the present review, IRTKS
amplifications and gene fusions have been recognized as drivers of
oncogenesis. This knowledge promotes our understanding of
carcinogenic mechanisms and may aid the identification of novel
treatment targets associated with IRTKS.
The tumorigenic role of upregulated circRNA_102231,
a noncoding RNA with a covalently closed ring structure, has been
revealed in GC tissue (78). In
addition, mechanistic analyses revealed that circRNA_102231 binds
to IRTKS, thereby increasing IRTKS protein stability and promoting
GC progression (78).
Stem cell therapy is currently being evaluated as a
promising approach for oncotherapy. A study in which human amniotic
mesenchymal stromal cells (hAMSCs) were cocultured with HT-29 colon
cancer cells revealed that the hAMSC secretome restrains the growth
and invasion of the HT-29 cells. This effect is mediated via the
downregulation of EGFR/c-Src/IRTKS expression, as well as the
reduced phosphorylation of p38/ERK1/2. Also, the secretome induces
the apoptosis of HT-29 cells, as evidenced by the upregulation of
Bax and downregulation of Bcl2 (66).
IRTKS and embryogenesis
Most studies on IRTKS function have focused on its
role in adult tissues and cells, and investigation into its
involvement in embryogenesis is limited. The embryos of IRSp53
knockout mice display pleiotropic phenotypes, including
developmental delay and oligodactyly, and do not survive due to
severely impaired cardiac and placental development (79). Although mice with IRTKS knockout
alone do not exhibit developmental and phenotypical abnormalities,
the concurrent deletion of IRSp53 and IRTKS results in exacerbated
placental abnormalities, particularly affecting spongiotrophoblast
differentiation and development, which results in an increased
embryonic lethality ratio (53,79).
Therefore, IRSp53 and IRTKS appear to have redundant genetic
interactions in placental formation, with IRTKS being essential for
proper placental development during embryogenesis (53,79).
IRTKS and antiviral immunity
IRTKS deficiency contributes to insulin resistance,
which may play a role in clinical infections and immune regulations
(77). In addition, an association
between IRTKS and inflammation has been implicated in the disease
progression of rheumatoid arthritis (RA), as evidenced by
correlations between IRTKS and C-reactive protein, a routinely
assessed marker of RA activity, in fibroblast-like synovial cells
obtained from patients with RA (80).
In 2015, Xia et al (81) identified IRTKS as an inhibitory
modulator of innate immune responses against RNA viruses. During
RNA virus invasion, viral RNA is recognized primarily by RIG-I-like
receptors (RLRs), a family of nucleic acid sensors that includes
retinoic acid-inducible gene I (RIG-I), melanoma
differentiation-associated protein 5 (MDA5) and laboratory of
genetics and physiology 2 (82).
RIG-I and MDA5 subsequently activate the mitochondrial adaptor
protein mitochondrial antiviral signaling protein (MAVS). These
events activate IFN regulatory factors and NF-κB, which in turn
stimulate the transcriptional induction of IFN-I/-III and other
proinflammatory cytokines, thereby facilitating the eradication of
RNA viruses (83).
Extracellular viral RNA triggers RLR activation,
whereas a number of regulatory mechanisms are in place to prevent
‘self-reactivity’ and maintain the immune homeostasis of host RNA.
The host RNA-binding protein poly(rC) binding protein 2 (PCBP2), a
negative regulator of MAVS, has been shown to be involved in host
cell mRNA stability (84). Xia
et al (81) confirmed that
IRTKS recruits Ubc9, an E2 ligase, to sumoylate PCBP2 at Lys37 in
the nucleus, which causes PCBP2 to translocate to the cytoplasm.
The cytoplasmic sumoylated PCBP2 initiates MAVS degradation,
leading to the downregulation of host immune responses. The study
demonstrated that IRTKS acts as an inhibitory regulator of the
RIG-I-MAVS signaling pathway, and its deficiency augments the
innate immune response of mice against RNA viruses, but not against
DNA viruses or bacteria (81).
Therefore, IRTKS appears to be a negative modulator
of antiviral immunity, which is crucial for balancing the
inflammation induced by viral infection and preventing cell damage
due to excessive inflammation.
IRTKS and hypothermia
Hypothermia, characterized by a core body
temperature <35°C, can be caused by excessive exposure to cold,
drug poisoning, and metabolic or nervous system dysfunction
(85). Although postmortem
biochemical explorations have revealed several biomarkers of
hypothermia, including catecholamines, cortisol, ketone bodies and
free fatty acids, death by hypothermia remains a diagnosis by
exclusion, as definitive and specific biomarkers are lacking
(85,86).
Exposure to cold triggers a series of stress
reactions in the body, including increased cortisol production and
lipolysis, which specifically promotes ketogenesis to maintain the
core temperature (85,87). Postmortem metabolomics has shown
potential in providing valuable insights and improving diagnostic
accuracy in cases of hypothermia. IRTKS-mediated insulin signaling
strongly affects glucose and lipid metabolism, suggesting that
IRTKS may be a good candidate as a forensic biomarker of
hypothermia. In a study of hypothermic mice continuously exposed to
cold water compared with mice maintained under standard conditions,
total of 4,094 differentially expressed genes were identified.
Among them, IRTKS was the most downregulated, suggesting that it is
a promising candidate biomarker of hypothermia (88).
As aforementioned, IRTKS deficiency contributes to
insulin resistance, which involves various metabolic reactions in
the body, including impaired glycolysis, increased lipolysis and
consequently ketogenesis, which are similar to those associated
with hypothermia induction and implies the relevance of IRTKS to
hypothermia. However, further studies are required to establish the
dependency relationships between IRTKS and hypothermia. In
addition, issues associated with postmortem stability, the use of
biological fluids, and the specificity and limitations of IRKS as a
novel forensic biomarker of hypothermia require further
investigation.
Conclusions and future prospects
IRTKS was initially identified as a substrate for
the IR. While studies have shown that IRTKS positively regulates
IR-IRS-PI3K-AKT signaling, little is known about the role of IRTKS
in downstream pathways, molecular events and clinical disease
manifestations.
Under physiological conditions, the IR-IRS-PI3K-AKT
signaling pathway is triggered by various factors, including growth
factors, cytokines and hormones, and mediates a number of cellular
processes, such as cell proliferation, cell cycle regulation and
metabolic homeostasis. Aberrant activation of the IR-IRS-PI3K-AKT
pathway is commonly associated with the occurrence of cancers,
metabolic disorders and autoimmune and inflammatory diseases, and
the progression of multiple malignancies (89,90).
Therapies that target specific cellular mechanisms, including
tyrosine kinase inhibitors (TKIs), have shown great potential in
the treatment of key human diseases such as hematological
malignancies, solid tumors and autoimmune disorders (91,92).
The tyrosine kinase family has emerged as one of the most important
drug targets in the 21st century, with >60 small-molecule
therapeutic drugs that target protein-tyrosine kinases being
approved by the US Food and Drug Administration in 2024, and TKIs
gaining prominence as effective pathway-directed agents (93).
Most small-molecule TKIs exert their effects by
targeting and binding to the enzymatic domain and competitively
blocking the ATP-binding pocket or inhibiting relevant signaling
cascades. However, due to the complex cross-connections and
compensatory mechanisms within signal transduction pathways, as
well as the dynamic evolution of genetic profiles, single-target
TKIs may not be clinically effective, and resistance to TKIs is
emerging (94,95). Although treatments with TKIs have
been demonstrated to prolong the overall survival of patients,
unsatisfactory treatment efficiency, drug resistance and side
effects limit their clinical application. Multikinase inhibitors
that target several protein-tyrosine kinases simultaneously, or the
combination of TKIs with other therapeutic approaches, such as
radiation and chemotherapy, show improved outcomes in disease
treatment (91). However,
multikinase inhibitors have both advantages and disadvantages. For
example, sunitinib and cabozantinib have potent off-target effects
on the Axl receptor protein tyrosine kinase, which can increase
their clinical efficacy (93,96).
However, the targeting of off-target kinases may also elicit
unwanted adverse effects, such as hypertension, heart failure, and
gastrointestinal and skin reactions (97). Accordingly, multikinase inhibition
appears to be ‘a double-edged sword’, which makes the choice of the
optimal treatment challenging (93).
Advancements in the understanding and monitoring of
protein-tyrosine kinases and their associated signaling cascades
could facilitate the development of new targeted therapies and
improved clinical outcomes. Gaining insights into the biological
and pathological functions of IRTKS may lead to the development of
new approaches for the treatment or prevention of cancers and other
diseases. Metabolic reprogramming is currently attracting
considerable attention in the study of tumor pathogenesis and
therapeutic development. Controlled by IRTKS, the conversion of
metabolic efficiency, its direction, and the expression and
activity of downstream effectors of IR-IRS-PI3K-AKT signaling,
particularly key enzymes and transporters involved in metabolism,
have emerged as promising areas for tumor research.
The role of IRTKS in various cell processes is of
increased interest to researchers. Evidence suggests that IRTKS
initiates and maintains membrane remodeling, induces the formation
of protrusions, triggers pathogen-driven actin assembly, positively
regulates insulin signaling, drives oncogenesis, participates in
embryonic development, and negatively modulates antiviral immunity.
The present review systematically summarizes advances in IRTKS
research, providing insights into its role in disease pathogenesis
and guiding the diagnosis and treatment of IRTKS-related diseases.
These findings have both basic and clinical implications, expanding
the range of therapeutic options. Understanding the regulatory
mechanisms and signaling pathways associated with IRTKS is likely
to remain an area of active research in the coming years.
Acknowledgements
Not applicable.
Funding
This study was supported by the Natural Science Foundation of
Gansu Province (grant no. 24JRRA417).
Availability of data and materials
Not applicable.
Authors' contributions
XZ contributed to acquisition, analysis,
interpretation of the literature and drafted the manuscript. ZZ
participated in revising the manuscript. Both authors read and
approved the final version of the manuscript. Data authentication
is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
A/E
|
attaching and effacing
|
|
BAIAP2
|
brain-specific angiogenesis inhibitor
1-associated protein 2
|
|
BAIAP2L1
|
BAIAP2-like 1
|
|
BAR
|
Bin-amphiphysin-Rvs
|
|
CRC
|
colorectal cancer
|
|
EHEC
|
Enterohemorrhagic E. coli
|
|
EGFR
|
epidermal growth factor receptor
|
|
Eps8
|
EGFR kinase substrate 8
|
|
EspFU
|
E. coli-secreted protein
F-like protein encoded on prophage U
|
|
ERK
|
extracellular regulated protein
kinase
|
|
F-actin
|
fibrous actin
|
|
F-BARs
|
Fes/CIP4 homology-BAR
|
|
FGFR
|
fibroblast growth factor receptor
|
|
FMNL2
|
formin-like 2
|
|
GBD
|
GTPase binding domain
|
|
GC
|
gastric cancer
|
|
GSK-3β
|
glycogen synthase kinase-3β
|
|
hAMSC
|
human amniotic mesenchymal stromal
cell
|
|
HCC
|
hepatocellular carcinoma
|
|
H3K9me3
|
histone H3 lysine 9
trimethylation
|
|
HP1α
|
heterochromatin protein 1-a
|
|
I-BARs
|
inverse-BAR
|
|
IFN
|
interferon
|
|
IMD
|
IRSp53/MIM homology domain
|
|
IR
|
insulin receptor
|
|
IRSp53
|
IR tyrosine kinase substrate p53
|
|
IRTKS
|
IR tyrosine kinase substrate
|
|
MAVS
|
mitochondrial antiviral signaling
protein
|
|
MDA5
|
melanoma differentiation-associated
protein 5
|
|
MIM
|
missing-in-metastasis
|
|
mTOR
|
mammalian target of rapamycin
|
|
N-WASP
|
neuronal WASP
|
|
OC
|
ovarian cancer
|
|
PC
|
pancreatic cancer
|
|
PCBP2
|
poly(rC) binding protein 2
|
|
PI3K
|
phosphatidylinositol 3-kinase
|
|
PIP3
|
phosphatidylinositol
3,4,5-triphosphate
|
|
RA
|
rheumatoid arthritis
|
|
RLR
|
RIG-I-like receptor
|
|
RIG-I
|
retinoic acid-inducible gene I
|
|
SH3
|
Src homology 3
|
|
SHIP2
|
SH2 containing inositol polyphosphate
5-phosphatase-2
|
|
Tir
|
translocated intimin receptor
|
|
VASP
|
vasodilator-stimulated
phosphoprotein
|
|
WASP
|
Wiskott-Aldrich syndrome protein
|
|
WAVE
|
WASP family verprolin-homologous
protein
|
|
WH2
|
WASP-homology 2
|
References
|
1
|
Millard TH, Dawson J and Machesky LM:
Characterisation of IRTKS, a novel IRSp53/MIM family actin
regulator with distinct filament bundling properties. J Cell Sci.
120:1663–1672. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Saarikangas J, Zhao HX, Pykäläinen A,
Laurinmäki P, Mattila PK, Kinnunen PK, Butcher SJ and Lappalainen
P: Molecular mechanisms of membrane deformation by I-BAR domain
proteins. Curr Biol. 19:95–107. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yamagishi A, Masuda M, Ohki T, Onishi H
and Mochizuki N: A novel actin bundling/filopodium-forming domain
conserved in insulin receptor tyrosine kinase substrate p53 and
missing in metastasis protein. J Biol Chem. 279:14929–14936. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fagerberg L, Hallstrom BM, Oksvold P,
Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S,
Danielsson A, Edlund K, et al: Analysis of the human
tissue-specific expression by genome-wide integration of
transcriptomics and antibody-based proteomics. Mol Cell Proteomics.
13:397–406. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hu RM, Han ZG, Song HD, Peng YD, Huang QH,
Ren SX, Gu YJ, Huang CH, Li YB, Jiang CL, et al: Gene expression
profiling in the human hypothalamus-pituitary-adrenal axis and
full-length cDNA cloning. Proc Natl Acad Sci USA. 97:9543–9548.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Peter BJ, Kent HM, Mills IG, Vallis Y,
Butler PJ, Evans PR and McMahon HT: BAR domains as sensors of
membrane curvature: The amphiphysin BAR structure. Science.
303:495–499. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhao HX, Pykäläinen A and Lappalainen P:
I-BAR domain proteins: Linking actin and plasma membrane dynamics.
Curr Opin Cell Biol. 23:14–21. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Frost A, Unger VM and De Camilli P: The
BAR domain superfamily: Membrane-molding macromolecules. Cell.
137:191–196. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Qualmann B, Koch D and Kessels MM: Let's
go bananas: Revisiting the endocytic BAR code. EMBO J.
30:3501–3515. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Suetsugu S, Toyooka K and Senju Y:
Subcellular membrane curvature mediated by the BAR domain
superfamily proteins. Semin Cell Dev Biol. 21:340–349. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mullins RD, Heuser JA and Pollard TD: The
interaction of Arp2/3 complex with actin: Nucleation, high affinity
pointed end capping, and formation of branching networks of
filaments. Proc Natl Acad Sci USA. 95:6181–6186. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bompard G and Caron E: Regulation of
WASP/WAVE proteins: Making a long story short. J Cell Biol.
166:957–962. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Machesky LM and Insall RH: Scar1 and the
related Wiskott-Aldrich syndrome protein, WASP, regulate the actin
cytoskeleton through the Arp2/3 complex. Curr Biol. 8:1347–1356.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pollard TD and Borisy GG: Cellular
motility driven by assembly and disassembly of actin filaments.
Cell. 112:453–465. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bear JE, Svitkina TM, Krause M, Schafer
DA, Loureiro JJ, Strasser GA, Maly IV, Chaga OY, Cooper JA, Borisy
GG and Gertler FB: Antagonism between Ena/VASP proteins and actin
filament capping regulates fibroblast motility. Cell. 109:509–521.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lebrand C, Dent EW, Strasser GA, Lanier
LM, Krause M, Svitkina TM, Borisy GG and Gertler FB: Critical role
of Ena/VASP proteins for filopodia formation in neurons and in
function downstream of netrin-1. Neuron. 42:37–49. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pellegrin S and Mellor H: The Rho family
GTPase Rif induces filopodia through mDia2. Curr Biol. 15:129–133.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Revenu C, Athman R, Robine S and Louvard
D: The co-workers of actin filaments: From cell structures to
signals. Nat Rev Mol Cell Biol. 5:635–646. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vignjevic D, Kojima S, Aratyn Y, Danciu O,
Svitkina T and Borisy GG: Role of fascin in filopodial protrusion.
J Cell Biol. 174:863–875. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Krugmann S, Jordens I, Gevaert K,
Driessens M, Vandekerckhove J and Hall A: Cdc42 induces filopodia
by promoting the formation of an IRSp53:Mena complex. Curr Biol.
11:1645–1655. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Disanza A, Mantoani S, Hertzog M, Gerboth
S, Frittoli E, Steffen A, Berhoerster K, Kreienkamp HJ, Milanesi F,
Di Fiore PP, et al: Regulation of cell shape by Cdc42 is mediated
by the synergic actin-bundling activity of the Eps8-IRSp53 complex.
Nat Cell Biol. 8:1337–1347. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Suetsugu S, Kurisu S, Oikawa T, Yamazaki
D, Oda A and Takenawa T: Optimization of WAVE2 complex-induced
actin polymerization by membrane-bound IRSp53, PIP(3), and Rac. J
Cell Biol. 173:571–585. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Goh WI, Lim KB, Sudhaharan T, Sem KP, Bu
W, Chou AM and Ahmed S: mDia1 and WAVE2 proteins interact directly
with IRSp53 in filopodia and are involved in filopodium formation.
J Biol Chem. 287:4702–4714. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Miki H, Yamaguchi H, Suetsugu S and
Takenawa T: IRSp53 is an essential intermediate between Rac and
WAVE in the regulation of membrane ruffling. Nature. 408:732–735.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fujiwara T, Mammoto A, Kim Y and Takai Y:
Rho small G-protein-dependent binding of mDia to an Src homology 3
domain-containing IRSp53/BAIAP2. Biochem Biophys Res Commun.
271:626–629. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chou AM, Sem KP, Wright GD, Sudhaharan T
and Ahmed S: Dynamin1 is a novel target for IRSp53 protein and
works with mammalian enabled (Mena) protein and Eps8 to regulate
filopodial dynamics. J Biol Chem. 289:24383–24396. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sudhaharan T, Sem KP, Liew HF, Yu YH, Goh
WI, Chou AM and Ahmed S: The Rho GTPase Rif signals through IRTKS,
Eps8 and WAVE2 to generate dorsal membrane ruffles and filopodia. J
Cell Sci. 129:2829–2840. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Crepin VF, Girard F, Schüller S, Phillips
AD, Mousnier A and Frankel G: Dissecting the role of the Tir:Nck
and Tir:IRTKS/IRSp53 signalling pathways in vivo. Mol Microbiol.
75:308–323. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li T, Cheng Z, Wang K, Chen F, Han Z and
Zhang X: Cloning and expressing of IRTKS and its effect on cell
morphology. J Med Mol Biol. 6:214–218. 2009.
|
|
30
|
Fox S, Tran A, Trinkle-Mulcahy L and
Copeland JW: Cooperative assembly of filopodia by the formin FMNL2
and I-BAR domain protein IRTKS. J Biol Chem. 298:1025122022.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tapon N and Hall A: Rho, Rac and Cdc42
GTPases regulate the organization of the actin cytoskeleton. Curr
Opin Cell Biol. 9:86–92. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hall A: Ras-related GTPases and the
cytoskeleton. Mol Biol Cell. 3:475–479. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Campellone KG, Brady MJ, Alamares JG, Rowe
DC, Skehan BM, Tipper DJ and Leong JM: Enterohaemorrhagic
Escherichia coli Tir requires a C-terminal 12-residue
peptide to initiate EspF-mediated actin assembly and harbours
N-terminal sequences that influence pedestal length. Cell
Microbiol. 8:1488–1503. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kühn S, Erdmann C, Kage F, Block J,
Schwenkmezger L, Steffen A, Rottner K and Geyer M: The structure of
FMNL2-Cdc42 yields insights into the mechanism of lamellipodia and
filopodia formation. Nat Commun. 6:70882015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Young LE, Heimsath EG and Higgs HN: Cell
type-dependent mechanisms for formin-mediated assembly of
filopodia. Mol Biol Cell. 26:4646–4659. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Veltman DM, Auciello G, Spence HJ,
Machesky LM, Rappoport JZ and Insall RH: Functional analysis of
Dictyostelium IBARa reveals a conserved role of the I-BAR domain in
endocytosis. Biochem J. 436:45–52. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li LS, Baxter SS, Zhao P, Gu N and Zhan X:
Differential interactions of missing in metastasis and insulin
receptor tyrosine kinase substrate with RAB proteins in the
endocytosis of CXCR4. J Biol Chem. 294:6494–6505. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Postema MM, Grega-Larson NE, Neininger AC
and Tyska MJ: IRTKS (BAIAP2L1) elongates epithelial microvilli
using EPS8-dependent and independent mechanisms. Curr Biol.
28:2876–2888.e4. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Meenderink LM, Gaeta IM, Postema MM,
Cencer CS, Chinowsky CR, Krystofiak ES, Millis BA and Tyska MJ:
Actin dynamics drive microvillar motility and clustering during
brush border assembly. Dev Cell. 50:545–556.e4. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gaeta IM, Meenderink LM, Postema MM,
Cencer CS and Tyska MJ: Direct visualization of epithelial
microvilli biogenesis. Curr Biol. 31:2561–2575.e6. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shifrin DA Jr, McConnell RE, Nambiar R,
Higginbotham JN, Coffey RJ and Tyska MJ: Enterocyte
microvillus-derived vesicles detoxify bacterial products and
regulate epithelial-microbial interactions. Curr Biol. 22:627–631.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Vallance BA, Chan C, Robertson ML and
Finlay BB: Enteropathogenic and enterohemorrhagic Escherichia
coli infections: Emerging themes in pathogenesis and
prevention. Can J Gastroenterol. 16:771–778. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kaper JB, Nataro JP and Mobley HL:
Pathogenic Escherichia coli. Nat Rev Microbiol. 2:123–140.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Campellone KG and Leong JM: Tails of two
Tirs: Actin pedestal formation by enteropathogenic E. coli
and enterohemorrhagic E. coli O157:H7. Curr Opin Microbiol.
6:82–90. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Allen-Vercoe E, Waddell B, Toh MC and
DeVinney R: Amino acid residues within enterohemorrhagic
Escherichia coli O157:H7 Tir involved in phosphorylation,
alpha-actinin recruitment, and Nck-independent pedestal formation.
Infect Immun. 74:6196–6205. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Brady MJ, Campellone KG, Ghildiyal M and
Leong JM: Enterohaemorrhagic and enteropathogenic Escherichia
coli Tir proteins trigger a common Nck-independent actin
assembly pathway. Cell Microbiol. 9:2242–2253. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Coburn B, Sekirov I and Finlay BB: Type
III secretion systems and disease. Clin Microbiol Rev. 20:535–549.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Loquet A, Sgourakis NG, Gupta R, Giller K,
Riedel D, Goosmann C, Griesinger C, Kolbe M, Baker D, Becker S and
Lange A: Atomic model of the type III secretion system needle.
Nature. 486:276–279. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Campellone KG: Cytoskeleton-modulating
effectors of enteropathogenic and enterohaemorrhagic Escherichia
coli: Tir, EspFU and actin pedestal assembly. FEBS J.
277:2390–2402. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Vingadassalom D, Kazlauskas A, Skehan B,
Cheng HC, Magoun L, Robbins D, Rosen MK, Saksela K and Leong JM:
Insulin receptor tyrosine kinase substrate links the E. coli
O157:H7 actin assembly effectors Tir and EspF(U) during pedestal
formation. Proc Natl Acad Sci USA. 106:6754–6759. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Aitio O, Hellman M, Kazlauskas A,
Vingadassalom DF, Leong JM, Saksela K and Permi P: Recognition of
tandem PxxP motifs as a unique Src homology 3-binding mode triggers
pathogen-driven actin assembly. Proc Natl Acad Sci USA.
107:21743–21748. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Aitio O, Hellman M, Skehan B, Kesti T,
Leong JM, Saksela K and Permi P: Enterohaemorrhagic Escherichia
coli exploits a tryptophan switch to hijack host f-actin
assembly. Structure. 20:1692–1703. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Huang LY, Wang YP, Wei BF, Yang J, Wang
JQ, Wu BH, Zhang ZZ, Hou YY, Sun WM, Hu RM, et al: Deficiency of
IRTKS as an adaptor of insulin receptor leads to insulin
resistance. Cell Res. 23:1310–1321. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wu CC, Cui XF, Huang LY, Shang X, Wu B,
Wang N, He K and Han Z: IRTKS promotes insulin signaling
transduction through inhibiting SHIP2 phosphatase activity. Int J
Mol Sci. 20:28342019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wang S, Liu Z, Ma YM, Guan X, Jiang Z, Sun
P, Liu ER, Zhang YK, Wang HY and Wang XS: Upregulated insulin
receptor tyrosine kinase substrate promotes the proliferation of
colorectal cancer cells via the bFGF/AKT signaling pathway.
Gastroenterol Rep (Oxf). 9:166–175. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chao A, Tsai CL, Jung SM, Chuang WC, Kao
C, Hsu A, Chen SH, Lin CY, Lee YC, Lee YS, et al: BAI1-associated
protein 2-like 1 (BAIAP2L1) is a potential biomarker in ovarian
cancer. PLoS One. 10:e01330812015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Huang LY, Wang XF, Cui XF, Li H, Zhao J,
Wu CC, Min L, Zhou Z, Wan L, Wang YP, et al: IRTKS is correlated
with progression and survival time of patients with gastric cancer.
Gut. 67:1400–1409. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wang YP, Huang LY, Sun WM, Zhang ZZ, Fang
JZ, Wei BF, Wu BH and Han ZG: Insulin receptor tyrosine kinase
substrate activates EGFR/ERK signalling pathway and promotes cell
proliferation of hepatocellular carcinoma. Cancer Lett. 337:96–106.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lu Y, Zhou XY, Zhou CL, Liu J, Yong T, Fan
Y and Wang C: Insulin receptor tyrosine kinase substrate (IRTKS)
promotes the tumorigenesis of pancreatic cancer via PI3K/AKT
signaling. Hum Cell. 35:1885–1899. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhang H, Ma H, Yang X, Fan L, Tian S, Niu
R, Yan M, Zheng M and Zhang S: Cell fusion-related proteins and
signaling pathways, and their roles in the development and
progression of cancer. Front Cell Dev Biol. 9:8096682022.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Oikawa T and Matsuo K: Possible role of
IRTKS in Tks5-driven osteoclast fusion. Commun Integr Biol.
5:511–515. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Li M, Brooks CL, Wu-Baer F, Chen D, Baer R
and Gu W: Mono-versus polyubiquitination: Differential control of
p53 fate by Mdm2. Science. 302:1972–1975. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Li LS, Liu HY, Baxter SS, Gu N, Ji M and
Zhan X: The SH3 domain distinguishes the role of I-BAR proteins
IRTKS and MIM in chemotactic response to serum. Biochem Biophys Res
Commun. 479:787–792. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Bromann PA, Korkaya H and Courtneidge SA:
The interplay between Src family kinases and receptor tyrosine
kinases. Oncogene. 23:7957–7968. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Chen G, Li T, Zhang L, Yi M, Chen F, Wang
Z and Zhang X: Src-stimulated IRTKS phosphorylation enhances cell
migration. FEBS Lett. 585:2972–2978. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Ebadi Zavieh S and Safari F: The antitumor
activity of hAMSCs secretome in HT-29 colon cancer cells through
downregulation of EGFR/c-Src/IRTKS expression and p38/ERK1/2
phosphorylation. Cell Biochem Biophys. 80:395–402. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Wang KS, Chen G, Shen HL, Li TT, Chen F,
Wang QW, Wang ZQ, Han ZG and Zhang X: Insulin receptor tyrosine
kinase substrate enhances low levels of MDM2-mediated p53
ubiquitination. PLoS One. 6:e235712011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
D'Amours D, Sallmann FR, Dixit VM and
Poirier GG: Gain-of-function of poly(ADP-ribose) polymerase-1 upon
cleavage by apoptotic proteases: Implications for apoptosis. J Cell
Sci. 114:3771–3778. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Hickman ES, Moroni MC and Helin K: The
role of p53 and pRB in apoptosis and cancer. Curr Opin Genet Dev.
12:60–66. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Nakanishi Y, Akiyama N, Tsukaguchi T,
Fujii T, Satoh Y, Ishii N and Aoki M: Mechanism of oncogenic signal
activation by the novel fusion kinase FGFR3-BAIAP2L1. Mol Cancer
Ther. 14:704–712. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Williams SV, Hurst CD and Knowles MA:
Oncogenic FGFR3 gene fusions in bladder cancer. Hum Mol Genet.
22:795–803. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Wu YM, Su F, Kalyana-Sundaram S, Khazanov
N, Ateeq B, Cao X, Lonigro RJ, Vats P, Wang R, Lin SF, et al:
Identification of targetable FGFR gene fusions in diverse cancers.
Cancer Discov. 3:636–647. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Wu G, Barnhill RL, Lee S, Li Y, Shao Y,
Easton J, Dalton J, Zhang J, Pappo A and Bahrami A: The landscape
of fusion transcripts in spitzoid melanoma and biologically
indeterminate spitzoid tumors by RNA sequencing. Mod Pathol.
29:359–369. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Guo R, Luo J, Chang J, Rekhtman N, Arcila
M and Drilon A: MET-dependent solid tumours-molecular diagnosis and
targeted therapy. Nat Rev Clin Oncol. 17:569–587. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Stransky N, Cerami E, Schalm S, Kim JL and
Lengauer C: The landscape of kinase fusions in cancer. Nat Commun.
5:48462014. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Cui XF, Shang XY, Xie J, Xie C, Tang Z,
Luo Q, Wu C, Wang G, Wang N, He K, et al: Cooperation between IRTKS
and deubiquitinase OTUD4 enhances the SETDB1-mediated H3K9
trimethylation that promotes tumor metastasis via suppressing
E-cadherin expression. Cancer Lett. 575:2164042023. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Xie J, Lu ZN, Bai SH, Cui XF, Lian HY, Xie
CY, Wang N, Wang L and Han ZG: Heterochromatin formation and
remodeling by IRTKS condensates counteract cellular senescence.
EMBO J. 43:4542–4577. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Yuan GF, Ding WW, Sun BJ, Zhu L, Gao YW
and Chen LL: Upregulated circRNA_102231 promotes gastric cancer
progression and its clinical significance. Bioengineered.
12:4936–4945. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Chou AM, Sem KP, Lam WJ, Ahmed S and Lim
CY: Redundant functions of I-BAR family members, IRSp53 and IRTKS,
are essential for embryonic development. Sci Rep. 7:404852017.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Galligan CL, Baig E, Bykerk V, Keystone EC
and Fish EN: Distinctive gene expression signatures in rheumatoid
arthritis synovial tissue fibroblast cells: Correlates with disease
activity. Genes Immun. 8:480–491. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Xia P, Wang S, Xiong Z, Ye B, Huang LY,
Han ZG and Fan Z: IRTKS negatively regulates antiviral immunity
through PCBP2 sumoylation-mediated MAVS degradation. Nat Commun.
6:81322015. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Rehwinkel J and Gack MU: RIG-I-like
receptors: Their regulation and roles in RNA sensing. Nat Rev
Immunol. 20:537–551. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Naesens L, Haerynck F and Gack MU: The RNA
polymerase III-RIG-I axis in antiviral immunity and inflammation.
Trends Immunol. 44:435–449. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Makeyev AV and Liebhaber SA: The
poly(C)-binding proteins: a multiplicity of functions and a search
for mechanisms. RNA. 8:265–278. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Rousseau G, Reynier P, Jousset N,
Rougé-Maillart C and Palmiere C: Updated review of postmortem
biochemical exploration of hypothermia with a presentation of
standard strategy of sampling and analyses. Clin Chem Lab Med.
56:1819–1827. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Elmsjö A, Ward LJ, Horioka K, Watanabe S,
Kugelberg FC, Druid H and Green H: Biomarker patterns and
mechanistic insights into hypothermia from a postmortem
metabolomics investigation. Sci Rep. 14:189722024. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Bańka K, Teresiński G and Buszewicz G:
Free fatty acids as markers of death from hypothermia. Forensic Sci
Int. 234:79–85. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Takamiya M, Saigusa K and Dewa K: DNA
microarray analysis of hypothermia-exposed murine lungs for
identification of forensic biomarkers. Leg Med (Tokyo).
48:1017892021. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Peng Y, Wang Y, Zhou C, Mei W and Zeng C:
PI3K/Akt/mTOR pathway and its role in cancer therapeutics: Are we
making headway? Front Oncol. 12:8191282022. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Ramasubbu K and Devi Rajeswari V:
Impairment of insulin signaling pathway PI3K/Akt/mTOR and insulin
resistance induced AGEs on diabetes mellitus and neurodegenerative
diseases: A perspective review. Mol Cell Biochem. 478:1307–1324.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Ebrahimi N, Fardi E, Ghaderi H, Palizdar
S, Khorram R, Vafadar R, Ghanaatian M, Rezaei-Tazangi F, Baziyar P,
Ahmadi A, et al: Receptor tyrosine kinase inhibitors in cancer.
Cell Mol Life Sci. 80:1042023. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Fares A, Carracedo Uribe C, Martinez D,
Rehman T, Silva Rondon C and Sandoval-Sus J: Bruton's tyrosine
kinase inhibitors: Recent updates. Int J Mol Sci. 25:22082024.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Roskoski R Jr: Properties of FDA-approved
small molecule protein kinase inhibitors: A 2024 update. Pharmacol
Res. 200:1070592024. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Nammour HM, Madrigal K, Starling CT and
Doan HQ: Advancing treatment options for merkel cell carcinoma: A
review of tumor-targeted therapies. Int J Mol Sci. 25:110552024.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Bae WH, Maraka S and Daher A: Challenges
and advances in glioblastoma targeted therapy: The promise of drug
repurposing and biomarker exploration. Front Oncol. 14:14414602024.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Myers SH, Brunton VG and Unciti-Broceta A:
AXL inhibitors in cancer: A medicinal chemistry perspective. J Med
Chem. 59:3593–3608. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Shyam Sunder S, Sharma UC and Pokharel S:
Adverse effects of tyrosine kinase inhibitors in cancer therapy:
Pathophysiology, mechanisms and clinical management. Signal
Transduct Target Ther. 8:2622023. View Article : Google Scholar : PubMed/NCBI
|














